
Characterization of Aurintricarboxylic Acid (ATA) Interactions with Plasma Transporter Protein and SARS-CoV-2 Viral Targets: Correlation of Functional Activity and Binding Energetics

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Experimental Strategy
2.2. Biophysical Properties and Binding Energetics
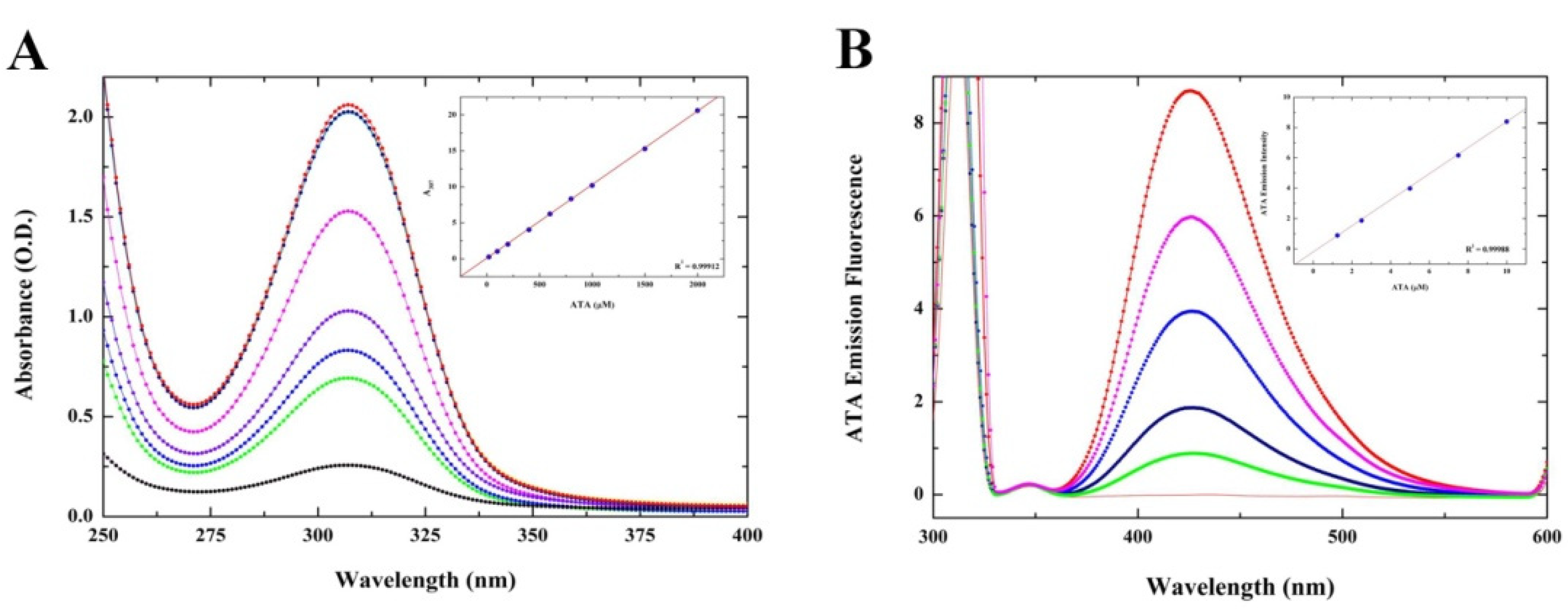
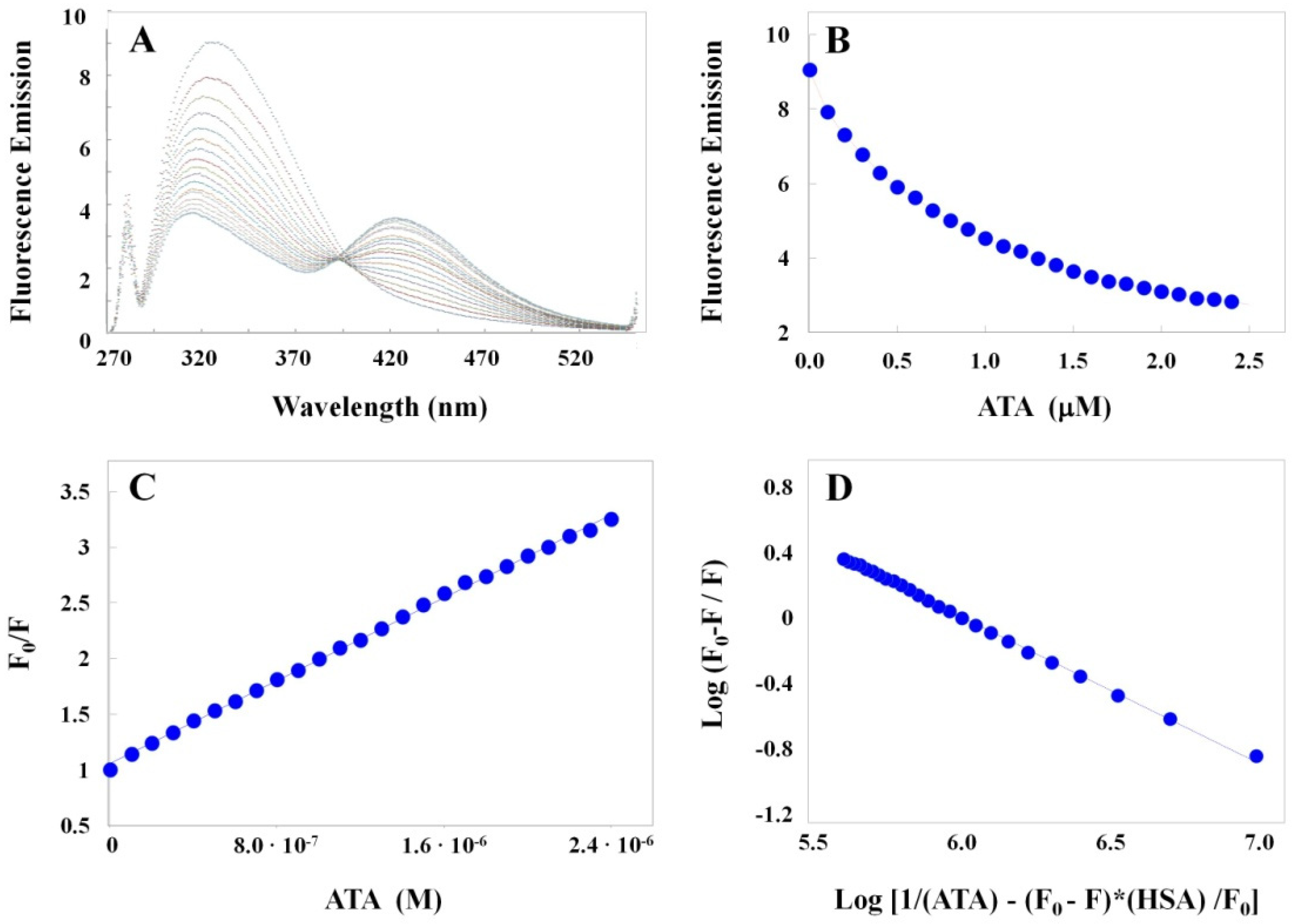
2.2.1. Optical Characterization of ATA–HSA Interactions: Binding Is Accompanied by Fluorescence Quenching and Energy Transfer
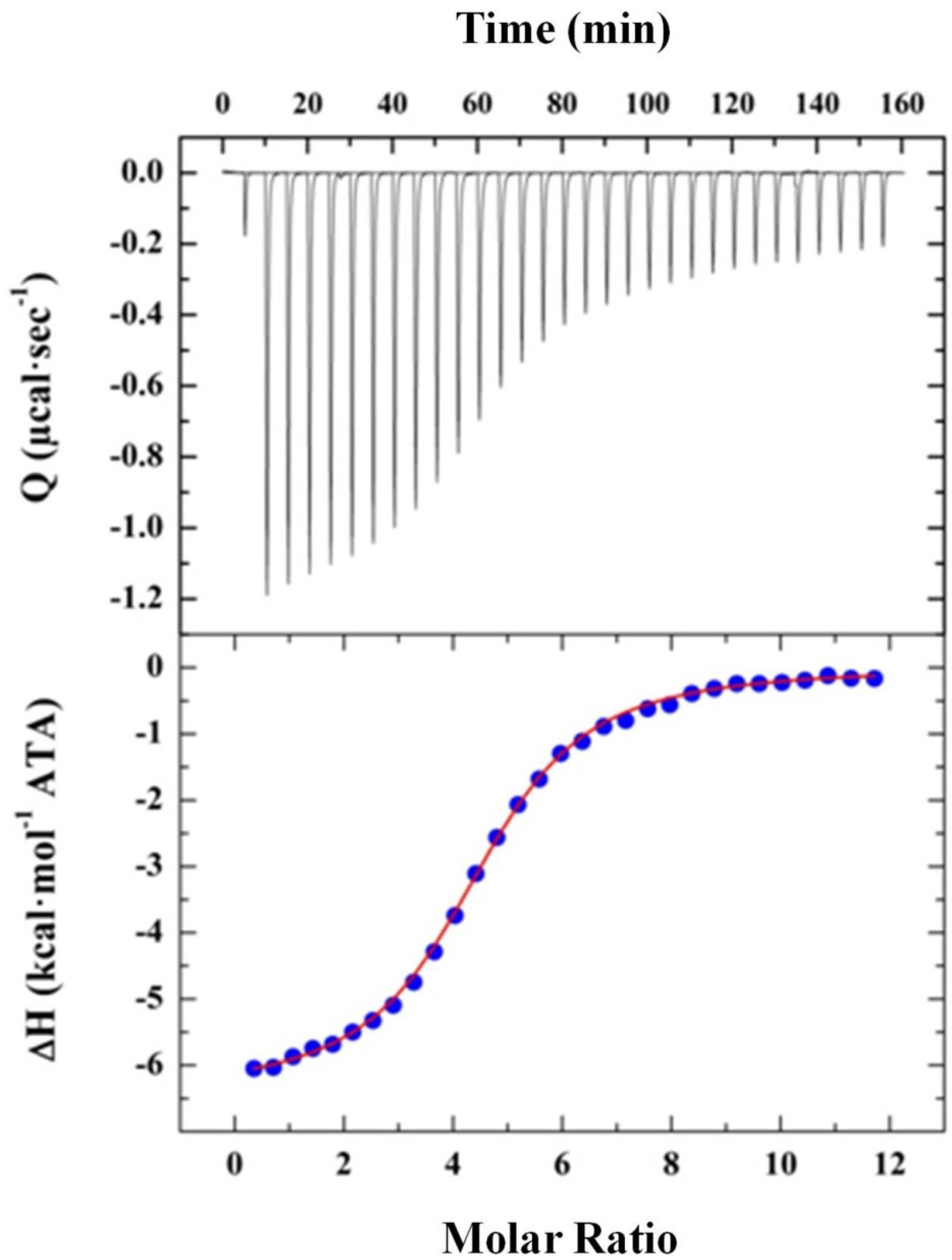
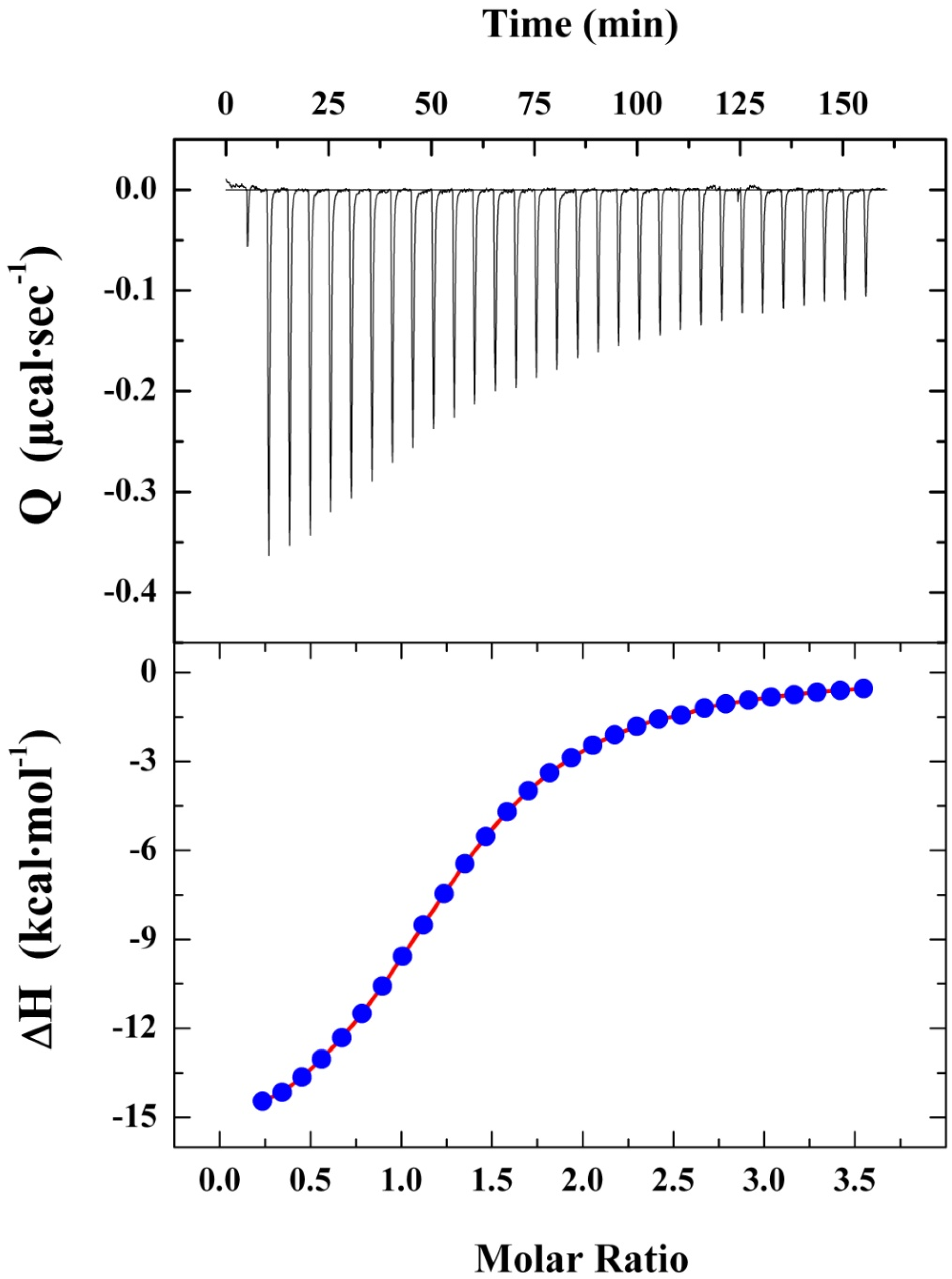
2.2.2. Elucidating ATA–HSA Binding Energetics via ITC: Protein-Ligand Complex Formation Is Enthalpy-Driven with Favorable Entropic Contributions
2.2.3. Characterizing HSA Binding of a Dichlorohexamer ATA Analog via ITC: Protein-Ligand Complex Formation Is Enthalpy-Driven with Unfavorable Entropic Contributions
2.2.4. Interaction of ATA-Derivatives with Host and/or Viral SARS-CoV-2 Targets: A Synthetic Chlorinated Hexamer Binds Ribosomal Particles with Moderate Affinity
2.3. Biological Properties and Functional Activity
2.3.1. Heterogeneous ATA and a Synthetic Chlorinated Hexamer Inhibit Protein Synthesis in Rabbit Reticulocyte Lysates
2.3.2. Heterogeneous ATA and Synthetic Chlorinated Hexamer Inhibit SARS-CoV-2 RNA Dependent RNA Polymerase
2.3.3. Heterogeneous ATA Is Non-Toxic and Exhibits Limited Activity as a Protein Synthesis Inhibitor in Cultured Vero E6 Cells
3. Discussion
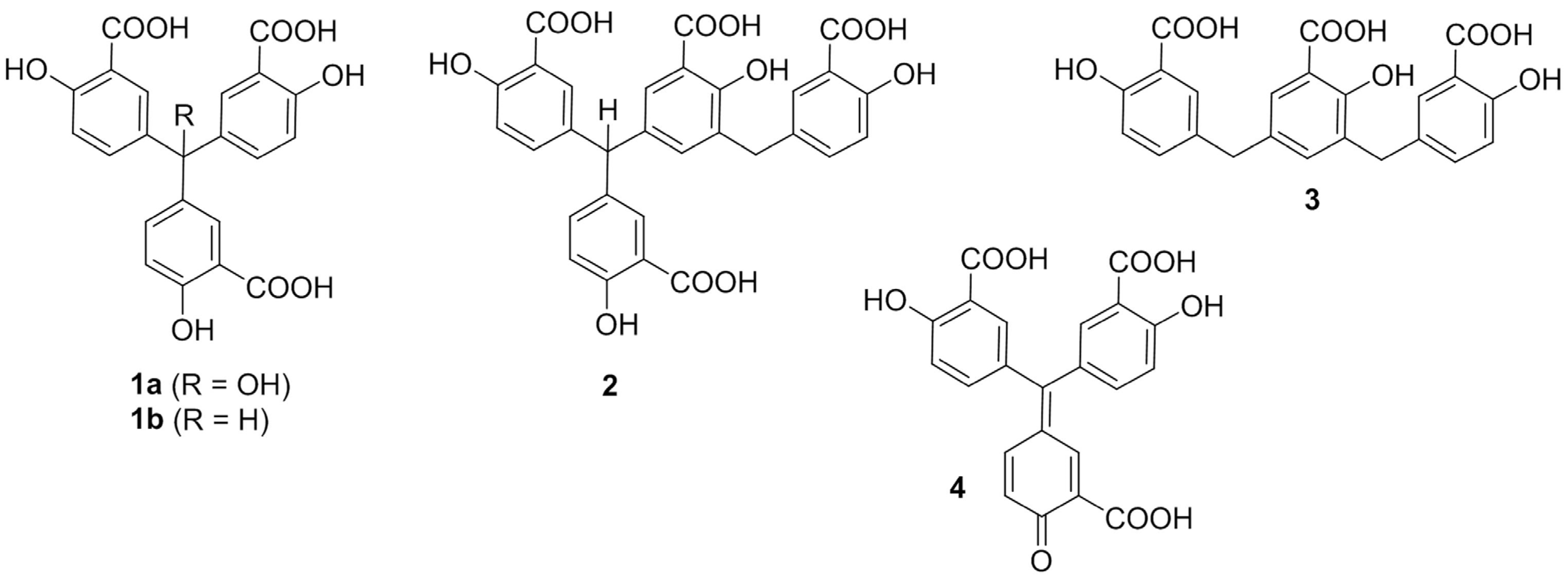
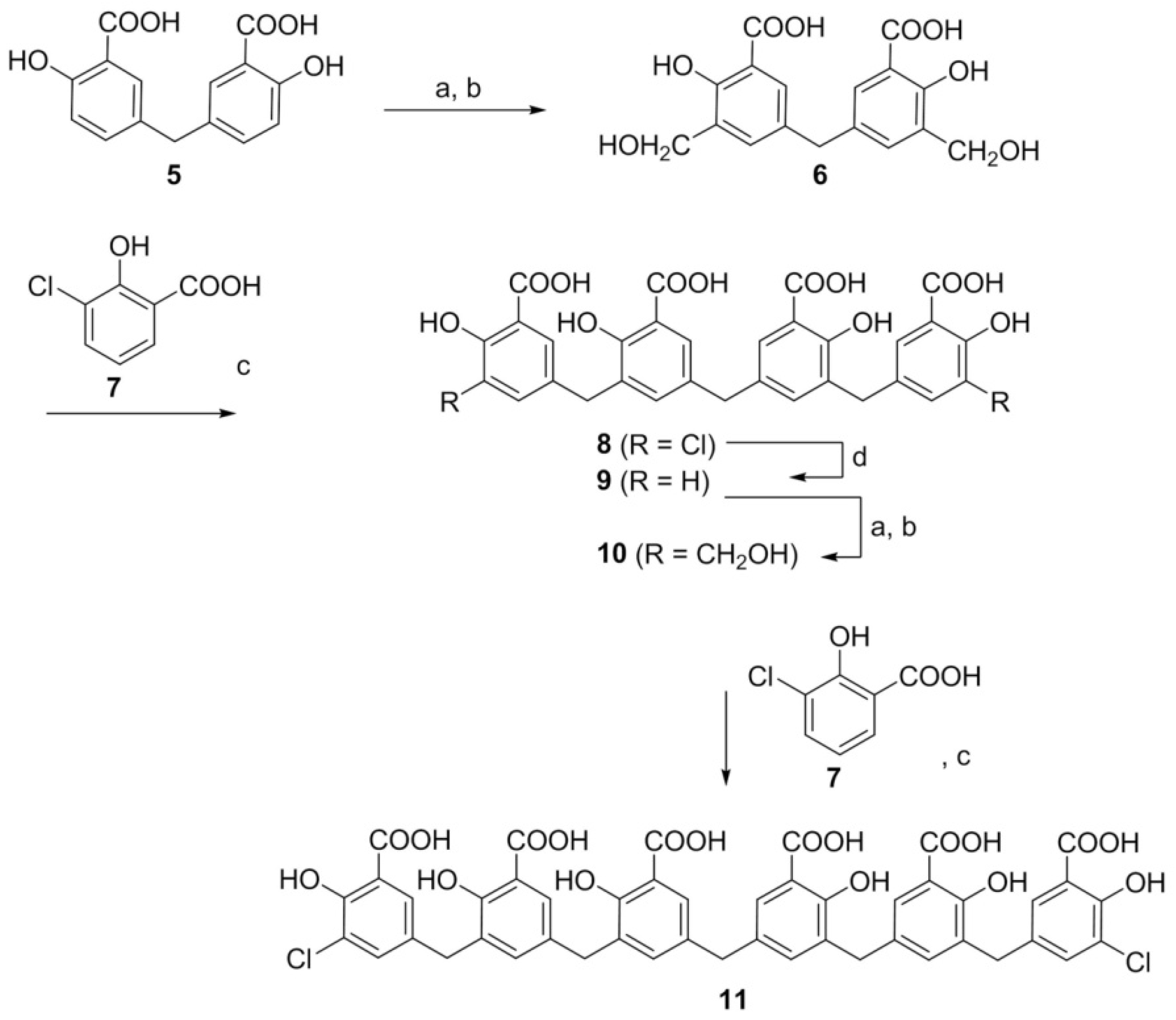
3.1. Biophysical Studies Assist Synthetic Efforts in Selecting a Representative Oligomeric ATA Analog
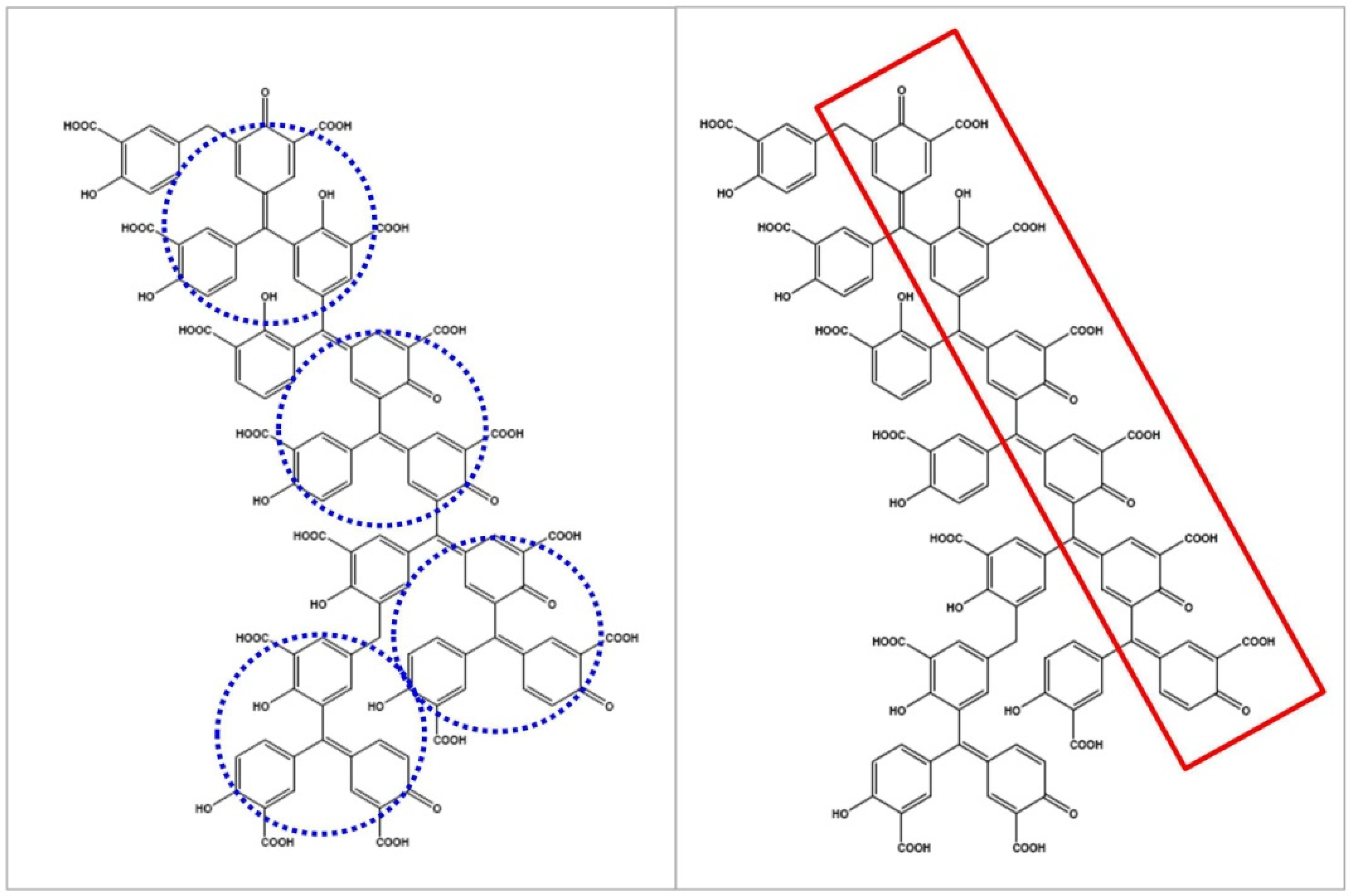
3.2. Characterization of ATA Intrinsic Biophysical Properties
3.3. Binding Profiles of Polymeric ATA versus the Synthetic Dichlorohexamer
3.4. ATA Derivatives Bind and Inhibit SARS-CoV-2 Targets
3.5. ATA Derivatives Bind Host Ribosomes and Inhibit Protein Synthesis Initiation
3.6. ATA Derivatives Bind and Inhibit SARS-CoV-2 RdRp
3.7. ATA Analog Bioactive Conformation as RdRp Inhibitor
3.8. ATA and Derivatives in the Cellular Context: Permeability and Toxicity
3.9. Structure-Activity Considerations regarding ATA Derived Lead Compounds
4. Concluding Remarks
5. Materials and Methods
5.1. Materials
5.2. UV/Vis Absorbance and Fluorescence Spectroscopy
5.2.1. Characterization of HSA and ATA Optical Properties
5.2.2. Characterization of HSA and ATA Fluorescence Properties
5.2.3. Characterization of ATA–HSA Binding via Fluorescence Quenching
5.2.4. Characterization of Ligand–Ribosome Binding via Fluorescence Anisotropy
