
Personalized Management and Treatment of Alzheimer’s Disease

 ,
, 

Abstract
:1. Introduction
2. Diagnostic Procedures
3. Phenotypic Features
4. Biomarkers
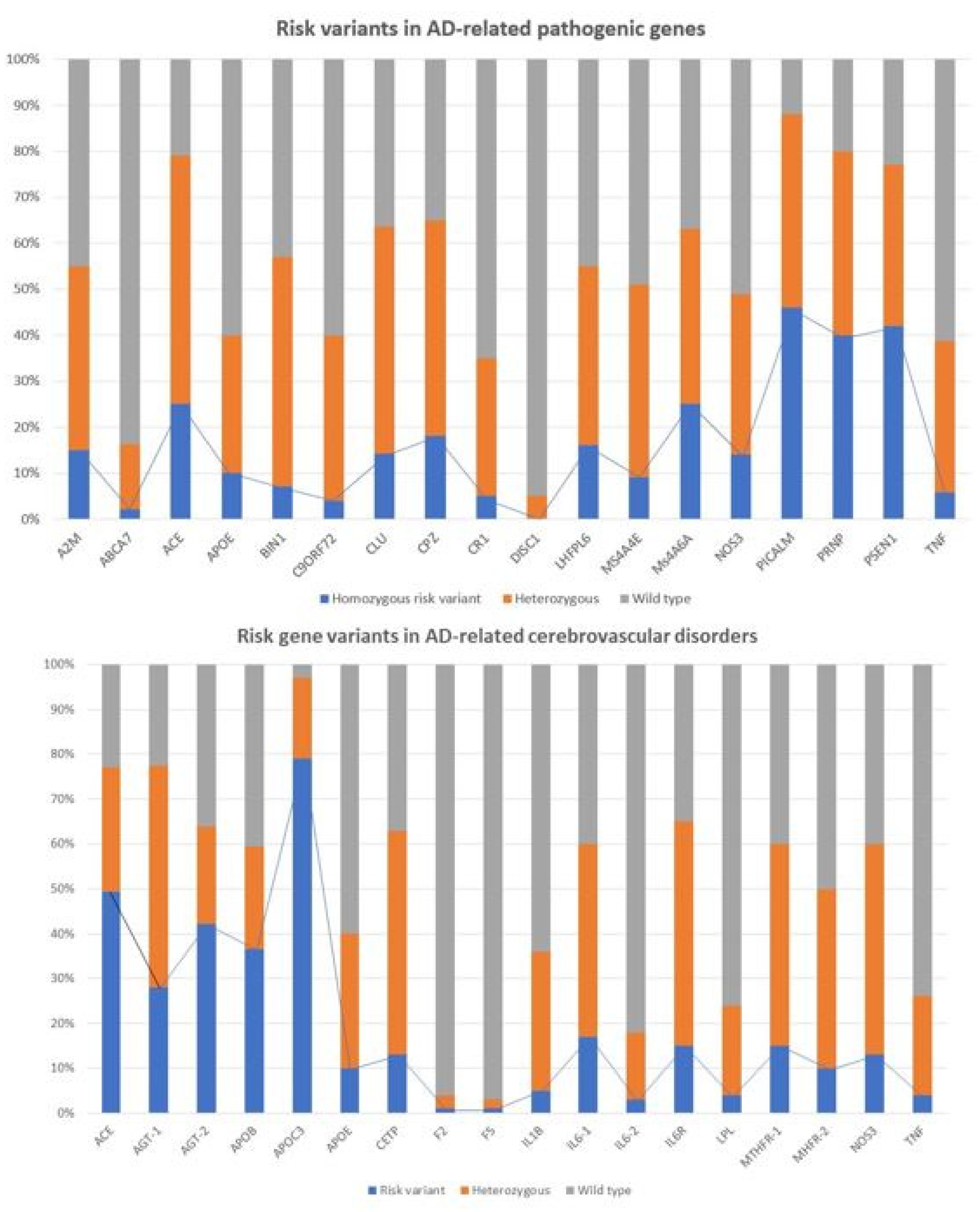
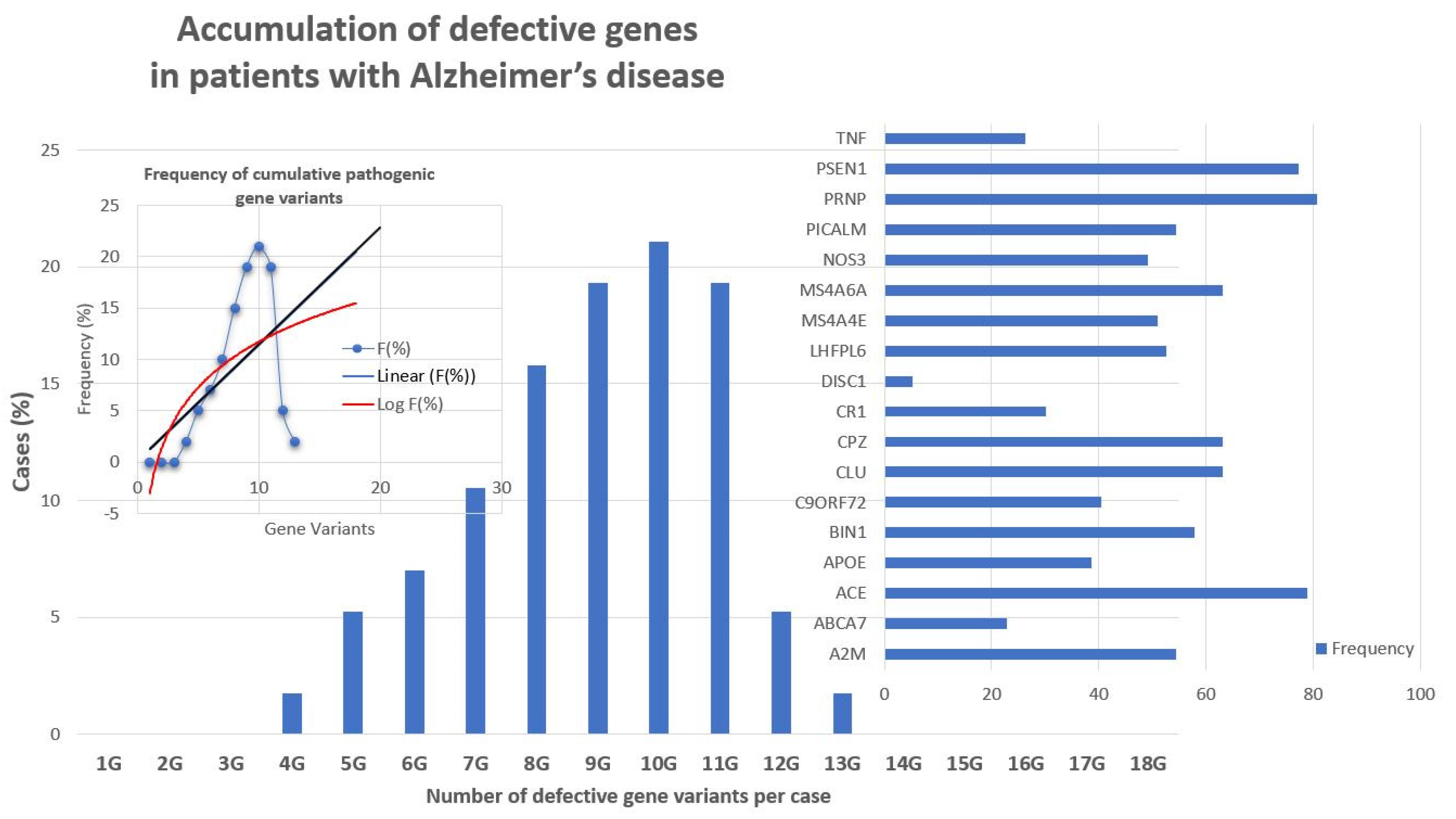
4.1. Genomic Markers
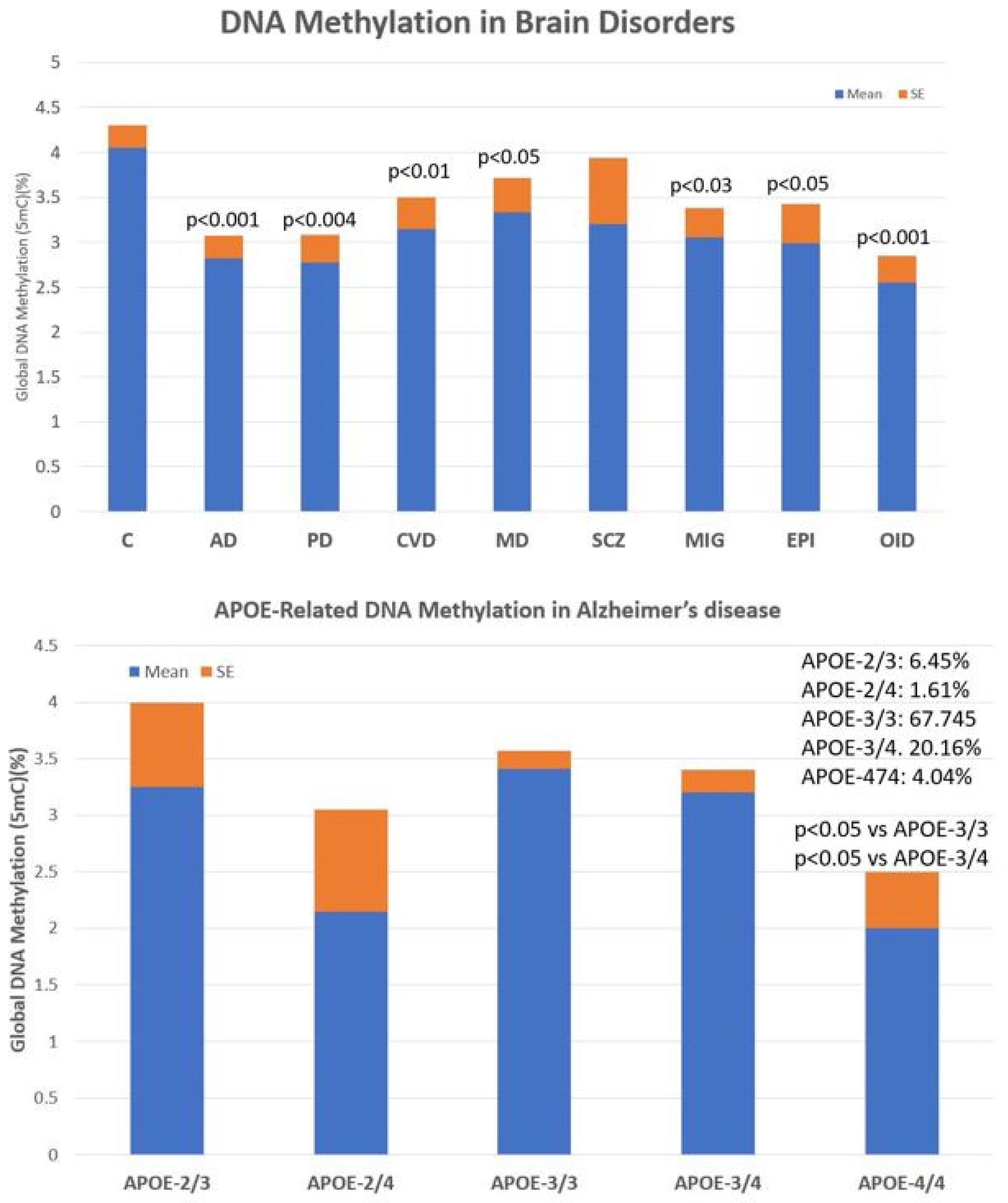
4.2. Epigenetic Markers
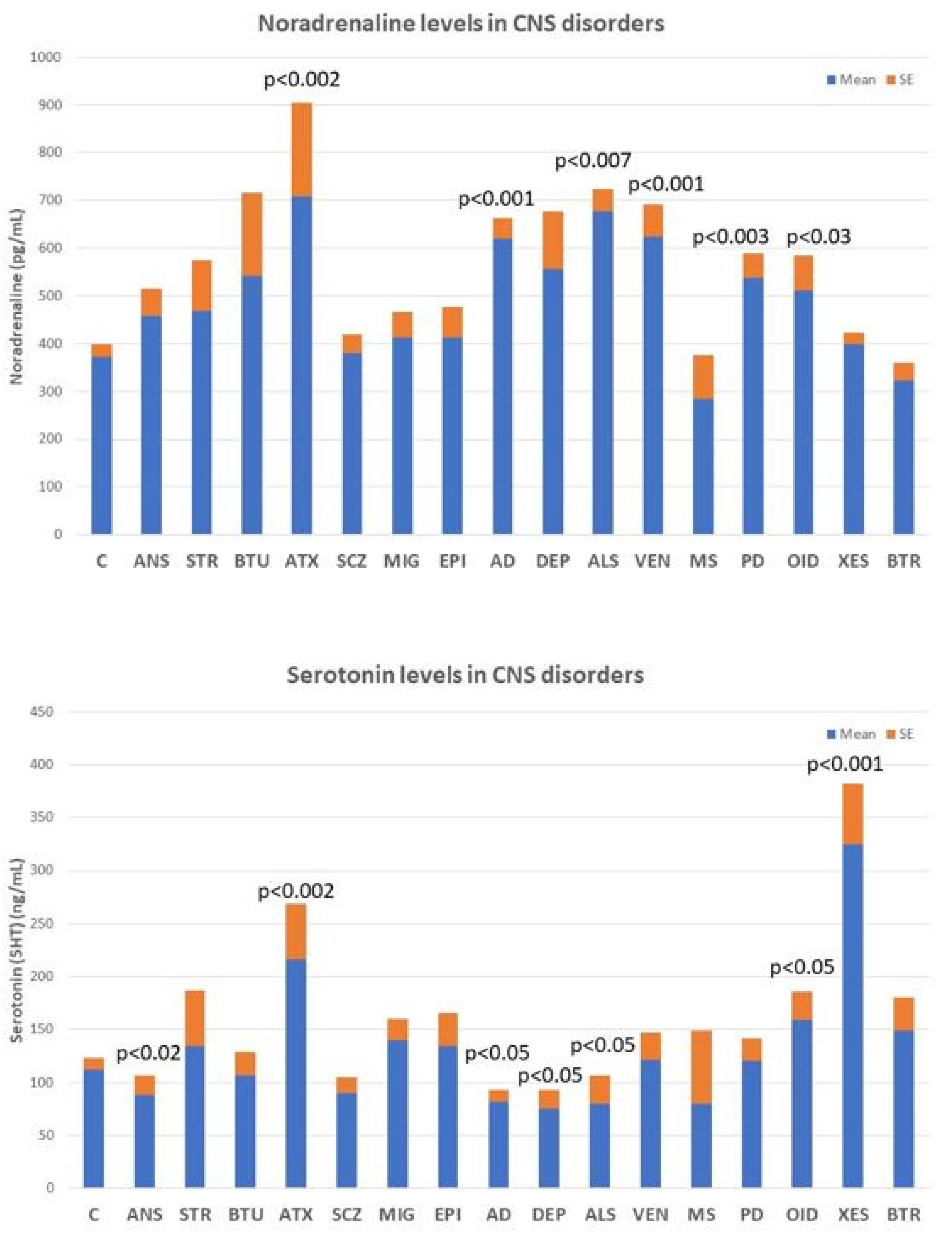
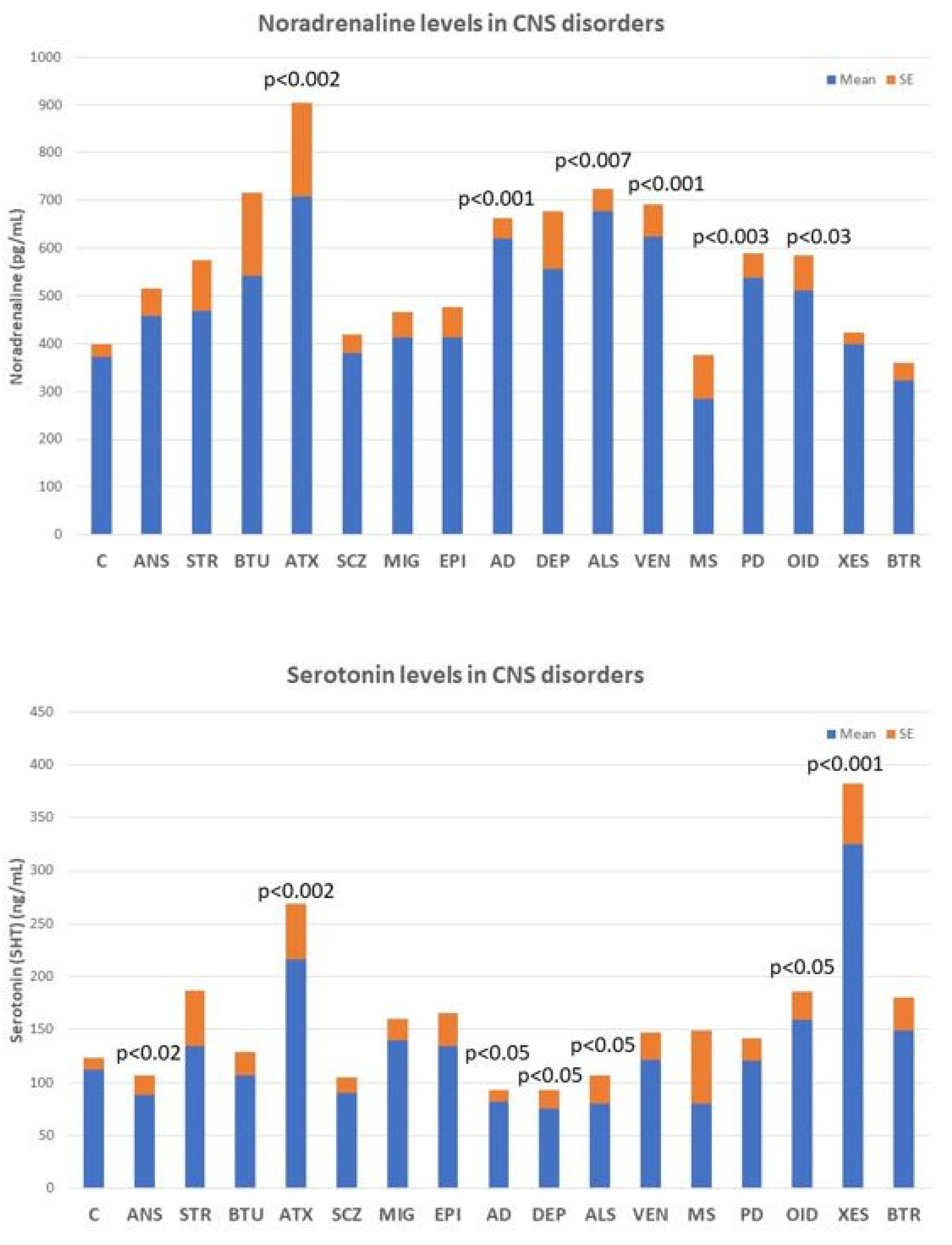
4.3. Neurotransmitters
4.4. Aβ/Tau Levels
5. Concomitant Disorders and Phenotype-Modifying Treatments
6. Alzheimer’s Disease Therapeutics and Drug Development
Immunotherapy
7. Pharmacogenomics
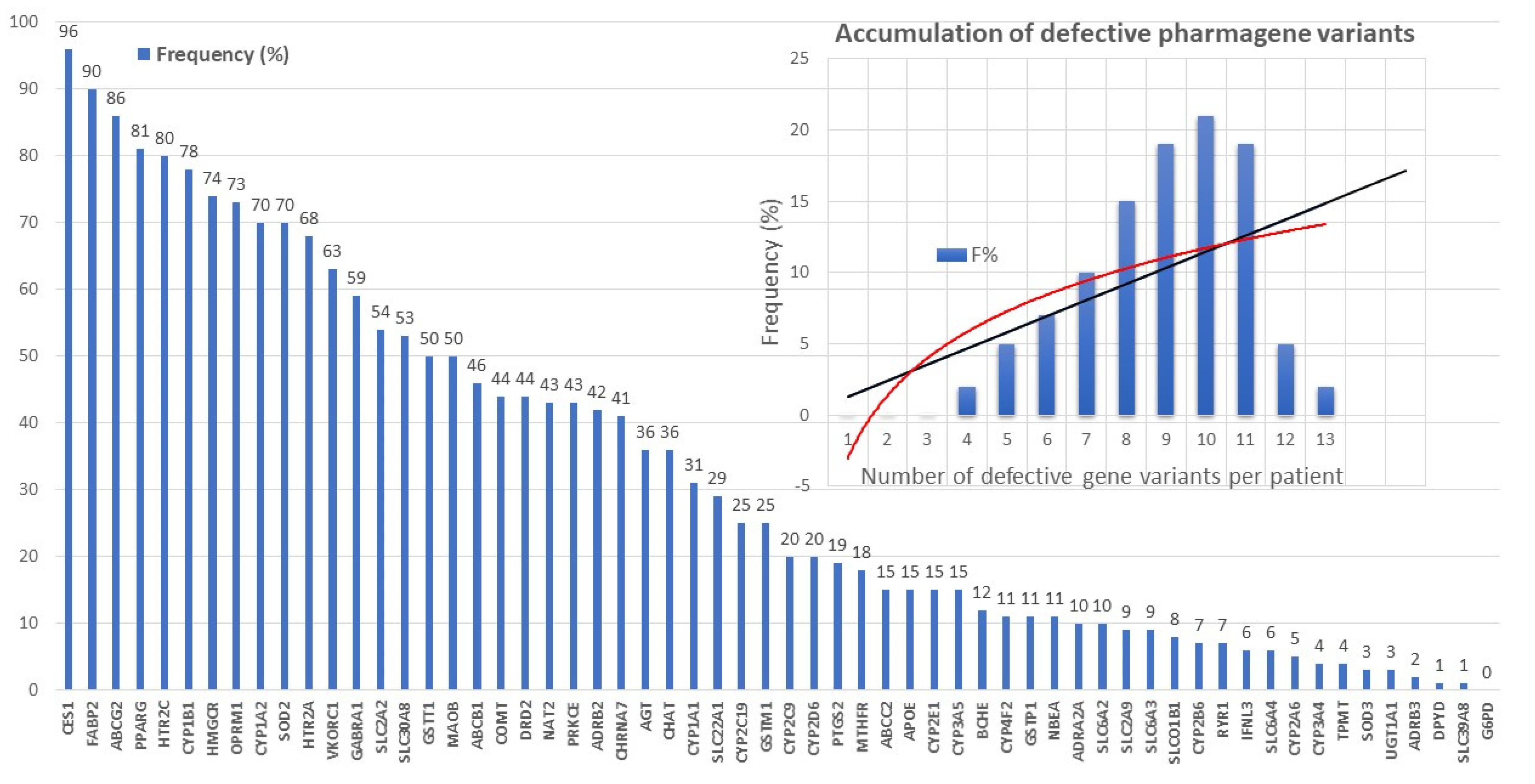
7.1. The Pharmacogenomic Machinery in Alzheimer’s Disease
7.2. Mechanistic Genes Involved in Cholinergic Neurotransmission
7.3. Metabolic Genes
7.4. Transporter Genes
7.5. Pharmacogenetics of Acetylcholinesterase Inhibitors
7.5.1. Donepezil
7.5.2. Galantamine
7.5.3. Rivastigmine
7.5.4. Huperzine A
7.6. Pharmacogenetics of Memantine
7.7. Pharmacogenetics of Aducanumab
7.8. Pharmacogenetics of Multifactorial Treatments
7.9. Pharmacoepigenetics
7.10. Pharmacogenomics of Mood Disorders and Anxiety
8. Future Trends
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cantarero-Prieto, D.; Leon, P.L.; Blazquez-Fernandez, C.; Sanchez Juan, P.; Sarabia Cobo, C. The economic cost of dementia: A systematic review. Dementia 2020, 19, 2637–2657. [Google Scholar] [CrossRef] [PubMed]
- Sado, M.; Ninomiya, A.; Shikimoto, R.; Ikeda, B.; Baba, T.; Yoshimura, K.; Mimura, M. The estimated cost of dementia in Japan, the most aged society in the world. PLoS ONE 2018, 13, e0206508. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 3 February 2022).