5.3. Characterization of Binding Energetics via Isothermal Titration Calorimetry
5.4. Biochemical and Biological Assays
5.4.1. Inhibition of SARS-CoV-2 RNA Dependent RNA Polymerase Active Complex
5.4.2. Inhibition of Protein Synthesis in Rabbit Reticulocyte Lysates
5.4.3. Cell Culture
5.4.4. Protein Synthesis in Cultured Cells: Puromycin Pulse Labeling
5.4.5. Protein Preparation and Immunoblotting to Detect Puromycilated Proteins
5.4.6. Evaluation of Growth Inhibition and Toxicity in Vero E6 Cells
5.4.7. Isolation of 80S Yeast Ribosomes
5.5. Synthesis of Defined ATA-Derived Analogs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Vesnaver, G.; Chang, C.N.; Eisenberg, M.; Grollman, A.P.; Breslauer, K.J. Influence of Abasic and Anucleosidic Sites on the Stability, Conformation, and Melting Behavior of a DNA Duplex—Correlations of Thermodynamic and Structural Data. Proc. Natl. Acad. Sci. USA 1989, 86, 3614–3618. [Google Scholar] [CrossRef] [Green Version]
- Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. Influence of the oxidatively damaged adduct 8-oxodeoxyguanosine on the conformation, energetics, and thermodynamic stability of a DNA duplex. Biochemistry 1995, 34, 16148–16160. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, C.A.; Plum, G.E.; Grollman, A.P.; Johnson, F.; Breslauer, K.J. The impact of an exocyclic cytosine adduct on DNA duplex properties: Significant thermodynamic consequences despite modest lesion-induced structural alterations. Biochemistry 1998, 37, 12507–12512. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, C.A.; Plum, G.E.; Mielewczyk, S.; Remeta, D.P.; Breslauer, K.J. A quantitative method for evaluating the stabilities of nucleic acids. Proc. Natl. Acad. Sci. USA 1999, 96, 6113–6118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minetti, C.A.S.A.; Remeta, D.P.; Dickstein, R.; Breslauer, K.J. Energetic signatures of single base bulges: Thermodynamic consequences and biological implications. Nucleic Acids Res. 2010, 38, 97–116. [Google Scholar] [CrossRef] [Green Version]
- Minetti, C.A.S.A.; Remeta, D.P.; Johnson, F.; Iden, C.R.; Breslauer, K.J. Impact of alpha-Hydroxy-Propanodeoxyguanine Adducts on DNA Duplex Energetics: Opposite Base Modulation and Implications for Mutagenicity and Genotoxicity. Biopolymers 2010, 93, 370–382. [Google Scholar]
- Lukin, M.; Minetti, C.A.S.A.; Remeta, D.P.; Attaluri, S.; Johnson, F.; Breslauer, K.J.; de los Santos, C. Novel post-synthetic generation, isomeric resolution, and characterization of Fapy-dG within oligodeoxynucleotides: Differential anomeric impacts on DNA duplex properties. Nucleic Acids Res. 2011, 39, 5776–5789. [Google Scholar] [CrossRef] [Green Version]
- Minetti, C.A.S.A.; Remeta, D.P.; Iden, C.R.; Johnson, F.; Grollman, A.P.; Breslauer, K.J. Impact of thymine glycol damage on DNA duplex energetics: Correlations with lesion-induced biochemical and structural consequences. Biopolymers 2015, 103, 491–508. [Google Scholar] [CrossRef]
- Minetti, C.A.; Sun, J.Y.; Jacobs, D.P.; Kang, I.; Remeta, D.P.; Breslauer, K.J. Impact of bistrand abasic sites and proximate orientation on DNA global structure and duplex energetics. Biopolymers 2018, 109, e23098. [Google Scholar] [CrossRef] [Green Version]
- Minetti, C.A.S.A.; Remeta, D.P.; Zharkov, D.O.; Plum, G.E.; Johnson, F.; Grollman, A.P.; Breslauer, K.J. Energetics of lesion recognition by a DNA repair protein: Thermodynamic characterization of formamidopyrimidine-glycosylase (Fpg) interactions with damaged DNA duplexes. J. Mol. Biol. 2003, 328, 1047–1060. [Google Scholar] [CrossRef]
- Minetti, C.A.S.A.; Remeta, D.P.; Miller, H.; Gelfand, C.A.; Plum, G.E.; Grollman, A.P.; Breslauer, K.J. The thermodynamics of template-directed DNA synthesis: Base insertion and extension enthalpies. Proc. Natl. Acad. Sci. USA 2003, 100, 14719–14724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minetti, C.A.S.A.; Remeta, D.P.; Breslauer, K.J. A continuous hyperchromicity assay to characterize the kinetics and thermodynamics of DNA lesion recognition and base excision. Proc. Natl. Acad. Sci. USA 2008, 105, 70–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslauer, K.J. The shaping of a molecular linguist: How a career studying DNA energetics revealed the language of molecular communication. J. Biol. Chem. 2021, 296, 100522. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The next ten stories on antiviral drug discovery (part E): Advents, advances, and adventures. Med. Res. Rev. 2011, 31, 118–160. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Potential antivirals and antiviral strategies against SARS coronavirus infections. Expert Rev. Anti. Infect. Ther. 2006, 4, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Caro, N. Über Oxyaurin und Oxyaurincarbonsäuren (About oxyaurine and oxyaurine carboxylic acids). Ber. Dtsch. Chem. Ges. 1892, 25, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Grollman, A.P. Emetine: New Uses for an Old Drug. Ohio State Med. J. 1970, 66, 257–259. [Google Scholar]
- Grollman, A.P. Structural Basis for Inhibition of Protein Synthesis by Emetine and Cycloheximide Based on an Analogy between Ipecac Alkaloids and Glutarimide Antibiotics. Proc. Natl. Acad. Sci. USA 1966, 56, 1867–1874. [Google Scholar] [CrossRef] [Green Version]