- Bachman, D.L.; Wolf, P.A.; Linn, R.; Knoefel, J.E.; Cobb, J.; Belanger, A.; D’Agostino, R.B.; White, L.R. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology 1992, 42, 115–119. [Google Scholar] [CrossRef]
- Cacabelos, R.; Fernández-Novoa, L.; Lombardi, V.; Kubota, Y.; Takeda, M. Molecular genetics of Alzheimer’s disease and aging. Methods Find. Exp. Clin. Pharmacol. 2005, 27, 1–573. [Google Scholar]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.R.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Shah, R.C.; Bennett, D.A. Diagnosis and Management of Dementia: Review. JAMA 2019, 322, 1589–1599. [Google Scholar] [CrossRef]
- Hersi, M.; Irvine, B.; Gupta, P.; Gomes, J.; Birkett, N.; Krewski, D. Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. Neurotoxicology 2017, 61, 143–187. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Asien, P.; Andrieu, S.; Bakardjan, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Cacabelos, P.; Torrellas, C.; Tellado, I.; Carril, J.C. Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Methods Mol. Biol. 2014, 1175, 323–556. [Google Scholar] [CrossRef]
- Cacabelos, R.; Carril, J.C.; Cacabelos, P.; Teijido, O.; Goldgaber, D. Pharmacogenomics of Alzheimer’s Disease: Genetic determinants of phenotypic variation and therapeutic outcome. J. Genom. Med. Pharmacogenom. 2016, 1, 151–209. [Google Scholar]
- Cacabelos, R.; Carril, J.C.; Cacabelos, N.; Kazantsev, A.G.; Vostrov, A.V.; Corzo, L.; Cacabelos, P.; Goldgaber, D. Sirtuins in Alzheimer’s Disease: SIRT2-Related GenoPhenotypes and Implications for PharmacoEpiGenetics. Int. J. Mol. Sci. 2019, 20, 1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, R. Population-level pharmacogenomics for precision drug development in dementia. Expert Rev. Precis. Med. Drug Develop. 2018, 3, 163–188. [Google Scholar] [CrossRef]
- Cacabelos, R. Pharmacogenomics of Cognitive Dysfunction and Neuropsychiatric Disorders in Dementia. Int. J. Mol. Sci. 2020, 21, 3059. [Google Scholar] [CrossRef] [PubMed]
- Matej, R.; Tesar, A.; Rusina, R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin. Biochem. 2019, 73, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Cacabelos, N.; Carril, J.C. The role of pharmacogenomics in adverse drug reactions. Expert Rev. Clin. Pharmacol. 2019, 12, 407–442. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [Green Version]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.A.; Kirson, N.Y.; Desai, U.; Cumming, A.K.G.; Faries, D.E.; Birnbaum, H.G. Medical costs of Alzheimer’s disease misdiagnosis among US Medicare beneficiaries. Alzheimers Dement. 2015, 11, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R.; Goldgaber, D.; Vostrov, A.; Matsuki, H.; Torrellas, C.; Corzo, L.; Carril, J.C.; Roses, A.D. APOE-TOMM40 in the Pharmacogenomics of demetia. J. Pharmacogen. Pharmacoproteom. 2014, 5, 135. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R. Pharmacogenomic of drugs to treat brain disorders. Expert Rev. Precis. Med. Drug Develop. 2020, 5, 181–234. [Google Scholar] [CrossRef]
- Cacabelos, R.; Carril, J.C.; Corzo, L.; Fernández-Novoa, L.; Pego, R.; Cacabelos, N.; Cacabelos, P.; Alcaraz, M.; Tellado, I.; Naidoo, V. Influence of pathogenic and metabolic genes on the pharmacogenetics of mood disorders in Alzheimer’s disease. Pharmaceuticals 2021, 14, 366. [Google Scholar] [CrossRef] [PubMed]
- Bature, F.; Guinn, B.A.; Pang, D.; Pappas, Y. Signs and symptoms preceding the diagnosis of Alzheimer’s disease: A systematic scoping review of literature from 1937 to 2016. BMJ Open 2017, 7, e015746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnemann, C.; Lang, U.E. Pathways Connecting Late-Life Depression and Dementia. Front. Pharmacol. 2020, 11, 279. [Google Scholar] [CrossRef]
- Bennett, S.; Thomas, A.J. Depression and dementia: Cause, consequence or coincidence? Maturitas 2014, 79, 184–190. [Google Scholar] [CrossRef]
- Gutzmann, H.; Qazi, A. Depression associated with dementia. Z. Gerontol. Geriatr. 2015, 48, 305–311. [Google Scholar] [CrossRef]
- Baruch, N.; Burgess, J.; Pillai, M.; Allan, C.L. Treatment for depression comorbid with dementia. Evid. Based Ment. Health 2019, 22, 167–171. [Google Scholar] [CrossRef]
- Bingham, K.S.; Flint, A.J.; Mulsant, B.H. Management of Late-Life Depression in the Context of Cognitive Impairment: A Review of the Recent Literature. Curr. Psychiatry Rep. 2019, 21, 74. [Google Scholar] [CrossRef]
- Kratz, T. The Diagnosis and Treatment of Behavioral Disorders in Dementia. Dtsch. Arztebl. Int. 2017, 114, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Aarsland, D. Epidemiology and Pathophysiology of Dementia-Related Psychosis. J. Clin. Psychiatry 2020, 81, AD19038BR1C. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H. MRI morphometry in Alzheimer’s disease. Ageing Res. Rev. 2016, 30, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Homma, A.; Mok, V.C.; Krishnamoorthy, E.; Alladi, S.; Meguro, K.; Abe, K.; Dominguez, J.; Marasigan, S.; Kandiah, N.; et al. Alzheimer’s disease with cerebrovascular disease: Current status in the Asia-Pacific region. J. Intern. Med. 2016, 280, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Dervenoulas, G.; Politis, M. on behalf of Alzheimer’s Disease Neuroimaging Initiative. Magnetic resonance imaging in Alzheimer’s disease and mild cognitive impairment. J. Neurol. 2019, 266, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- Whitwell, J.L. Alzheimer’s disease neuroimaging. Curr. Opin. Neurol. 2018, 31, 396–404. [Google Scholar] [CrossRef]
- Cacabelos, R. Pharmacogenomic Biomarkers in Neuropsychiatry: The Path to Personalized Medicine in Mental Disorders. In The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes. Molecular Genetic and Genomic Markers; Ritsner, M.S., Ed.; Springer: Amsterdam, The Netherlands, 2009; Volume 4, pp. 3–63. [Google Scholar]
- Caroli, A.; Frisoni, G.B.; Alzheimer’s Disease Neuroimaging Initiative. The dynamics of Alzheimer’s disease biomarkers in the Alzheimer’s Disease Neuroimaging Initiative cohort. Neurobiol. Aging 2010, 31, 1263–1274. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Peterson, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Wigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [Green Version]
- Zetterberg, H.; Bendlin, B.B. Biomarkers for Alzheimer’s disease: Preparing for a new era of disease-modifying therapies. Mol. Psychiatry 2021, 26, 296–308. [Google Scholar] [CrossRef]
- Márquez, F.; Yassa, M.A. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 21. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R. Genomic characterization of Alzheimer’s disease and genotype-related phenotypic analysis of biological markers in dementia. Pharmacogenomics 2004, 5, 1049–1105. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Corzo, L.; Amado, L.; Pichel, V.; Lombardi, V.; Kubota, Y. Phenotypic profiles and functional genomics in Alzheimer’s disease and in dementia with a vascular component. Neurol. Res. 2004, 26, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Fernández-Novoa, L.; Corzo, L.; Pichel, V.; Lombardi, V.; Kubota, Y. Genomics and phenotypic profiles in dementia: Implications for pharmacological treatment. Methods Find. Exp. Clin. Pharmacol. 2004, 26, 421–444. [Google Scholar] [PubMed]
- Cacabelos, R.; Lombardi, V.; Fernández-Novoa, L.; Kubota, Y.; Corzo, L.; Pichel, V.; Takeda, M. A functional genomics approach to the analysis of biological markers in Alzheimer disease. In Molecular Neurobiology of Alzheimer’s Disease and Related Disorders; Takeda, M., Tanaka, T., Cacabelos, R., Eds.; Karger: Basel, Switzerland, 2004; pp. 236–285. [Google Scholar]
- Cacabelos, R.; Martínez-Bouza, R.; Carril, J.C.; Fernández-Novoa, L.; Lombardi, V.; Carrera, I.; Corzo, L.; McKay, A. Genomics and pharmacogenomics of brain disorders. Curr. Pharm. Biotechnol. 2012, 13, 674–725. [Google Scholar] [CrossRef]
- Cacabelos, R.; Martínez-Bouza, R. Genomics and pharmacogenomics of dementia. CNS Neurosc. Ther. 2011, 17, 566–576. [Google Scholar] [CrossRef]
- Cai, Y.; An, S.S.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, R.J.; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Giau, V.V.; Bagyinszky, E.; Yang, Y.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cauwenberghe, C.; van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertram, L. Next Generation Sequencing in Alzheimer’s Disease. Syst. Biol. Alzheimers Dis. 2016, 1303, 281–297. [Google Scholar] [CrossRef]
- Zhu, J.B.; Tan, C.C.; Tan, L.; Yu, J.T. State of Play in Alzheimer’s Disease Genetics. J. Alzheimers Dis. 2017, 58, 631–659. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R.; Torrellas, C.; Teijido, O.; Carril, J.C. Pharmacogenetic considerations in the treatment of Alzheimer’s disease. Pharmacogenomics 2016, 17, 1041–1074. [Google Scholar] [CrossRef]
- Cacabelos, R.; Meyyazhagan, A.; Carril, J.C.; Cacabelos, P.; Teijido, O. Pharmacogenetics of vascular risk factors in Alzheimer’s Disease. J. Pers. Med. 2018, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R. Pharmacogenomics of central nervous system (CNS) drugs. Drug Dev. Res. 2012, 73, 461–476. [Google Scholar] [CrossRef]
- Cacabelos, R.; Takeda, M. Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Future 2006, 3, 5–146. [Google Scholar] [CrossRef]
- Cacabelos, R. Psychogeriatric research: A conceptual introduction to aging and geriatric neuroscience. Psychogeriatrics 2001, 1, 158–188. [Google Scholar] [CrossRef]
- Cacabelos, R. The application of functional genomics to Alzheimer’s disease. Pharmacogenomics 2003, 4, 597–621. [Google Scholar] [CrossRef]