- Grollman, A.P. Inhibitors of Protein Biosynthesis.V. Effects of Emetine on Protein and Nucleic Acid Biosynthesis in Hela Cells. J. Biol. Chem. 1968, 243, 4089–4094. [Google Scholar] [CrossRef]
- Grollman, A.P.; Stewart, M.L. Inhibition of Attachment of Messenger Ribonucleic Acid to Ribosomes. Proc. Natl. Acad. Sci. USA 1968, 61, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.L.; Grollman, A.P.; Huang, M.T. Aurintricarboxylic acid: Inhibitor of initiation of protein synthesis. Proc. Natl. Acad. Sci. USA 1971, 68, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grollman, A.P.; Horwitz, S.B. Rational Design of Antiviral Agents. In Molecular Pharmacology—Drug Design; Ariens, E.F.E., Ed.; Academic Press: Cambridge, MA, USA, 1971; pp. 261–276. [Google Scholar]
- Huang, M.; Grollman, A.P. Effects of Aurintricarboxylic Acid on Ribosomes and Biosynthesis of Globin in Rabbit Reticulocytes. Mol. Pharmacol. 1972, 8, 111–127. [Google Scholar] [PubMed]
- Grollman, A.P.; Jarkovsky, Z. Emetine and Related Alkaloides. In Mechansim of Action of Antimicrobial and Antitumor Agents Antibiotics; Springer: Berlin/Heidelberg, Germany, 1975; pp. 420–435. [Google Scholar]
- Liao, L.L.; Horwitz, S.B.; Huang, M.T.; Grollman, A.P.; Steward, D.; Martin, J. Triphenylmethane Dyes as Inhibitors of Reverse-Transcriptase, Ribonucleic-Acid Polymerase, and Protein-Synthesis-Structure-Activity-Relationships. J. Med. Chem. 1975, 18, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Steward, D.L.; Martin, J.; Grollman, A.P. Inhibition of influenza virus by triphenylmethane compounds. Ann. N. Y. Acad. Sci. 1977, 284, 638–649. [Google Scholar] [CrossRef]
- Grollman, A.P. Mechanism of Action of Emetine-Demonstration of Its Inhibitory Action on Protein Synthesis. J. Clin. Investig. 1966, 45, 1018. [Google Scholar]
- Blumenthal, T.; Landers, T.A. The inhibition of nucleic acid-binding proteins by aurintricarboxylic acid. Biochem. Biophys. Res. Commun. 1973, 55, 680–688. [Google Scholar] [CrossRef]
- Givens, J.F.; Manly, K.F. Inhibition of RNA-directed DNA polymerase by aurintricarboxylic acid. Nucleic Acids. Res. 1976, 3, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, S.; Kuwano, M.; Yamamoto, M.; Endo, H. Effect of Aurintricarboxylic Acid on RNA-Polymerase from Rat-Liver. J. Biochem. 1977, 81, 135–141. [Google Scholar] [CrossRef]
- Grollman, A. Inhibition of messenger ribonucleic acid attachment to ribosomes. II. Proposed mechanisms for the design of novel antiviral agents. Antimicrob. Agents Chemother. 1968, 8, 36. [Google Scholar]
- Polatnick, J.; Bachrach, H.L. Effect of zinc and other chemical agents on foot-and-mouth-disease virus replication. Antimicrob. Agents Chemother. 1978, 13, 731–734. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Adonov, A.; Traykova-Adonova, M.; Cao, J.; Cutts, T.; Grudesky, E.; Deschambaul, Y.; Berry, J.; Drebot, M.; Li, X. Potent and selective inhibition of SARS coronavirus replication by aurintricarboxylic acid. Biochem. Biophys. Res. Commun. 2004, 320, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Ben David, A.; Diamant, E.; Dor, E.; Barnea, A.; Natan, N.; Levin, L.; Chapman, S.; Mimran, L.C.; Epstein, E.; Zichel, R.; et al. Identification of SARS-CoV-2 Receptor Binding Inhibitors by In Vitro Screening of Drug Libraries. Molecules 2021, 26, 3213. [Google Scholar] [CrossRef] [PubMed]
- Cushman, M.; Wang, P.L.; Stowell, J.G.; Schols, D.; De Clercq, E. Structural Investigation and Anti-Hiv Activities of High-Molecular-Weight ATA Polymers. J. Org. Chem. 1992, 57, 7241–7248. [Google Scholar] [CrossRef]
- Gonzalez, R.G.; Blackburn, B.J.; Schleich, T. Fractionation and Structural Elucidation of the Active Components of Aurintricarboxylic Acid, a Potent Inhibitor of Protein Nucleic-Acid Interactions. Biochim. Biophys. Acta 1979, 562, 534–545. [Google Scholar] [CrossRef]
- Gonzalez, R.G.; Haxo, R.S.; Schleich, T. Mechanism of action of polymeric aurintricarboxylic acid, a potent inhibitor of protein-nucleic acid interactions. Biochemistry 1980, 19, 4299–4303. [Google Scholar] [CrossRef]
- Cushman, M.; Kanamathareddy, S. Synthesis of the Covalent Hydrate of the Incorrectly Assumed Structure of Aurintricarboxylic Acid (Ata). Tetrahedron 1990, 46, 1491–1498. [Google Scholar] [CrossRef]
- Cushman, M.; Kanamathareddy, S.; De Clercq, E.; Schols, D.; Goldman, M.E.; Bowen, J.A. Synthesis and anti-HIV activities of low molecular weight aurintricarboxylic acid fragments and related compounds. J. Med. Chem. 1991, 34, 337–342. [Google Scholar] [CrossRef]
- Cushman, M.; Wang, P.L.; Chang, S.H.; Wild, C.; De Clercq, E.; Schols, D.; Goldman, M.E.; Bowen, J.A. Preparation and anti-HIV activities of aurintricarboxylic acid fractions and analogues: Direct correlation of antiviral potency with molecular weight. J. Med. Chem. 1991, 34, 329–337. [Google Scholar] [CrossRef]
- Tsutsui, K.; Seki, S.; Tsutsui, K.; Oda, T. Fractionation of Aurintricarboxylic Acid and Effects of Its Components on Nuclear Swelling and Nucleic-Acid Synthesis. Biochim. Biophys. Acta 1978, 517, 14–23. [Google Scholar] [CrossRef]
- Cushman, M.; Kanamathareddy, S. Synthesis and Evaluation of a Triphenylcarbinol Related to the Incorrectly Assumed Structure of Aurintricarboxylic Acid. Ann. N. Y. Acad. Sci. 1990, 616, 499–502. [Google Scholar] [CrossRef]