- Cacabelos, R. Pharmacogenomics in Alzheimer’s disease. Pharm. Drug Discov. Dev. 2008, 448, 213–357. [Google Scholar]
- Cacabelos, R.; Fernández-Novoa, L.; Martínez-Bouza, R.; McKay, A.; Carril, J.C.; Lombardi, V.; Corzo, L.; Carrera, I.; Tellado, I.; Nebril, L.; et al. Future trends in the pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6 variants. Pharmaceuticals 2010, 3, 3040–3100. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R. Molecular pathology and pharmacogenomics in Alzheimer’s disease: Polygenic-related effects of multifactorial treatments on cognition, anxiety, and depression. Methods Find. Expert Clin. Pharmacol. 2007, 29, 1–91. [Google Scholar]
- Sabbagh, M.N.; Malek-Ahmadi, M.; Dugger, B.N.; Lee, K.; Sue, L.I.; Serrano, G.; Walker, D.G.; Davis, K.; Jacobson, S.A.; Beach, T.G. The influence of Apolipoprotein E genotype on regional pathology in Alzheimer’s disease. BMC Neurol. 2013, 13, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, R.E.; Cutter, G.R.; Schneider, L.S. Effect of APOE genotype status on targeted clinical trials outcomes and efficiency in dementia and mild cognitive impairment resulting from Alzheimer’s disease. Alzheimers Dement. 2014, 10, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R.; Torrellas, C.; López-Muñoz, F. Epigenomics of Alzheimer’s disease. J. Exp. Med. 2014, 6, 75–82. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Epigenetic drug discovery for Alzheimer’s disease. Expert Opin. Drug Discov. 2014, 9, 1059–1086. [Google Scholar] [CrossRef]
- Cacabelos, R. (Ed.) Pathoepigenetics: The role of epigenetic biomarkers in disease pathogenesis. In Pharmacoepigenetics, 1st ed.; Academic Press: Oxford, UK, 2019; pp. 139–189. [Google Scholar]
- Qazi, T.J.; Quan, Z.; Mir, A.; Qing, H. Epigenetics in Alzheimer’s Disease: Perspective of DNA Methylation. Mol. Neurobiol. 2018, 55, 1026–1044. [Google Scholar] [CrossRef]
- Ciceri, F.; Rotllant, D.; Maes, T. Understanding Epigenetic Alterations in Alzheimer’s and Parkinson’s Disease: Towards Targeted Biomarkers and Therapies. Curr. Pharm. Des. 2017, 23, 839–857. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6156–6167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.; Bhattacharjee, S.; Sharma, A.R.; Sharma, G.; Lee, S.S.; Chakraborty, C. miRNAs in Alzheimer Disease—A Therapeutic Perspective. Curr. Alzheimer Res. 2017, 14, 1198–1206. [Google Scholar] [CrossRef]
- Silvestro, S.; Bramanti, P.; Mazzon, E. Role of miRNAs in Alzheimer’s Disease and Possible Fields of Application. Int. J. Mol. Sci. 2019, 20, 3979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetterberg, H.; Burnham, S.C. Blood-based molecular biomarkers for Alzheimer’s disease. Mol. Brain 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G. RNA and Oxidative Stress in Alzheimer’s Disease: Focus on microRNAs. Oxidative Med. Cell. Longev. 2020, 2020, 2638130. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R.; Carril, J.C.; Sanmartín, A.; Cacabelos, P. Pharmacoepigenetic processors: Epigenetic drugs, Drug resistance, Toxicoepigenetics, and Nutriepigenetics. In Pharmacoepigenetics, 1st ed.; Cacabelos, R., Ed.; Academic Press: Oxford, UK, 2019; pp. 191–424. [Google Scholar]
- Cacabelos, R.; Cacabelos, P.; Carril, J.C. Epigenetics and pharmacoepigenetics of age-related neurodegenerative disorders. In Pharmacoepigenetics, 1st ed.; Cacabelos, R., Ed.; Academic Press: Oxford, UK, 2019; pp. 903–950. [Google Scholar]
- Cacabelos, R. Epigenomic networking in drug development: From pathogenic mechanisms to pharmacogenomics. Drug Dev. Res. 2014, 75, 348–365. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.W. Alzheimer’s disease: Early diagnosis and treatment. Hong Kong Med. J. 2012, 18, 228–237. [Google Scholar]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Barbe, C.; Jolly, D.; Morrone, I.; Wolak-Thierry, A.; Dramé, M.; Novella, J.L.; Mahmoudi, R. Factors associated with quality of life in patients with Alzheimer’s disease. BMC Geriatr. 2018, 18, 159. [Google Scholar] [CrossRef] [Green Version]
- Ettcheto, M.; Olloquequi, J.; Sánchez-López, E.; Busquets, O.; Cano, A.; Manzine, P.R.; Beas-Zarate, C.; Castro-Torres, R.D.; García, M.L.; Bulló, M.; et al. Benzodiazepines and Related Drugs as a Risk Factor in Alzheimer’s Disease Dementia. Front. Aging Neurosci. 2020, 11, 344. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, E.D.; Kamat, A.; Gahan, W.P.; Ourso, V.; Woodard, K.; Kerwin, D.R.; Binder, E.F.; Burns, J.M.; Cullum, M.; Hydan, L.S.; et al. Baseline Prevalence of Polypharmacy in Older Hypertensive Study Subjects with Elevated Dementia Risk: Findings from the Risk Reduction for Alzheimer’s Disease Study (rrAD). J. Alzheimers Dis. 2020, 77, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Andin, U.; Passant, U.; Gustafson, L.; Englund, E. Alzheimer’s disease (AD) with and without white matter pathology-clinical identification of concurrent cardiovascular disorders. Arch. Gerontol. Geriatr. 2007, 44, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, B.; Suppiah, S.; Piersson, A.D.; Razali, R.M.; Mohamad, M.; Hassan, H.A.; Ibrahim, N. Cardiovascular risk factors of Alzheimer’s disease and other neurocognitive disorders in Malaysia. Med. J. Malays. 2021, 76, 291–297. [Google Scholar]
- Contois, J.H.; Anamani, D.E.; Tsongalis, G.J. The underlying molecular mechanism of apolipoprotein E polymorphism: Relationships to lipid disorders, cardiovascular disease, and Alzheimer’s disease. Clin. Lab. Med. 1996, 16, 105–123. [Google Scholar] [CrossRef]
- Cacabelos, R.; Fernández-Novoa, L.; Lombardi, V.; Corzo, L.; Pichel, V.; Kubota, Y. Cerebrovascular risk factors in Alzheimer’s disease: Brain hemodynamics and pharmacogenomic implications. Neurol. Res. 2003, 25, 567–580. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar] [CrossRef]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Chornenkyy, Y.; Wang, W.X.; Wei, A.; Nelson, P.T. Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 2019, 29, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Baglietto-Vargas, D.; Shi, J.; Yaeger, D.M.; Ager, R.; LaFerla, F.M. Diabetes and Alzheimer’s disease crosstalk. Neurosci. Biobehav. Rev. 2016, 64, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Pharmacogenetic considerations when prescribing cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug Metab. Toxicol. 2020, 16, 673–701. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Have there been improvement in Alzheimer’s disease drug discovery over the past 5 years? Exp. Opin. Drug Discov. 2018, 13, 523–538. [Google Scholar] [CrossRef]
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. Biomed Res. Int. 2020, 2020, 5120230. [Google Scholar] [CrossRef]
- Cacabelos, R. How plausible is an Alzheimer’s disease vaccine? Expert Opin. Drug Discov. 2020, 15, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Hkan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Foroutan, N.; Hopkins, R.B.; Tarride, J.E.; Florez, I.D.; Levine, M. Safety and efficacy of active and passive immunotherapy in mild-to-moderate Alzheimer’s disease: A systematic review and network meta-analysis. Clin. Investig. Med. 2019, 42, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Janus, C.; Pearson, J.; McLaurin, J.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Chishti, M.A.; Horne, P.; Heslin, D.; French, J.; et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 2000, 408, 979–982. [Google Scholar] [CrossRef]
- Herline, K.; Drummond, E.; Wisniewski, T. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev. Vaccines 2018, 17, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Zilka, N.; Zilkova, M.; Kovacech, B.; Shrabana, R.; Ondrus, M.; Fialova, L.; Kontsekova, E.; Otto, M.; Novak, M. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimers Dis. 2019, 6, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, J.L.; Sabbagh, M.N.; Al-Hasan, Y.; Decourt, B. Tau immunotherapies for Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Fernandez-Novoa, L.; Aliev, G.; Vigo, C.; Cacabelos, R. Validating Immunotherapy in Alzheimer’s Disease: The EB101 Vaccine. Curr. Pharm. Des. 2016, 22, 849–858. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Zhu, K.; Meng, Y.; Ding, L.; Wang, J.C.; Yin, W.C.; Yan, Y.; Cao, Y.P. Reduction of amyloid beta by Aβ3-10-KLH vaccine also decreases tau pathology in 3×Tg-AD mice. Brain Res. Bull. 2018, 142, 233–240. [Google Scholar] [CrossRef]
- Rosenberg, R.N.; Fu, M.; Lambracht-Washington, D. Active full-length DNA Aβ42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimers Res. Ther. 2018, 10, 115. [Google Scholar] [CrossRef]
- Oddo, S.; Billings, L.; Kesslak, J.P.; Cribbs, D.H.; LaFerla, F.M. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004, 43, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Lalli, G.; Schott, J.M.; Hardy, J.; De Strooper, B. Aducanumab: A new phase in therapeutic development for Alzheimer’s disease? EMBO Mol. Med. 2021, 13, e14781. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Banerjee, D. A Primer on the Evolution of Aducanumab: The First Antibody Approved for Treatment of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 83, 1537–1552. [Google Scholar] [CrossRef]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, J.; Williams, L.; Stella, H.; Leitermann, K.; Mikulskis, A.; O’Gorman, J.; Sevigny, J. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2016, 2, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Van de Vrede, L.; Gibbs, D.M.; Koestler, M.; La Joie, R.; Ljubenkov, P.A.; Provost, K.; Soleimani-Meiggoni, D.; Strom, A.; Tsoy, E.; Rabinovici, G.D.; et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement. 2020, 12, e12101. [Google Scholar] [CrossRef]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.W.; Chen, X.W.; Sneed, K.B.; Yang, Y.X.; Zhang, X.; He, Z.X.; Chow, K.; Yang, T.; Duan, W.; Zhou, S.F. Clinical association between pharmacogenomics and adverse drug reactions. Drugs 2015, 75, 589–631. [Google Scholar] [CrossRef]