- Wang, P.; Kozlowski, J.; Cushman, M. Isolation and Structure Elucidation of Low-Molecular-Weight Components of Aurintricarboxylic Acid (Ata). J. Org. Chem. 1992, 57, 3861–3866. [Google Scholar] [CrossRef]
- Smith, T.J. Aurintricarboxylic Acid-Derived Polysalicylates as Platforms for Drug Development: A Mini-Review. IOSR J. Pharm. Biol. Sci. 2018, 13, 44–47. [Google Scholar]
- Zhang, Q.B.; Qian, M.X.; Wu, Y.; Wang, Y.P.; Shangguan, W.W.; Lu, J.G.; Zhao, W.J.; Feng, J. Design and biological evaluation of novel long-acting adalimumab Fab conjugated with the albumin binding domain. Eur. J. Pharmacol. 2021, 904, 174152. [Google Scholar] [CrossRef] [PubMed]
- Tayyab, S.; Feroz, S.R. Serum albumin: Clinical significance of drug binding and development as drug delivery vehicle. Adv. Protein Chem. Struct. Biol. 2021, 123, 193–218. [Google Scholar] [PubMed]
- Gao, Y.; Kuang, Y.; Du, X.W.; Zhou, J.; Chandran, P.; Horkay, F.; Xu, B. Imaging Self-Assembly Dependent Spatial Distribution of Small Molecules in a Cellular Environment. Langmuir 2013, 29, 15191–15200. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M-pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Liang, F.B.; Huang, Z.H.; Lee, S.Y.; Liang, J.; Ivanov, M.I.; Alonso, A.; Bliska, J.B.; Lawrence, D.S.; Mustelin, T.; Zhang, Z.Y. Aurintricarboxylic acid blocks in vitro and in vivo activity of YopH, an essential virulent factor of Yersinia pestis, the agent of plague. J. Biol. Chem. 2003, 278, 41734–41741. [Google Scholar] [CrossRef] [Green Version]
- Balzarini, J.; Mitsuya, H.; De Clercq, E.; Broder, S. Aurintricarboxylic acid and Evans Blue represent two different classes of anionic compounds which selectively inhibit the cytopathogenicity of human T-cell lymphotropic virus type III/lymphadenopathy-associated virus. Biochem. Biophys. Res. Commun. 1986, 136, 64–71. [Google Scholar] [CrossRef]
- Shadrick, W.R.; Mukherjee, S.; Hanson, A.M.; Sweeney, N.L.; Frick, D.N. Aurintricarboxylic acid modulates the affinity of hepatitis C virus NS3 helicase for both nucleic acid and ATP. Biochemistry 2013, 52, 6151–6159. [Google Scholar] [CrossRef] [Green Version]
- Cushman, M.; Sherman, P. Inhibition of HIV-1 integration protein by aurintricarboxylic acid monomers, monomer analogs, and polymer fractions. Biochem. Biophys. Res. Commun. 1992, 185, 85–90. [Google Scholar] [CrossRef]