- Cacabelos, R. Pleiotropy and promiscuity in pharmacogenomics for the treatment of Alzheimer’s disease and related risk factors. Future Neurol. 2018, 13. [Google Scholar] [CrossRef]
- Shapira, M.; Tur-Kaspa, I.; Bosgraaf, L.; Livni, N.; Grant, A.D.; Grisaru, D.; Korner, M.; Ebstrin, R.P.; Soreq, H. A transcription-activating polymorphism in the ACHE promoter associated with acute sensitivity to anti-acetylcholinesterases. Hum. Mol. Genet. 2000, 9, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Lane, R.; Feldman, H.H.; Meyer, J.; He, Y.; Ferris, S.H.; Nordberg, A.; Darreh-Shori, T.; Soininen, H.; Pirttila, T.; Farlow, M.R.; et al. Synergistic effect of apolipoprotein E epsilon4 and butyrylcholinesterase K-variant on progression from mild cognitive impairment to Alzheimer’s disease. Pharmacogenet. Genom. 2008, 18, 289–928. [Google Scholar] [CrossRef]
- Cuddy, L.K.; Seah, C.; Pasternak, S.H.; Rylett, R.J. Amino-Terminal β-Amyloid Antibody Blocks β-Amyloid-Mediated Inhibition of the High-Affinity Choline Transporter CHT. Front. Mol. Neurosci. 2017, 10, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payette, D.J.; Xie, J.; Guo, Q. Reduction in CHT1-mediated choline uptake in primary neurons from presenilin-1 M146V mutant knock-in mice. Brain Res. 2007, 1135, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhou, Z.; Tan, H.; Zhu, S.; Wang, Y.; Sun, Y.; Li, X.M.; Wang, J.F. Nitrosylation of Vesicular Transporters in Brain of Amyloid Precursor Protein/Presenilin 1 Double Transgenic Mice. J. Alzheimers Dis. 2017, 55, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.M.; Aubert, I. Overexpression of the vesicular acetylcholine transporter increased acetylcholine release in the hippocampus. Neuroscience 2012, 218, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kolisnyk, B.; Al-Onaizi, M.A.; Xu, J.; Parfitt, G.M.; Ostapchenko, V.G.; Hanin, G.; Soreeq, H.; Prado, M.A.M.; Prado, V.F. Cholinergic Regulation of hnRNPA2/B1 Translation by M1 Muscarinic Receptors. J. Neurosci. 2016, 36, 6287–6296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolejší, E.; Liraz, O.; Rudajev, V.; Zimcuk, P.; Dolezal, V.; Michaelson, D.M. Apolipoprotein E4 reduces evoked hippocampal acetylcholine release in adult mice. J. Neurochem. 2016, 136, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albin, R.L.; Bohnen, N.I.; Muller, M.L.T.M.; Dauer, W.T.; Sarter, M.; Frey, K.A.; Koeppe, R.A. Regional vesicular acetylcholine transporter distribution in human brain: A [18 F]fluoroethoxybenzovesamicol positron emission tomography study. J. Comp. Neurol. 2018, 526, 2884–2897. [Google Scholar] [CrossRef]
- Wallace, T.L.; Bertrand, D. Importance of the nicotinic acetylcholine receptor system in the prefrontal cortex. Biochem. Pharmacol. 2013, 85, 1713–1720. [Google Scholar] [CrossRef]
- Ma, K.G.; Qian, Y.H. Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Talebi, M.; Mahmoudi, J.; Babri, S.; Shanehbandi, D. Selective activation of α7 nicotinic acetylcholine receptor by PHA-543613 improves Aβ25-35-mediated cognitive deficits in mice. Neuroscience 2015, 298, 81–93. [Google Scholar] [CrossRef]
- Vauthier, V.; Housset, C.; Falguières, T. Targeted pharmacotherapies for defective ABC transporters. Biochem. Pharmacol. 2017, 136, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, R.; Llovo, R.; Fraile, C.; Fernández-Novoa, L. Pharmacogenetic aspects of therapy with cholinesterase inhibitors: The role of CYP2D6 in Alzheimer’s disease pharmacogenetics. Curr. Alzheimer Res. 2007, 4, 479–500. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, R. Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 2007, 3, 303–333. [Google Scholar]
- Brewster, J.T.; Dell’Acqua, S.; Thach, D.Q.; Sessler, J.L. Classics in Chemical Neuroscience: Donepezil. ACS Chem. Neurosci. 2019, 10, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, M.; Eap, C.B. Pharmacodynamic, pharmacokinetic and pharmacogenetic aspects of drugs used in the treatment of Alzheimer’s disease. Clin. Pharmacokinet. 2013, 52, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, M.; Guidi, M.; Ebbing, K.; Eyer, S.; Wilhelm, L.; Michon, A.; Thomazic, V.; Stancu, I.; Alnawaqil, A.M.; Bula, C.; et al. Population pharmacokinetic approach to evaluate the effect of CYP2D6, CYP3A, ABCB1, POR and NR1I2 genotypes on donepezil clearance. Br. J. Clin. Pharmacol. 2014, 78, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Cacabelos, R. World Guide for Drug Use and Pharmacogenomics; EuroEspes Publishing: A Corunna, Spain, 2012. [Google Scholar]
- Xiao, T.; Jiao, B.; Zhang, W.; Tang, B.; Shen, L. Effect of the CYP2D6 and APOE Polymorphisms on the Efficacy of Donepezil in Patients with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. CNS Drugs 2016, 30, 899–907. [Google Scholar] [CrossRef]
- Sokolow, S.; Li, X.; Chen, L.; Taylor, K.D.; Rotter, J.I.; Rissman, R.A.; Aisen, P.S.; Apostolova, L.G. Deleterious Effect of Butyrylcholinesterase K-Variant in Donepezil Treatment of Mild Cognitive Impairment. J. Alzheimers Dis. 2017, 56, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Russo, P.; Kisialiou, A.; Moroni, R.; Prinzi, G.; Fini, M. Effect of Genetic Polymorphisms (SNPs) in CHRNA7 Gene on Response to Acetylcholinesterase Inhibitors (AChEI) in Patients with Alzheimer’s Disease. Curr. Drug Targets 2017, 18, 1179–1190. [Google Scholar] [CrossRef]
- Noetzli, M.; Guidi, M.; Ebbing, K.; Eyer, S.; Zumbach, S.; Giannakopoulos, P.; von Gunten, A.; Csajka, C.; Eap, C.B. Relationship of CYP2D6, CYP3A, POR, and ABCB1 genotypes with galantamine plasma concentrations. Ther. Drug Monit. 2013, 35, 270–275. [Google Scholar] [CrossRef]
- Birks, J.S.; Grimley, E.J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, 10, CD001191. [Google Scholar] [CrossRef]
- Gul, A.; Bakht, J.; Mehmood, F. Huperzine-A response to cognitive impairment and task switching deficits in patients with Alzheimer’s disease. J. Chin. Med. Assoc. 2019, 82, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.P.; Li, X.N.; Yuan, F.; Chen, W.L.; Yang, M.J.; Xu, H.R. Evaluation of the in vitro and in vivo metabolic pathway and cytochrome P450 inhibition/induction profile of Huperzine A. Biochem. Biophys. Res. Commun. 2016, 480, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, M.; Guidi, M.; Ebbing, K.; Eyer, S.; Wilhelm, L.; Michon, A.; Thomazic, V.; Alnawaqil, A.M.; Maurer, S.; Zumbach, S.; et al. Population pharmacokinetic study of memantine: Effects of clinical and genetic factors. Clin. Pharmacokinet. 2013, 52, 211–223. [Google Scholar] [CrossRef]
- Bastrup, J.; Hansen, K.H.; Poulsen, T.B.G.; Kastaniegaard, K.; Asuni, A.A.; Christensen, S.; Belling, D.; Helboe, L.; Stensballe, A.; Volbracht, C. Anti-Aβ Antibody Aducanumab Regulates the Proteome of Senile Plaques and Closely Surrounding Tissue in a Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 79, 249–265. [Google Scholar] [CrossRef]
- Serretti, A. Genetics and pharmacogenetics of mood disorders. Psychiatr. Pol. 2017, 51, 197–203. [Google Scholar] [CrossRef]
- Wyska, E. Pharmacokinetic considerations for current state-of-the-art antidepressants. Expert Opin. Drug Metab. Toxicol. 2019, 15, 831–847. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Souza, R.P.; Müller, D.J. Pharmacogenetics of anxiolytic drugs. J. Neural. Transm. 2009, 116, 667–677. [Google Scholar] [CrossRef]
- Torrellas, C.; Risso, A.; Carril, J.C.; Cacabelos, R. Optimización del uso de antidepresivos con estrategias farmacogenéticas. Gen.-T. 2015, 10, 17–26. [Google Scholar]
- David, S.; Geldmacher, P.J. Whitehouse. Differential diagnosis of Alzheimer’s disease. Neurology 1997, 48, 2S–9S. [Google Scholar] [CrossRef]
- Salmon, E.; Sadzot, B.; Maquet, P.; Degueldre, C.; Lemaire, C.; Rigo, P.; Comar, D.; Franck, G. Differential diagnosis of Alzheimer’s disease with PET. J. Nucl. Med. 1994, 35, 391–398. [Google Scholar] [PubMed]
- Swainson, R.; Hodges, J.R.; Galton, C.J.; Semle, J.; Michael, A.; Dunn, B.D.; Iddon, J.L.; Robbins, T.W. Early Detection and Differential Diagnosis of Alzheimer’s Disease and Depression with Neuropsychological Tasks. Dement. Geriatr. Cogn. Disord. 2001, 12, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Storey, E.; Slavin, M.J.; Kinsella, G.J. Patterns of cognitive impairment in Alzheimer’s disease: Assessment and differential diagnosis. Front. Biosci. 2002, 7, e155–e184. [Google Scholar] [CrossRef] [Green Version]
- Karkou, V.; Meekums, B. Dance movement therapy for dementia. Cochrane Database Syst. Rev. 2017, 2, CD011022. [Google Scholar] [CrossRef]
- Ruths, S.; Straand, J.; Nygaard, H.A. Psychotropic drug use in nursing homes: Diagnostic indications and variations between institutions. Eur. J. Clin. Pharmacol. 2001, 57, 523–528. [Google Scholar] [CrossRef]
- Holmquist, I.B.; Svensson, B.; Höglund, P. Perceived anxiety, depression, and sleeping problems in relation to psychotropic drug use among elderly in assisted-living facilities. Eur. J. Clin. Pharmacol. 2005, 61, 215–224. [Google Scholar] [CrossRef]
- Zahirovic, I.; Torisson, G.; Wattmo, C.; Londos, E. Psychotropic and anti-dementia treatment in elderly persons with clinical signs of dementia with Lewy bodies: A cross-sectional study in 40 nursing homes in Sweden. BMC Geriatr. 2018, 18, 50. [Google Scholar] [CrossRef] [Green Version]
- Gulla, C.; Selbaek, G.; Flo, E.; Kjome, R.; Kirkevold, I.; Husebo, B.S. Multi-psychotropic drug prescription and the association to neuropsychiatric symptoms in three Norwegian nursing home cohorts between 2004 and 2011. BMC Geriatr. 2016, 16, 115. [Google Scholar] [CrossRef] [Green Version]
- Janus, S.I.; van Manen, J.G.; IJzerman, M.J.; Zuidema, S.U. Psychotropic drug prescriptions in Western European nursing homes. Int. Psychogeriatr. 2016, 28, 1775–1790. [Google Scholar] [CrossRef]
- Helvik, A.S.; Šaltytė Benth, J.; Wu, B.; Engedal, K.; Selbaek, G. Persistent use of psychotropic drugs in nursing home residents in Norway. BMC Geriatr. 2017, 17, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J. Disease modification is not all—We need symptomatic therapies for Alzheimer disease. Nat. Rev. Neurol. 2022, 18, 3–4. [Google Scholar] [CrossRef] [PubMed]
- van der Steen, J.T.; van Soest-Poortvliet, M.C.; van der Wouden, J.C.; Bruinsma, M.S.; Sholten, R.J.; Vink, A.C. Music-based therapeutic interventions for people with dementia. Cochrane Database Syst Rev. 2017, 5, CD003477. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; Amos, E.C.; Canick, J.; Ackerley, M.; Raji, C.; Fiala, M.; Ahdidan, J. Reversal of cognitive decline in Alzheimer’s disease. Aging 2016, 8, 1250–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, P.; Youngberg, W. Clinical Lifestyle Medicine Strategies for Preventing and Reversing Memory Loss in Alzheimer’s. Am. J. Lifestyle Med. 2018, 12, 391–395. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levalahti, E.; Ahtiluoto, S.; Antikainen, R.; Backman, L.; Hanninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]
- Blackman, J.; Swirski, M.; Clynes, J.; Harding, S.; Leng, Y.; Coulthard, E. Pharmacological and non-pharmacological interventions to enhance sleep in mild cognitive impairment and mild Alzheimer’s disease: A systematic review. J. Sleep Res. 2021, 30, e13229. [Google Scholar] [CrossRef]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef]
- Braithwaite, S.P.; Stock, J.B.; Lombroso, P.J.; Nairn, A.C. Protein phosphatases and Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2012, 106, 343–379. [Google Scholar] [CrossRef] [Green Version]
- Pathak, A.; Rohilla, A.; Gupta, T.; Akhtar, M.J.; Haider, M.R.; Sharma, K.; Haider, K.; Yar, M.S. DYRK1A kinase inhibition with emphasis on neurodegeneration: A comprehensive evolution story-cum-perspective. Eur. J. Med. Chem. 2018, 158, 559–592. [Google Scholar] [CrossRef]
- Girotra, P.; Behl, T.; Sehgal, A.; Singh, S.; Bungau, S. Investigation of the Molecular Role of Brain-Derived Neurotrophic Factor in Alzheimer’s Disease. J. Mol. Neurosci. 2022, 72, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Dahlström, M.; Madjid, N.; Nordvall, G.; Halldin, M.M.; Vazquez-Juarez, E.; Lindskog, M.; Sandin, J.; Winblad, B.; Eriksdotter, M.; Forsell, P. Identification of Novel Positive Allosteric Modulators of Neurotrophin Receptors for the Treatment of Cognitive Dysfunction. Cells 2021, 10, 1871. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 2021, 296, 100105. [Google Scholar] [CrossRef] [PubMed]
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef]
- Ferrari, C.; Sorbi, S. The complexity of Alzheimer’s disease: An evolving puzzle. Physiol. Rev. 2021, 101, 1047–1081. [Google Scholar] [CrossRef]
- Cacabelos, R.; Naidoo, V.; Corzo, L.; Cacabelos, N.; Carril, J.C. Genophenotypic Factors and Pharmacogenomics in Adverse Drug Reactions. Int. J. Mol. Sci. 2021, 222, 3302. [Google Scholar] [CrossRef]
- Cacabelos, R. What have we learnt from past failures in Alzheimer’s disease drug discovery? Expert Opin. Drug Discov. 2022, 17, 1–15. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (Normal Range) | Total | Females | Males | Differences |
|---|---|---|---|---|
| N | 2701 | 1491 (55%) | 1210 (45%) | |
| Age (years) Range | 67.63 ± 0.19 50–96 | 68.26 ± 0.27 50–96 | 66.86 ± 0.28 50–94 | p < 0.001 |
| Systolic blood pressure (mm Hg) (120–160) | 141.39 ± 0.41 <120: 13.74% >160: 21.84% | 141.25 ± 0.56 14.75% 22.25% | 141.56 ± 0.61 12.37% 21.33% | p = 0.74 p(χ2) = 0.13 p(χ2) = 0.66 |
| Diastolic blood pressure (mm Hg) (70–85) | 79.38 ± 0.21 <70: 10.48% >85: 28.53% | 78.83 ± 0.28 11.27% 26.84% | 80.05 ± 0.33 9.54% 30.74% | p < 0.001 p(χ2) = 0.24 p(χ2) = 0.10 |
| Pulse (bpm) (60–100) | 68.29 ± 0.23 <60: 23.34% >100: 2.07% | 69.60 ± 0.31 19.05% 2.35% | 66.67 ± 0.35 28.75% 1.74% | p < 0.001 p(χ2) = 0.003 p(χ2) = 0.34 |
| Weight (Kg) | 72.23 ± 0.27 (36–127) | 66.41 ± 0.32 (36–112) | 79.41 ± 0.37 (42–127) | p < 0.001 |
| Hight (cm) | 160.71 ± 0.21 (120–188) | 154.96 ± 0.21 (140–182) | 167.79 ± 0.26 (120–188) | p < 0.001 |
| BMI (Kg/m2) Underweight (15–18.5) Normal weight (18.5–25) Overweight (25–30) Obese Class I (moderate) (30–35) Obese Class II (severe)(35–40) Obese Class III (very severe) (>40) | 29.93 ± 0.09 1.01% 25.03% 45.26% 21.25% 6.08% 1.33% | 27.76 ± 0.13 1.54% 29.15% 39.50% 20.85% 7.34% 1.62% | 28.15 ± 0.11 0.27% 19.86% 52.35% 21.75% 4.69% 1.08% | p < 0.002 p(χ2) < 0.003 p(χ2) < 0.001 p(χ2) < 0.001 p(χ2) = 0.69 p(χ2) < 0.01 p(χ2) = 0.34 |
| Glucose (mg/dL) (70–105) | 102.00 ± 0.55 <70: 0.41% >105: 26.55% | 99.17 ± 0.69 0.54% 21.63% | 105.49 ± 0.87 0.24% 32.70% | p < 0.001 p(χ2) = 0.38 p(χ2) < 0.001 |
| Cholesterol (mg/dL) (140–220) | 219.02 ± 0.90 <140: 3.60% >220: 40.54% | 228.61 ± 1.22 1.81% 56.27% | 207.20 ± 1.28 5.79% 21.16% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| HDL-cholesterol (mg/dL) (35–75) | 55.04 ± 0.28 <35: 5.37% >75: 10.11% | 59.98 ± 0.38 2.35% 15.49% | 48.95 ± 0.34 9.09% 3.47% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| LDL-cholesterol (mg/dL) (80–160) | 140.85 ± 0.77 <80: 5.15% >160: 29.54% | 146.08 ± 1.03 3.69% 33.67% | 134.41 ± 1.12 6.94% 24.46% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| Triglycerides (mg/dL) (50–150) | 113.83 ± 1.31 <50: 5.59% >150: 19.44% | 108.47 ± 1.50 5.97% 16.63% | 120.44 ± 2.26 5.12% 22.89% | p < 0.001 p(χ2) = 0.41 p(χ2) < 0.001 |
| Urea (mg/dL) (15–30) | 42.90 ± 0.26 <15: 0.34% >30: 88.49% | 41.79 ± 0.33 0.34% 86.45% | 44.27 ± 0.40 0.34% 90.99% | p < 0.001 p = 0.75 p(χ2) = 0.37 |
| Creatinine (mg/dL) (0.70–1.40) | 0.91 ± 0.006 <0.70: 14.03% >1.40: 3.59% | 0.81 ± 0.004 23.47% 1.74% | 1.03 ± 0.01 2.40% 8.26% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| Uric acid (mg/dL) (3.4–7.0) | 4.47 ± 0.03 <3.4: 23.77% >7.0: 6.03% | 3.90 ± 0.03 36.02% 2.48% | 5.17 ± 0.05 8.68% 10.41% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| Total Protein (g/dL) (6.5–8.0) | 6.93 ± 0.03 <6.5: 13.77% >8.0: 1.78% | 6.90 ± 0.01 12.54% 1.61% | 6.97 ± 0.07 15.29% 1.98% | p = 0.17 p(χ2) = 0.08 p(χ2) = 0.56 |
| Albumin (g/dL) (3.5–5.0) | 4.20 ± 0.007 <3.5: 1.40% >5.0: 0.95% | 4.18 ± 0.008 1.50% 0.58% | 4.21 ± 0.01 1.60% 1.60% | p = 0.02 p(χ2) = 0.99 p(χ2) = 0.03 |
| Calcium (mg/dL) (8.1–10.4) | 9.24 ± 0.009 <8.1: 0.41% >10.4: 2.04% | 9.27 ± 0.01 0.20% 2.35% | 9.19 ± 0.01 0.66% 1.65% | p < 0.001 p(χ2) = 0.12 p(χ2) = 0.26 |
| Phosphorus (mg/dL) (2.5–5.0) | 3.41 ± 0.01 <2.5: 2.41% >5.0: 0.67% | 3.52 ± 0.01 0.92% 0.81% | 3.27 ± 0.01 3.47% 0.49% | p < 0.001 p(χ2) < 0.002 p(χ2) = 0.46 |
| GOT/ASAT (IU/L) (10–40) | 21.70 ± 0.20 <10: 0.45% >40: 3.85% | 21.29 ± 0.42 0.60% 3.35% | 22.20 ± 0.39 0.25% 4.46% | p = 0.006 p(χ2) = 0.83 p(χ2) = 0.18 |
| GPT/ALAT (IU/L) (9–43) | 23.52 ± 0.36 <9: 2.81% >43: 7.07% | 21.51 ± 0.45 3.02% 4.96% | 26.00 ± 0.56 2.56% 9.67% | p < 0.001 p(χ2) = 0.56 p(χ2) < 0.001 |
| GGT (IU/L) (11–50) | 30.55 ± 0.79 <11: 7.55% >50: 11.81% | 26.37 ± 1.07 11.54% 8.52% | 35.69 ± 1.17 2.65% 15.87% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| Alkaline phosphatase (IU/L) (37–111) | 77.05 ± 0.62 <37: 2.07% >111: 10.37% | 79.41 ± 0.85 1.74% 10.39% | 74.14 ± 0.82 2.48% 10.83% | p < 0.001 p(χ2) = 0.24 p(χ2) = 0.98 |
| Bilirubin (mg/dL) (0.20–1.00) | 0.75 ± 0.02 <0.20: 0.30% >1.00: 15.07% | 0.71 ± 0.04 0.34% 10.33% | 0.80 ± 0.01 0.24% 20.91% | p < 0.001 p(χ2) = 0.95 p(χ2) < 0.001 |
| CPK (IU/L) (38–174) | 92.72 ± 2.00 <38: 8.22% >174: 7.29% | 87.20 ± 3.12 9.25% 5.03% | 99.52 ± 2.29 6.94% 10.08% | p < 0.001 p(χ2) < 0.05 p(χ2) < 0.001 |
| LDH (IU/L) (200–480) | 277.29 ± 1.52 <200: 13.59% >480: 1.74% | 289.77 ± 2.06 6.71% 2.21% | 261.88 ± 2.16 15.87% 1.16% | p < 0.001 p(χ2) < 0.008 p(χ2) < 0.05 |
| Na+ (mEq/L) (135–148) | 141.75 ± 0.04 <135: 0.70% >148: 0.89% | 141.86 ± 0.05 0.36% 0.80% | 141.62 ± 0.06 0.82% 0.99% | p < 0.002 p(χ2) = 0.65 p(χ2) = 0.76 |
| K+ (mEq/L) (3.5–5.3) | 4.33 ± 0.006 <3.5: 0.85% >5.3: 1.15% | 4.29 ± 0.009 1.07% 0.67% | 4.38 ± 0.009 0.57% 1.74% | p < 0.001 p(χ2) = 0.24 p(χ2) < 0.01 |
| Cl- (mEq/L) (98–107) | 104.06 ± 0.07 <98: 1.22% >107: 13.92% | 104.20 ± 0.13 1.07% 14.89% | 103.88 ± 0.07 1.40% 12.96% | p < 0.04 p(χ2) = 0.55 p(χ2) = 0.29 |
| Fe2+ (µg/dL) (35–160) | 86.60 ± 0.70 <35: 5.04% >160: 2.61% | 81.98 ± 0.89 7.01% 1.64% | 92.17 ± 1.10 3.80% 4.20% | p < 0.001 p(χ2) < 0.01 p(χ2) < 0.001 |
| Ferritin (ng/mL) (F: 11–307) (M: 24–336) | 121.05 ± 2.91 <11: 3.43% >307: 7.65% | 81.01 ± 2.48 <11: 5.55% >307: 2.09% | 169.39 ± 5.32 <24: 1.8% >336: 15.3% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| Folate (ng/mL) (>3.00) | 7.94 ± 0.08 <5: 27.14% | 8.31 ± 0.12 23.27% | 7.48 ± 0.12 31.90% | p < 0.001 p(χ2) < 0.001 |
| Vitamin B12 (pg/mL) (170–1000) | 481.98 ± 5.37 <200: 10.10% | 499.87 ± 7.60 6.71% | 459.99 ± 7.44 14.30% | p < 0.001 p(χ2) < 0.001 |