- Freire, E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov. Today 2008, 13, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Campoy, A.; Todd, M.J.; Freire, E. HIV-1 protease inhibitors: Enthalpic versus entropic optimization of the binding affinity. Biochemistry 2000, 39, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Tarcsay, A.; Keseru, G.M. Contributions of Molecular Properties to Drug Promiscuity Miniperspective. J. Med. Chem. 2013, 56, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Canal, B.; McClure, A.W.; Curran, J.F.; Wu, M.; Ulferts, R.; Weissmann, F.; Zeng, J.K.; Bertolin, A.P.; Milligan, J.C.; Basu, S.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp14/nsp10 exoribonuclease. Biochem. J. 2021, 478, 2445–2464. [Google Scholar] [CrossRef]
- Marcus, A.; Bewley, J.D.; Weeks, D.P. Aurintricarboxylic acid and initiation factors of wheat embryo. Science 1970, 167, 1735–1736. [Google Scholar] [CrossRef]
- Siegelman, F.; Apirion, D. Aurintricarboxylic acid, a preferential inhibitor of initiation of protein synthesis. J. Bacteriol. 1971, 105, 902–907. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, U.; Giri, K.; Bhattacharyya, N.P. Interaction of aurintricarboxylic acid (ATA) with four nucleic acid binding proteins DNase I, RNase A, reverse transcriptase and Taq polymerase. Spectrochim. Acta A 2009, 74, 1145–1151. [Google Scholar] [CrossRef]
- Hallick, R.B.; Chelm, B.K.; Gray, P.W.; Orozco, E.M., Jr. Use of aurintricarboxylic acid as an inhibitor of nucleases during nucleic acid isolation. Nucleic Acids. Res. 1977, 4, 3055–3064. [Google Scholar] [CrossRef] [Green Version]
- Nakane, H.; Balzarini, J.; De Clercq, E.; Ono, K. Differential inhibition of various deoxyribonucleic acid polymerases by Evans blue and aurintricarboxylic acid. Eur. J. Biochem. 1988, 177, 91–96. [Google Scholar] [CrossRef]
- Myskiw, C.; Deschambault, Y.; Jefferies, K.; He, R.; Cao, J. Aurintricarboxylic acid inhibits the early stage of vaccinia virus replication by targeting both cellular and viral factors. J. Virol. 2007, 81, 3027–3032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.N.; Zhao, J.Y.; Li, Q.J.; Wang, M.H.; Zhu, M.; Wang, J.X.; Cen, S.; Wang, Y.C. Discovery and optimization of 2-((1H-indol-3-yl)thio)-N-benzyl-acetamides as novel SARS-CoV-2 RdRp inhibitors. Eur. J. Med. Chem. 2021, 223, 113622. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Lee, D.Y.; Shrestha, S.; Shim, Y.S.; Kim, K.C.; Kim, M.-K.; Lee, K.-H.; Won, J.; Kang, J.-S. Aurintricarboxylic acid translocates across the plasma membrane, inhibits protein tyrosine phosphatase and prevents apoptosis in PC12 cells. Mol. Cells 2004, 18, 46–52. [Google Scholar] [PubMed]
- Zhou, H.; Fang, Y.; Xu, T.; Ni, W.J.; Shen, A.Z.; Meng, X.M. Potential therapeutic targets and promising drugs for combating SARS-CoV-2. Br. J. Pharmacol. 2020, 177, 3147–3161. [Google Scholar] [CrossRef] [PubMed]