| TSH (µIU/mL) (0.20–4.50) | 1.54 ± 0.04 <0.20: 2.55% >4.50: 2.37% | 1.67 ± 0.07 3.02% 2.88% | 1.38 ± 0.03 1.98% 1.73% | p < 0.001 p(χ2) = 0.12 p(χ2) = 0.07 |
| T4 (ng/mL) (0.54–1.40) | 0.94 ± 0.01 <0.54: 0.77% >1.40: 3.15% | 0.94 ± 0.02 1.07% 3.15% | 0.94 ± 0.01 0.41% 3.14% | p = 0.55 p(χ2) = 0.08 p(χ2) = 0.92 |
| PRL (ng/mL) (F: 1.9–25) (M: 2.5–17) | 10.18 ± 0.53 <1.9: 3.48% >25: 6.53% | 11.74 ± 0.89 <1.9: 2.08% >25: 6.69% | 8.19 ± 0.41 <2.5: 5.17% >17: 6.32% | p < 0.001 p(χ2) < 0.03 p(χ2) = 0.95 |
| Cortisol (µg/dL) (6.02–18.4) | 13.46 ± 0.18 <6: 10% >18: 26.25% | 13.31 ± 0.24 4.39% 17.32% | 13.64 ± 0.26 3.74% 15.80% | p = 0.30 p(χ2) = 0.79 p(χ2) = 0.70 |
| ACTH (pg/mL) (<46) | 23.31 ± 0.62 >50: 5.25% | 21.11 ± 0.78 3.69% | 26.10 ± 0.99 7.18% | p < 0.001 p(χ2) < 0.05 |
| GH (ng/mL) (F. 0.12–9.88) (M: 0.03–2.47) | 0.76 ± 0.04 <0.03–0.12: 11.14% >2.47–9.80: 4.26% | 0.77 ± 0.06 >0.12: 18.25% >9.80: 0.46% | 0.75 ± 0.06 <0.03: 2.3% >2.47: 8.91% | p < 0.04 p(χ2) < 0.001 p(χ2) < 0.001 |
| FSH (mIU/mL) (F: 21.7–153) (M: 0.7–11.1) | 41.37 ± 1.36 <0.7–21.7: 4.10% >11–1–153: 11.14% | 67.07 ± 1.57 <21: 6.93% >153: 0.92% | 9.39 ± 0.47 <0.7: 0.57% >11: 23.85% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| LH (mIU/mL) (F: 7.7–58.5) (M: 1.7–8.6) | 16.84 ± 0.53 <1.7–7.7: 6.79% >8.6–58.5: 7.30% | 26.04 ± 0.63 <7.7: 7.16% >58.5: 2.77% | 5.38 ± 0.20 <1.7: 6.32% >8.6: 12.93% | p < 0.001 p(χ2) = 0.77 p(χ2) < 0.001 |
| Estrogen (pg/mL) (20–30) | 26.23 ± 1.29 <20: 6.95% >30: 16.31% | |||
| Testosterone (ng/dL) (193–740) | 281.45 ± 11.49 <190: 31.15% >740: 1.87% | |||
| α-Amylase (U/L) (28–100) | 59.98 ± 1.26 >100: 6.70% | 57.94 ± 1.64 5.70% | 62.67 ± 1.95 7.77% | p = 0.05 p(χ2) = 0.43 |
| Lipase (U/L) (13–60) | 43.62 ± 0.71 >60: 10.58% | 42.98 ± 0.76 9.56% | 44.46 ± 1.29 11.94% | p = 0.75 p(χ2) = 0.42 |
| AFP (ng/mL) (0–7) | 3.17 ± 0.10 >7: 5.44% | 3.33 ± 0.10 5.88% | 2.97 ± 0.19 4.85% | p < 0.001 p(χ2) = 0.68 |
| CEA (ng/mL) (0–3.8) | 3.97 ± 1.40 >3.8: 16.76% | 2.32 ± 0.09 12.99% | 6.14 ± 2.26 21.75% | p < 0.05 p(χ2) < 0.01 |
| CA 19.9 (U/mL) (0–27) | 14.41 ± 2.05 >27: 8.94% | 11.60 ± 0.61 7.35% | 18.14 ± 4.70 11.04% | p = 0.27 p(χ2) = 0.15 |
| CA 72.4 (U/mL) (0–6.9) | 3.96 ± 0.86 >6.9: 13.01% | 4.86 ± 1.46 14.71% | 2.85 ± 0.66 10.91% | p = 0.18 p(χ2) = 0.78 |
| CA 125 (U/mL) (0–35) | 16.38 ± 1.67 >35: 5.97% | 15.82 ± 2.42 4.19% | 17.16 ± 2.14 8.50% | p = 0.87 p(χ2) = 0.16 |
| CYFRA 21.1 (ng/mL) (0–3.3) | 1.85 ± 0.08 >3.3: 10.39% | 1.76 ± 0.06 9.52% | 1.94 ± 0.15 11.32% | p = 0.83 p(χ2) = 0.76 |
| SCC (ng/mL) (0–2.3) | 1.30 ± 0.09 >2.3: 9.785 | 1.05 ± 0.05 6.59% | 1.55 ± 0.18 12.96% | p < 0.001 p(χ2) = 0.11 |
| NSE (ng/mL) (0–16.3) | 10.28 ± 0.17 >16.3: 3.95% | 10.77 ± 0.24 5.36% | 9.78 ± 0.23 2.48% | p < 0.001 p(χ2) = 0.31 |
| PSA (ng/mL) (<4) | 2.31 ± 0.16 >4: 13.45% | |||
| CA 15.3 (U/mL) (0–26.4) | 14.05 ± 0.58 >26.4: 7.25% | |||
| RBC (×106/µL) (3.80–5.50) | 4.62 ± 0.008 <3.80: 2.78% >5.50: 3.99% | 4.47 ± 0.01 4.09% 2.01% | 4.81 ± 0.01 1.16% 6.45% | p < 0.001 p(χ2) < 0.001 p(χ2) = 0.001 |
| HCT (%) (40.0–50.0) | 42.41 ± 0.23 <40.0: 29.58% >50.0: 3.07% | 40.69 ± 0.26 42.85% 0.54% | 44.53 ± 0.39 13.22% 6.19% | p < 0.001 p(χ2) = 0.001 p(χ2) = 0.001 |
| Hb (g/dL) (13.5–17.0) | 14.04 ± 0.02 <13.5: 32.06% >17.0: 2.29% | 13.47 ± 0.03 46.88% 0.13% | 14.74 ± 0.04 13.80% 4.95% | p < 0.001 p(χ2) = 0.001 p(χ2) = 0.001 |
| MCV (fL) (80–100) | 91.06 ± 0.10 <80: 2.29% >100: 3.59% | 90.48 ± 0.13 2.41% 2.55% | 91.77 ± 0.15 2.15% 4.88% | p < 0.001 p(χ2) = 0.75 p(χ2) < 0.003 |
| MCH (pg) (27.0–33.0) | 30.41 ± 0.03 <27.0: 3.62% >33.0: 5.48% | 30.19 ± 0.05 4.02% 5.23% | 30.69 ± 0.05 3.14% 8.26% | p < 0.001 p(χ2) = 0.28 p(χ2) < 0.004 |
| MCHC (g/dL) (31.0–35.0) | 33.37 ± 0.01 <31.0: 0.63% >35.0: 2.33% | 33.32 ± 0.02 0.80% 2.35% | 33.43 ± 0.02 0.41% 2.31% | p < 0.002 p(χ2) = 0.30 p(χ2) = 0.94 |
| ADE (RDW) (%) (11.0–15.0) | 12.95 ± 0.02 <11.0: 1.81% >15.0: 5.63% | 13.01 ± 0.03 1.61% 6.24% | 12.88 ± 0.03 2.07% 4.88% | p < 0.05 p(χ2) = 0.47 p(χ2) = 0.17 |
| WBC (×103/µL) (4.0–11.0) | 6.35 ± 0.03 <4.0: 5.41% >11.0: 2.55% | 6.18 ± 0.05 7.18% 2.28% | 6.56 ± 0.05 3.47% 2.89% | p < 0.001 p(χ2) < 0.001 p(χ2) = 0.39 |
| %Neu (45.0–70.0) | 60.15 ± 0.19 <45.0: 6.03% >70.0: 17.92% | 59.98 ± 0.25 7.04% 15.16% | 60.35 ± 0.30 4.79% 16.86% | p = 0.22 p(χ2) < 0.02 p(χ2) = 0.33 |
| %Lym (20.0–40.0) | 29.60 ± 0.22 <20: 13.44% >40: 11.51% | 30.22 ± 0.22 11.80% 12.81% | 28.83 ± 0.42 15.45% 9.92% | p < 0.001 p(χ2) < 0.02 p(χ2) < 0.04 |
| %Mon (3.0–10.0) | 7.24 ± 0.03 <3.0: 1.33% >10.0: 9.48% | 7.03 ± 0.05 1.67% 7.31% | 7.50 ± 0.06 0.91% 12.15% | p < 0.001 p(χ2) = 0.12 p(χ2) < 0.001 |
| %Eos (1.0–5.0) | 2.81 ± 0.05 <1.0: 5.73% >5.0: 23.92% | 2.61 ± 0.06 6.71% 24.21% | 3.05 ± 0.09 4.55% 23.55% | p < 0.001 p(χ2) < 0.001 p(χ2) < 0.001 |
| %Bas (0.0–1.0) | 0.69 ± 0.07 >1.0: 14.09% | 0.69 ± 0.14 9.09% | 0.69 ± 0.01 19.60% | p < 0.001 p(χ2) < 0.001 |
| PLT (×103/µL) (150–450) | 227.11 ± 1.25 <150: 7.59% >450: 0.66% | 239.85 ± 1.67 4.63% 0.55% | 211.29 ± 1.80 11.24% 0.85% | p < 0.001 p(χ2) < 0.001 p(χ2) = 0.49 |
| MPV (fL) (6.0–10.0) | 8.55 ± 0.01 <6.0: 0.18% >10.0: 9.03% | 8.55 ± 0.02 0.13% 8.58% | 8.55 ± 0.02 0.25% 9.58% | p = 0.99 p(χ2) = 0.49 p(χ2) = 0.44 |
| MMSE Score (0–30) <25/30 | 23.05 ± 0.13 47.38% | 22.07 ± 0.19 54.46% | 24.25 ± 0.19 38.68% | p < 0.001 p(χ2) < 0.001 |
| ADAS-Cog | 9.37 ± 0.21 | 9.94 ± 0.30 | 8.65 ± 0.31 | p < 0.001 |
| ADAS-Mem | 11.40 ± 0.12 | 11.67 ± 0.16 | 11.05 ± 0.17 | p < 0.02 |
| ADAS-Cog-T | 19.06 ± 0.30 | 19.91 ± 0.41 | 18.04 ± 0.43 | p < 0.002 |
| ADAS-NonCog | 4.75 ± 0.08 | 5.36 ± 0.12 | 3.98 ± 0.12 | p < 0.001 |
| ADAS-T | 23.18 ± 0.36 | 24.66 ± 0.49 | 21.34 ± 0.51 | p < 0.001 |
| HARS <10: Normal 11–17: Mild 18–24: Mild-Moderate 25–30: Moderate-Severe | 11.28 ± 0.11 39.97% 46.65% 11.36% 2.01% | 12.45 ± 0.15 30.73% 51.29% 15.04% 2.94% | 9.82 ± 0.15 51.37% 40.94% 6.81% 0.88% | p < 0.001 p < 0.001 p < 0.002 p < 0.001 p < 0.001 |
| HDRS 0–7: Normal 8–13: Mild 14–18: Moderate 19–22: Severe >23: Very Severe | 10.06 ± 0.10 34.60% 41.03% 17.18% 4.78% 2.41% | 11.02 ± 0.14 28.67% 42.65% 21.84% 6.54% 3.24% | 8.88 ± 0.15 42.93% 40.55% 12.19% 2.83% 1.50% | p < 0.001 p < 0.001 p = 0.52 p < 0.001 p < 0.001 p < 0.01 |
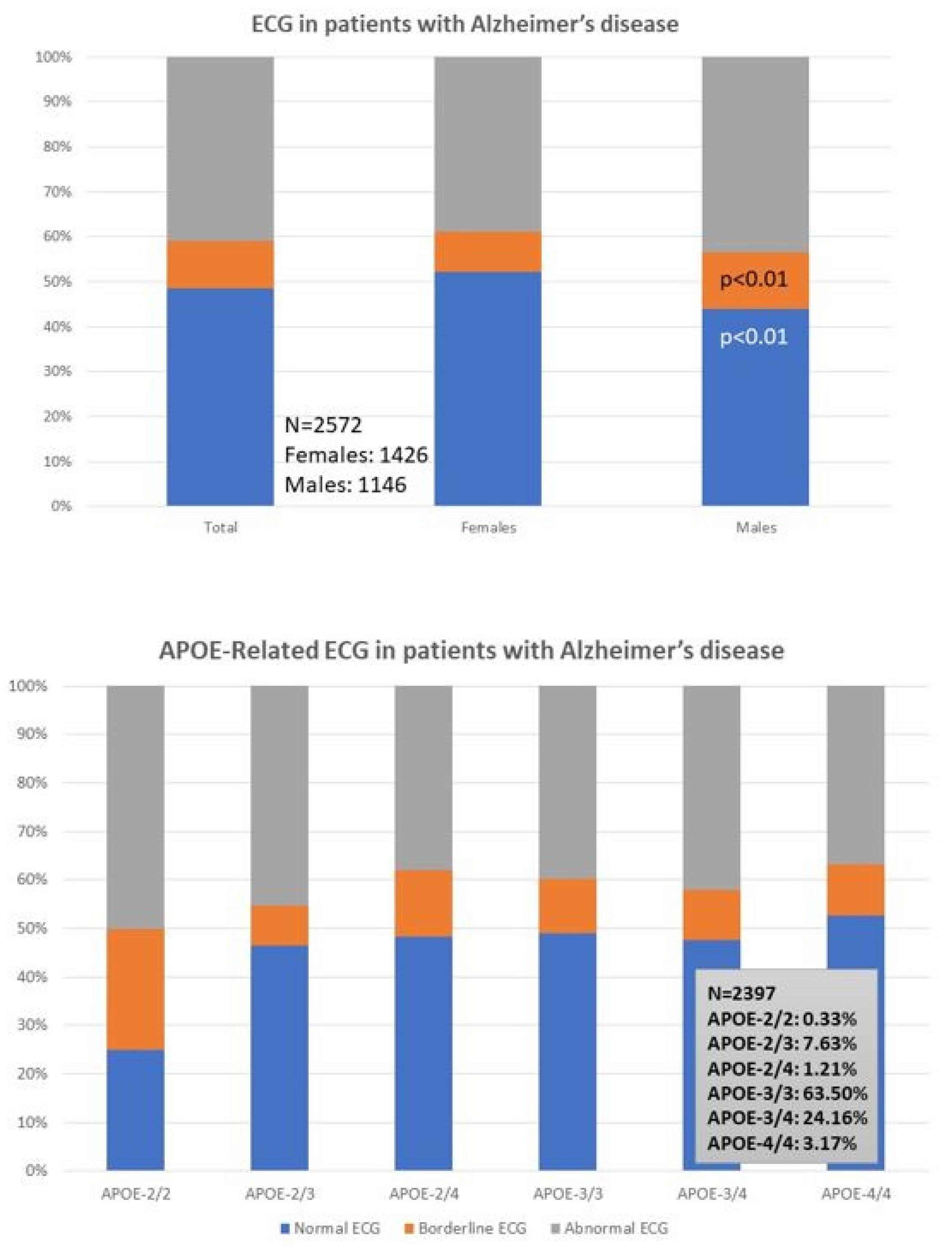
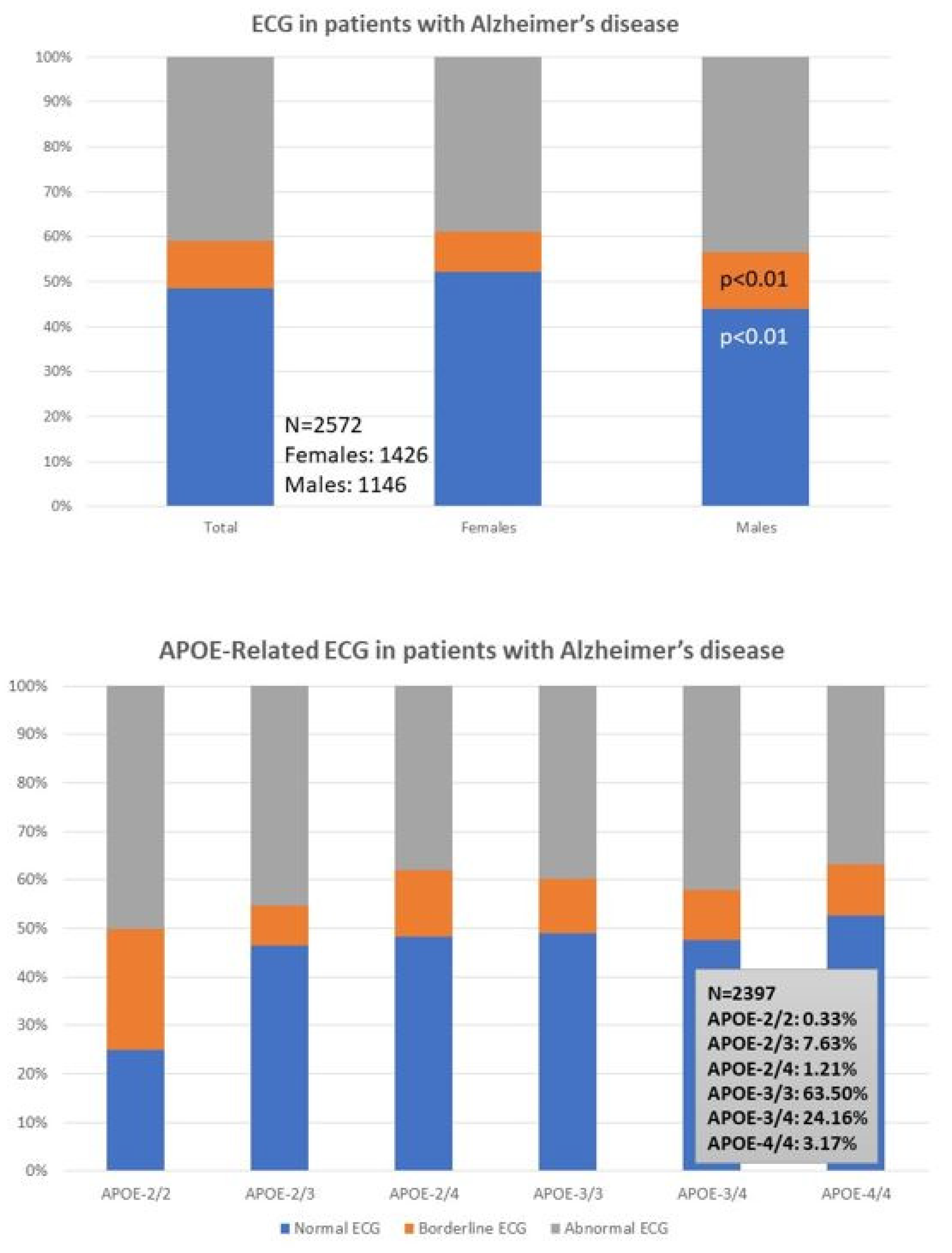
| ECG Normal Borderline Abnormal | 48.44% 10.73% 40.83% | 52.10% 9.12% 38.78% | 43.89% 12.74% 43.37% | p < 0.01 p < 0.01 p = 0.13 |
| MRI Normal Abnormal | 26.78% 73.22% | 29.00% 71.00% | 24.16% 75.84% | p = 0.52 p = 0.20 |
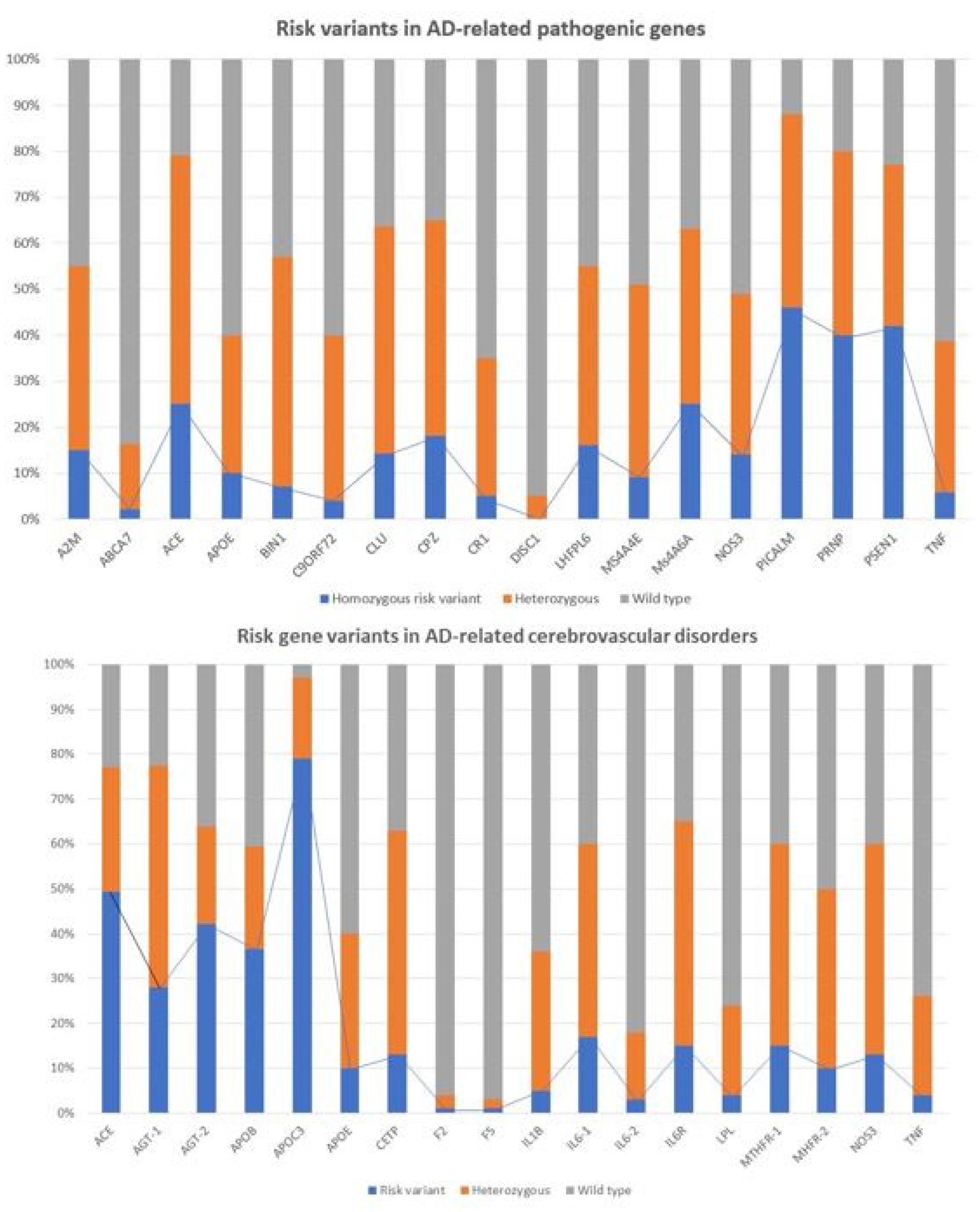
| Gene | Gene Name | OMIM | Location | dbSNP | Polymorphism | MAF | Genotype |
|---|---|---|---|---|---|---|---|
| AA: 45.61% | |||||||
| A2M | alpha-2-macroglobulin | 103,950 | chr12:9067708 | rs669 | c.2998A>G | 0.31 (G) | AG: 40.35% |
| GG: 14.04% | |||||||
| TT: 77.19% | |||||||
| ABCA7 | ATP binding cassette subfamily A member 7 | 605,414 | chr19:1046521 | rs3764650 | c.1622+115T>G | 0.20 (G) | TG: 21.05% |
| GG: 1.76% | |||||||
| CC: 21.05% | |||||||
| ACE | angiotensin I converting enzyme | 106,180 | chr17:63477060 | rs4332 | c.496-66T>C | 0.47 (T) | CT: 54.39% |
| TT: 24.56% | |||||||
| *2*2: <1% | |||||||
| *2*3: 1.75% | |||||||
| APOE | apolipoprotein E | 107,741 | chr19:44908822 | rs7412 | c.4070C>T | 0.08 (T) | *2*4: 1.75% |
| rs429358 | c.3932T>C | 0.15 (C) | *3*3: 59.65% | ||||
| *3*4: 33.34% | |||||||
| *4*4: 3.51% | |||||||
| AA: 42.11% | |||||||
| BIN1 | bridging integrator 1 | 601,248 | chr2:127137039 | rs744373 | g.127137039A>G | 0.36 (G) | AG: 50.88% |
| GG: 7.01% | |||||||
| CC: 59.65% | |||||||
| C9ORF72 | chromosome 9 open reading frame 72 | 614,260 | chr9:27543280 | rs3849942 | g.27543283T>C | 0.22 (T) | TC: 36.84% |
| TT: 3.51% | |||||||
| GG: 36.84% | |||||||
| CLU | Clusterin | 185,430 | chr8:27607002 | rs11136000 | c.247-478A>G | 0.38 (A) | AG: 49.12% |
| AA: 14.04% | |||||||
| CC: 17.54% | |||||||
| CPZ | carboxypeptidase Z | 603,105 | chr4:8650823 | rs7436874 | g.8649098C>T | 0.36 (C) | CT: 47.37% |
| TT: 35.09% | |||||||
| GG: 64.92% | |||||||
| CR1 | complement C3b/C4b receptor 1 | 120,620 | chr1:207611623 | rs3818361 | c.4946-54A>G | 0.25 (A) | AG: 29.82% |
| AA: 5.26% | |||||||
| TT: 94.74% | |||||||
| DISC1 | disrupted in schizophrenia 1 | 605,210 | chr1:232155150 | rs16856202 | c.2242-7030T>G | 0.03 (G) | TG: 5.26% |
| GG: <1% | |||||||
| AA: 45.61% | |||||||
| LHFPL6 | LHFPL tetraspan subfamily member 6 | 606,710 | chr13:39872236 | rs7995844 | g.39298100G>A | 0.35 (G) | GA: 38.60% |
| GG: 15.79% | |||||||
| CC: 49.12% | |||||||
| MS4A4E | membrane spanning 4-domains A4E | 608,401 | chr11:60204322 | rs670139 | c.279-2443C>A | 0.38 (A) | CA: 42.11% |
| AA: 8.77% | |||||||
| CC: 36.84% | |||||||
| MS4A6A | membrane spanning 4-domains A6A | 606,548 | chr11:60171834 | rs610932 | c.*149+175A>C | 0.45 (A) | CA: 38.60% |
| AA: 24.56% | |||||||
| GG: 50.88% | |||||||
| NOS3 | nitric oxyde synthse 3 | 163,729 | chr7:150991055 | rs1799983 | c.894G>T | 0.18 (T) | GT: 35.09% |
| TT: 14.03% | |||||||
| CC: 45.61% | |||||||
| PICALM | phosphatidylinositol binding clathrin assembly protein | 603,025 | chr11:86157598 | rs3851179 | g.85868640T>C | 0.31 (T) | CT: 42.11% |
| TT: 12.28% | |||||||
| GG: 19.30% | |||||||
| PRNP | prion protein | 176,640 | chr20:4686093 | rs1799990 | c.385A>G | 0.73 (A) | AG: 40.35% |
| AA: 40.35% | |||||||
| TT: 22.80% | |||||||
| PSEN1 | presenilin 1 | 104,311 | chr14:73136434 | rs165932 | c.856+16G>T | 0.43 (G) | GT: 35.09% |
| GG: 42.11% | |||||||
| GG: 73.68% | |||||||
| TNF | tumor necrosis factor | 191,160 | chr6:31575566 | rs1800629 | c.-308G>A | 0.09 (A) | GA: 22.81% |
| AA: 3.51% |