- Khursheed, A.; Jain, V.; Rasool, A.; Rather, M.A.; Malik, N.A.; Shalla, A.H. Molecular scaffolds from mother nature as possible lead compounds in drug design and discovery against coronaviruses: A landscape analysis of published literature and molecular docking studies. Microb Pathog. 2021, 157, 104933. [Google Scholar] [CrossRef] [PubMed]
- Tun, M.M.N.; Morita, K.; Ishikawa, T.; Urata, S. The Antiviral Effect of the Chemical Compounds Targeting DED/EDh Motifs of the Viral Proteins on Lymphocytic Choriomeningitis Virus and SARS-CoV-2. Viruses 2021, 13, 1220. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 1st ed.; Plenum Press: New York, NY, USA, 1983. [Google Scholar]
- Ingersoll, C.M.; Strollo, C.M. Steady-state fluorescence anisotropy to investigate flavonoids binding to proteins. J. Chem. Educ. 2007, 84, 1313–1315. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Lomakin, I.B.; Steitz, T.A. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 2013, 500, 307. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ka × 105 (M−1) | Kd (µM) | na | ΔG (kcal·mol−1) | ΔH (kcal·mol−1) | TΔS (kcal·mol−1) |
|---|---|---|---|---|---|---|
| ATA | 5.0 ± 0.1 | 6.7 ± 0.1 | 4.5 ± 0.5 | −7.6 ± 0.1 | −6.4 ± 0.2 | 1.2 ± 0.1 |
| Dichlorohexamer | 9.9 ± 0.5 | 1.0 ± 0.1 | 1.0 ±0.0 | −8.0 ± 0.2 | −15.1± 0.2 | −7.1 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minetti, C.A.; Remeta, D.P.; Hashimoto, K.; Bonala, R.; Chennamshetti, R.; Yin, X.; Garcia-Diaz, M.; Grollman, A.P.; Johnson, F.; Sidorenko, V.S. Characterization of Aurintricarboxylic Acid (ATA) Interactions with Plasma Transporter Protein and SARS-CoV-2 Viral Targets: Correlation of Functional Activity and Binding Energetics. Life 2022, 12, 872. https://doi.org/10.3390/life12060872
Minetti CA, Remeta DP, Hashimoto K, Bonala R, Chennamshetti R, Yin X, Garcia-Diaz M, Grollman AP, Johnson F, Sidorenko VS. Characterization of Aurintricarboxylic Acid (ATA) Interactions with Plasma Transporter Protein and SARS-CoV-2 Viral Targets: Correlation of Functional Activity and Binding Energetics. Life. 2022; 12(6):872. https://doi.org/10.3390/life12060872
Chicago/Turabian StyleMinetti, Conceição A., David P. Remeta, Keiji Hashimoto, Radha Bonala, Rajesh Chennamshetti, Xingyu Yin, Miguel Garcia-Diaz, Arthur P. Grollman, Francis Johnson, and Viktoriya S. Sidorenko. 2022. "Characterization of Aurintricarboxylic Acid (ATA) Interactions with Plasma Transporter Protein and SARS-CoV-2 Viral Targets: Correlation of Functional Activity and Binding Energetics" Life 12, no. 6: 872. https://doi.org/10.3390/life12060872