| Gene Symbol | Gene Name | OMIM | Location | dbSNP ID | Polymorphism | MAF | Genotype |
|---|---|---|---|---|---|---|---|
| CC: 18.70% | |||||||
| ACE | angiotensin I converting enzyme | 106,180 | chr17:63486920 | rs4332 | c.496-66T>C | 0.47 (T) | CT: 39.81% |
| TT: 41.49% | |||||||
| CC: 11.46% | |||||||
| AGT | Angiotensinogen | 106,150 | chr1:230710231 | rs4762 | c.620C>T | 0.10 (T) | CT: 21.96% |
| TT: 66.58% | |||||||
| TT: 21.97% | |||||||
| AGT | Angiotensinogen | 106,150 | chr1:230710048 | rs699 | c.803T>C | 0.30 (T) | TC: 56.48% |
| CC: 21.61% | |||||||
| CC: 29.03% | |||||||
| APOB | apolipoprotein B | 107,730 | chr2:21009323 | rs693 | c.2488C>T | 0.25 (T) | CT: 47.64% |
| TT: 23.33% | |||||||
| CC: 78.94% | |||||||
| APOC3 | apolipoprotein C-III | 107,720 | chr11:116832924 | rs5128 | c.3175C>G | 0.23 (C) | CG: 17.60% |
| GG: 3.46% | |||||||
| *2*2: 0.32% | |||||||
| *2*3: 7.62% | |||||||
| APOE | apolipoprotein E | 107,741 | chr19:44908822 | rs7412 | c.4070C>T | 0.08 (T) | *2*4: 1.28% |
| chr19:44908684 | rs429358 | c.3932T>C | 0.15 (C) | *3*3: 63.73% | |||
| *3*4:23.88% | |||||||
| *4*4: 3.17% | |||||||
| GG: 37.39% | |||||||
| CETP | cholesteryl ester transfer protein | 118,470 | chr16:56962376 | rs708272 | c.+279G>A | 0.38 (A) | GA: 49.42% |
| AA: 13.19% | |||||||
| GG: 96.41% | |||||||
| F2 | coagulation factor II, thrombin | 176,930 | chr11:46739505 | rs1799963 | c.20210G>A | 0.01 (A) | GA: 3.47% |
| AA: 0.12% | |||||||
| GG: 98.02% | |||||||
| F5 | coagulation factor V | 227,400 | chr1:169549811 | rs6025 | c.1691G>A | 0.01 (A) | GA: 1.61% |
| AA: 0.37% | |||||||
| TT: 4.59% | |||||||
| IL1B | interleukin 1 beta | 147,720 | chr2:112832813 | rs1143634 | c.3954T>C | 0.13 (T) | TC: 31.39% |
| CC: 64.02% | |||||||
| GG: 39.95% | |||||||
| IL6 | interleukin 6 | 147,620 | chr7:22727026 | rs1800795 | c.-174G>C | 0.14 (C) | GC: 43.55% |
| CC: 16.50% | |||||||
| GG: 81.12% | |||||||
| IL6 | interleukin 6 | 147,620 | chr7:22726627 | rs1800796 | c.-573G>C | 0.31 (C) | GC: 15.90% |
| CC: 2.98% | |||||||
| AA: 34.41% | |||||||
| IL6R | interleukin 6 receptor | 147,880 | chr1:154454494 | rs2228145 | c.1510A>C | 0.36 (C) | AC: 49.69% |
| CC: 15.90% | |||||||
| CC: 76.02% | |||||||
| LPL | lipoprotein lipase | 609,708 | chr8:19962213 | rs328 | c.1421C>G | 0.09 (G) | CG: 20.00% |
| GG: 3.98% | |||||||
| CC: 38.90% | |||||||
| MTHFR | methylenetetrahydrofolate reductase | 607,093 | chr1:11796321 | rs1801133 | c.665C>T | 0.25 (T) | CT: 45.84% |
| TT: 15.26% | |||||||
| AA: 50.36% | |||||||
| MTHFR | methylenetetrahydrofolate reductase | 607,093 | chr1:11794419 | rs1801131 | c.1286A>C | 0.25 (C) | AC: 39.90% |
| CC: 9.74% | |||||||
| GG: 39.54% | |||||||
| NOS3 | nitric oxyde synthse 3 | 163,729 | chr7:150991055 | rs1799983 | c.894G>T | 0.18 (T) | GT: 47.67% |
| TT: 12.79% | |||||||
| GG: 73.79% | |||||||
| TNF | tumor necrosis factor | 191,160 | chr6:31575566 | rs1800629 | c.-308G>A | 0.09 (A) | GA: 22.86% |
| AA: 3.35% |
| Drug | Properties | Pharmacogenetics |
|---|---|---|
 | Name:Aducanumab, BIIB-037, Aduhelm, 1384260-65-4 IUPAC Name: Immunoglobulin G1, anti-(human.beta.-amyloid) (human monoclonal biib037 heavy chain), disulfide with human monoclonal biib037.kappa.-chain, and dimer Molecular Formula: C6472H10028N1740O2014S46 Molecular Weight: 145910.3123 g/mol Category: Monoclonal antibody (mAb), anti-amyloid beta A4 protein Mechanism: Monoclonal IgG1 antibody that binds to amyloid-β, reducing amyloid plaques in the brain Effect: Anti-amyloid beta A4 protein slowing the rate of progression of Alzheimer’s disease and levels of p-tau in the cerebrospinal fluid | Pathogenic genes: APP, APOE, PSEN1, PSEN2 Mechanistic genes: Drug metabolism-related genes: -Substrate: -Inhibitor: Transporter genes: Pleiotropic genes: APOE, IL6, IL1B, TNF |
 | Name: Donepezil hydrochloride, Aricept, 120011-70-3, Donepezil HCl, BNAG, E-2020, and E2020 IUPAC Name: 2-[(1-benzylpiperidin-4-yl)methyl]-5,6-dimethoxy-2,3-dihydroinden-1-one;hydrochloride Molecular Formula: C24H30ClNO3 Molecular Weight: 415.9529 g/mol Category: Cholinesterase inhibitor Mechanism: Centrally active, reversible acetylcholinesterase inhibitor; increases the acetylcholine available for synaptic transmission in the CNS Effect: Nootropic agent, cholinesterase inhibitor, and parasympathomimetic effect | Pathogenic genes: APP, APOE, CHAT Mechanistic genes: ACHE, BCHE, CHAT, CHRNA7 Drug metabolism-related genes: -Substrate: CYP2D6 (major), CYP3A4 (major), UGTs, ACHE -Inhibitor: ABCB1, ACHE, BCHE, hERG Transporter genes: ABCB1, ABCA1, ABCG2, SCN1A Pleiotropic genes: APOE, PLP, MAG, MBP, CNPase, MOG |
 | Name: Galantamine hydrobromide: Galanthamine hydrobromide, 1953-04-4, Nivalin, Razadyne, UNII-MJ4PTD2VVW, and Nivaline IUPAC Name: (1S,12S,14R)-9-methoxy-4-methyl-11-oxa-4-azatetracyclo [8.6.1.0^{1,12}.0^{6,17}]heptadeca-6,8,10(17),15-tetraen-14-ol Molecular Formula: C17H22BrNO3 Molecular Weight: 368.26548 g/mol Category: Cholinesterase inhibitor Mechanism: Reversible and competitive acetylcholinesterase inhibition leading to an increased concentration of acetylcholine at cholinergic synapses; modulates nicotinic acetylcholine receptor; may increase glutamate and serotonin levels Effect: Nootropic agent, cholinesterase inhibitor, and parasympathomimetic effect | Pathogenic genes: APOE, APP Mechanistic genes: ACHE, BCHE, CHRNA4, CHRNA7, CHRNB2, SLC18A3 Drug metabolism-related genes: -Substrate: ABCB1, CYP2D6 (major), CYP3A4 (major), UGT1A1 -Inhibitor: ACHE, BCHE Transporter genes: ABCB1, SLC18A3 |
 | Name: Memantine Hydrochloride, 41100-52-1, Namenda, Memantine HCL, Axura, 3,5-Dimethyl-1-adamantanamine hydrochloride, and 3,5-dimethyladamantan-1-amine hydrochloride IUPAC Name: 3,5-dimethyladamantan-1-amine;hydrochloride Molecular Formula: C12H22ClN Molecular Weight: 215.76278 g/mol Category: N-Methyl-D-Aspartate receptor antagonist Mechanism: Binds preferentially to NMDA receptor-operated cation channels; may act by blocking actions of glutamate, mediated in part by NMDA receptors Effect: Dopamine agent, antiparkinson agent, excitatory amino acid antagonist, and antidyskinetic | Pathogenic genes: APOE, MAPT, PSEN1 Mechanistic genes: CHRFAM7A, DLGAP1, FOS, GRIN2A, GRIN2B, GRIN3A, HOMER1, HTR3A Drug metabolism-related genes: -Inhibitor: CYP1A2 (weak), CYP2A6 (weak), CYP2B6 (strong), CYP2C9 (weak), CYP2C19 (weak), CYP2D6 (strong), CYP2E1 (weak), CYP3A4 (weak), NR1I2 Transporter genes: NR1I2 Pleiotropic genes: APOE, MAPT, MT-TK, PSEN1 |
 | Name: Rivastigmine tartrate, 129101-54-8, SDZ-ENA 713, Rivastigmine hydrogentartrate, Rivastigmine Hydrogen Tartrate, ENA 713, and ENA-713 IUPAC Name: (2R,3R)-2,3-dihydroxybutanedioic acid;[3-[(1S)-1-(dimethylamino)ethyl]phenyl] N-ethyl-N-methylcarbamate Molecular Formula: C18H28N2O8 Molecular Weight: 400.42352 g/mol Category: Cholinesterase inhibitor Mechanism: Increases acetylcholine in CNS through reversible inhibition of its hydrolysis by acetylcholinesterase Effect: Neuroprotective agent, cholinesterase inhibitor, and cholinergic agent | Pathogenic genes: APOE, APP, CHAT Mechanistic genes: ACHE, BCHE, CHAT, CHRNA4, CHRNB2, SLC18A3 Drug metabolism-related genes: -Substrate: UGT1A9, UGT2B7 -Inhibitor: ACHE, BCHE Transporter genes: SLC18A3 Pleiotropic genes: APOE, MAPT |
 | Name: Tacrine Hydrochloride, Tacrine HCl, 1684-40-8, Hydroaminacrine, tacrine.HCl, 9-AMINO-1,2,3,4-TETRAHYDROACRIDINE HYDROCHLORIDE, and Tenakrin IUPAC Name: 1,2,3,4-tetrahydroacridin-9-amine;hydrochloride Molecular Formula: C13H15ClN2 Molecular Weight: 234.7246 g/mol Category: Cholinesterase inhibitor Mechanism: Elevates acetylcholine in cerebral cortex by slowing degradation of acetylcholine Effect: Nootropic agent, cholinesterase inhibitor, and parasympathomimetic effect | Pathogenic genes: APOE Mechanistic genes: ACHE, BCHE, CHRNA4, CHRNB2 Drug metabolism-related genes: -Substrate: CYP1A2 (major), CYP2D6 (minor), CYP3A4 (major), CES1, GSTM1, GSTT1 -Inhibitor: ACHE, BCHE, CYP1A2 (weak) Transporter genes: ABCB4, SCN1A Pleiotropic genes: APOE, LEPR, MTHFR |
 | Name: (-)-Huperazine A, Huperzine A; Huperzine-A; 102518-79-6; (-)-Huperzine A; (+/−)-Huperzine A IUPAC name: (1R,9R,13E)-1-amino-13-ethylidene-11-methyl-6-azatricyclo[7.3.1.02,7]trideca-2(7),3,10-trien-5-one Molecular Formula: C15H18N2O Molecular Weight: 242.32g/mol Category: Neuroprotectanct, Cholinesterase Inhibitor Mechanism: Increases acetylcholine in the brain by inhibiting acetylcholinesterase and slowing acetylcholine hydrolysis Effect: Neuroprotective, acetylcholinesterase inhibitor, cognitive enhancer, and antiepileptic | Pathogenic genes: APP, APOE Mechanistic genes: ACHE Drug metabolism-related genes: -Substrate: ABCB1, CYP1A2, CYP3A1, CYP3A2, CYP2C11, CYP2E1, CES1, CES2 -Inhibitor: ACHE -Inducer: CYP1A2 Transporter genes: ABCB1, ABCG2 Pleiotropic genes: APOE, BDNF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacabelos, R.; Naidoo, V.; Martínez-Iglesias, O.; Corzo, L.; Cacabelos, N.; Pego, R.; Carril, J.C. Personalized Management and Treatment of Alzheimer’s Disease. Life 2022, 12, 460. https://doi.org/10.3390/life12030460
Cacabelos R, Naidoo V, Martínez-Iglesias O, Corzo L, Cacabelos N, Pego R, Carril JC. Personalized Management and Treatment of Alzheimer’s Disease. Life. 2022; 12(3):460. https://doi.org/10.3390/life12030460
Chicago/Turabian StyleCacabelos, Ramón, Vinogran Naidoo, Olaia Martínez-Iglesias, Lola Corzo, Natalia Cacabelos, Rocío Pego, and Juan C. Carril. 2022. "Personalized Management and Treatment of Alzheimer’s Disease" Life 12, no. 3: 460. https://doi.org/10.3390/life12030460