
Spatial and Species Variations of Bacterial Community Structure and Putative Function in Seagrass Rhizosphere Sediment

Abstract
:1. Introduction
2. Materials and Methods
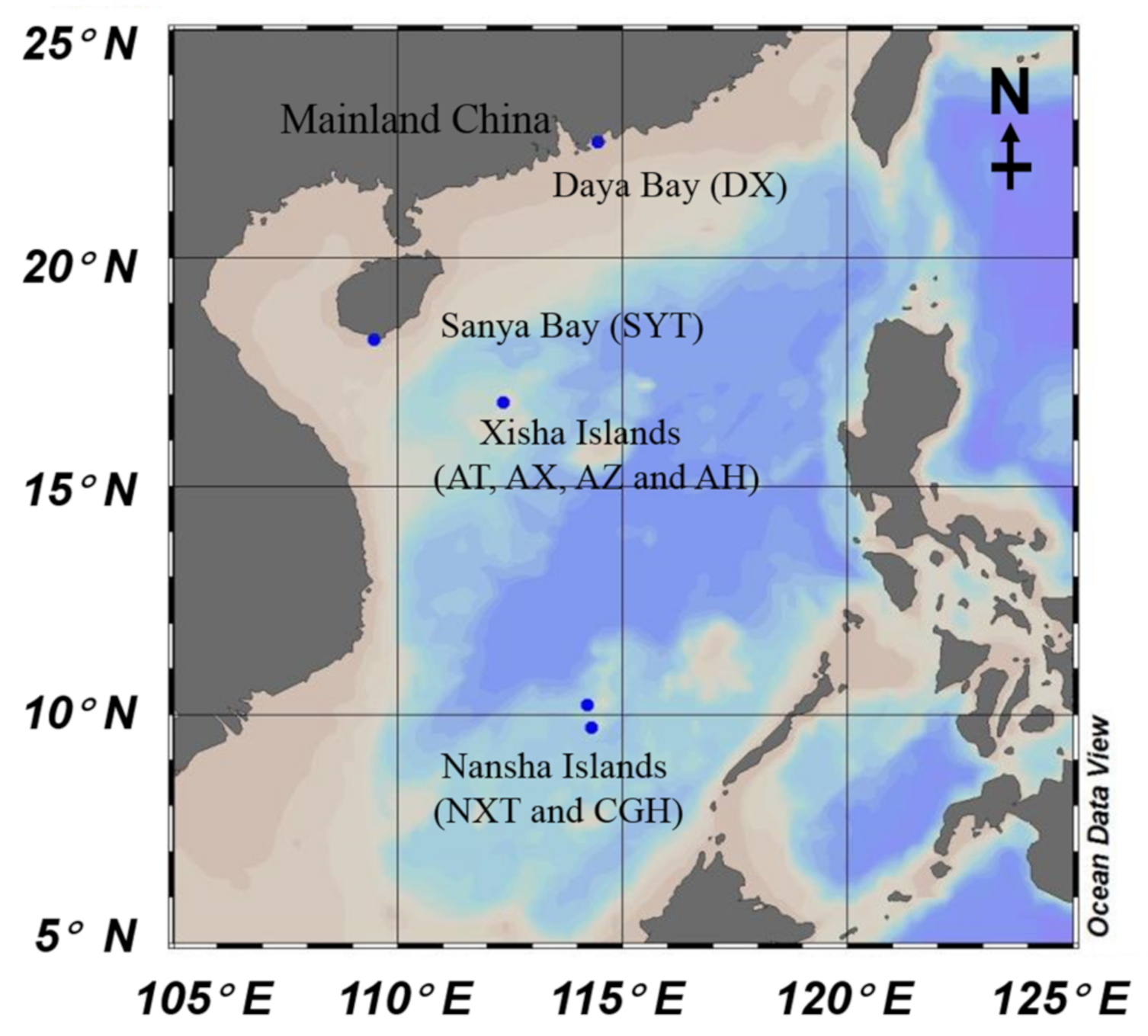
2.1. Description of Sampling Location
2.2. Sample Collection and Physic-Chemical Property Measurement
2.3. DNA Extraction, PCR, Library Preparation, and Illumina MiSeq Sequencing
2.4. Statistical Analysis
3. Results
3.1. Sampling Locations and Physicochemical Properties of the Seagrass Rhizosphere
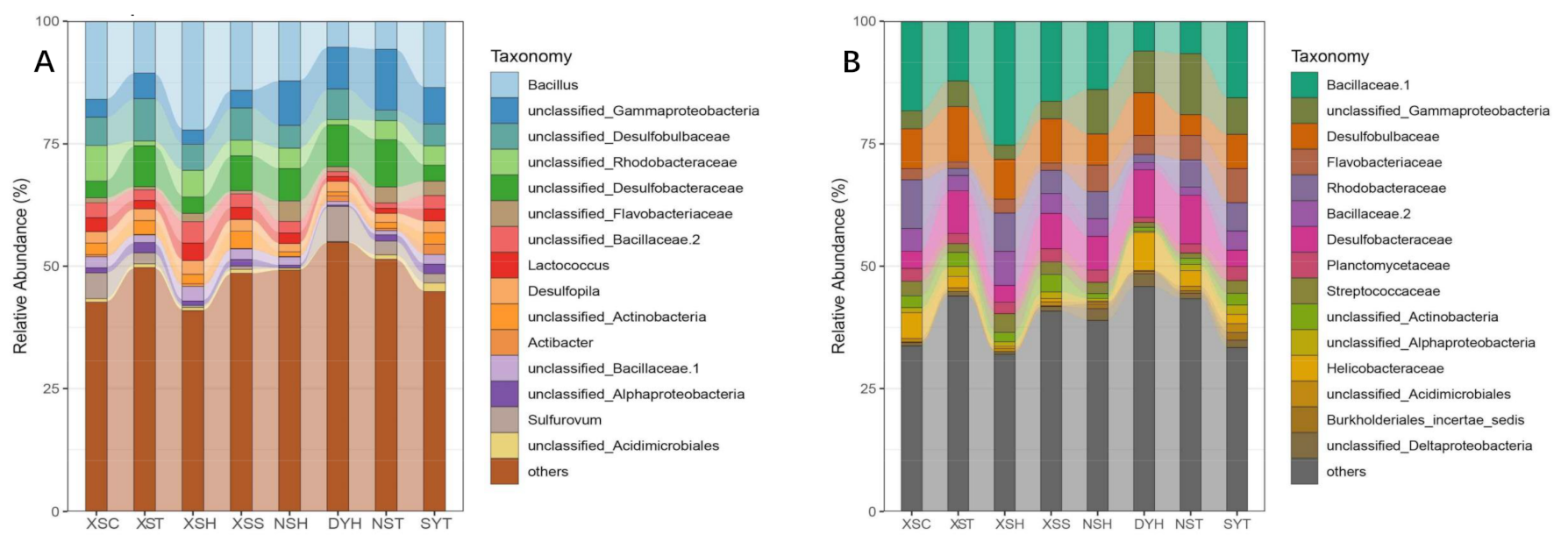
3.2. Taxonomy, Phylogenetic Diversity, and Composition of Rhizosphere Bacterial Communities of the Seagrass
3.3. Potential Functional Roles of Microbial Played in Seagrass Rhizosphere
3.4. Venn Diagram Analysis of the Variations in Taxonomy Species and Functional Groups
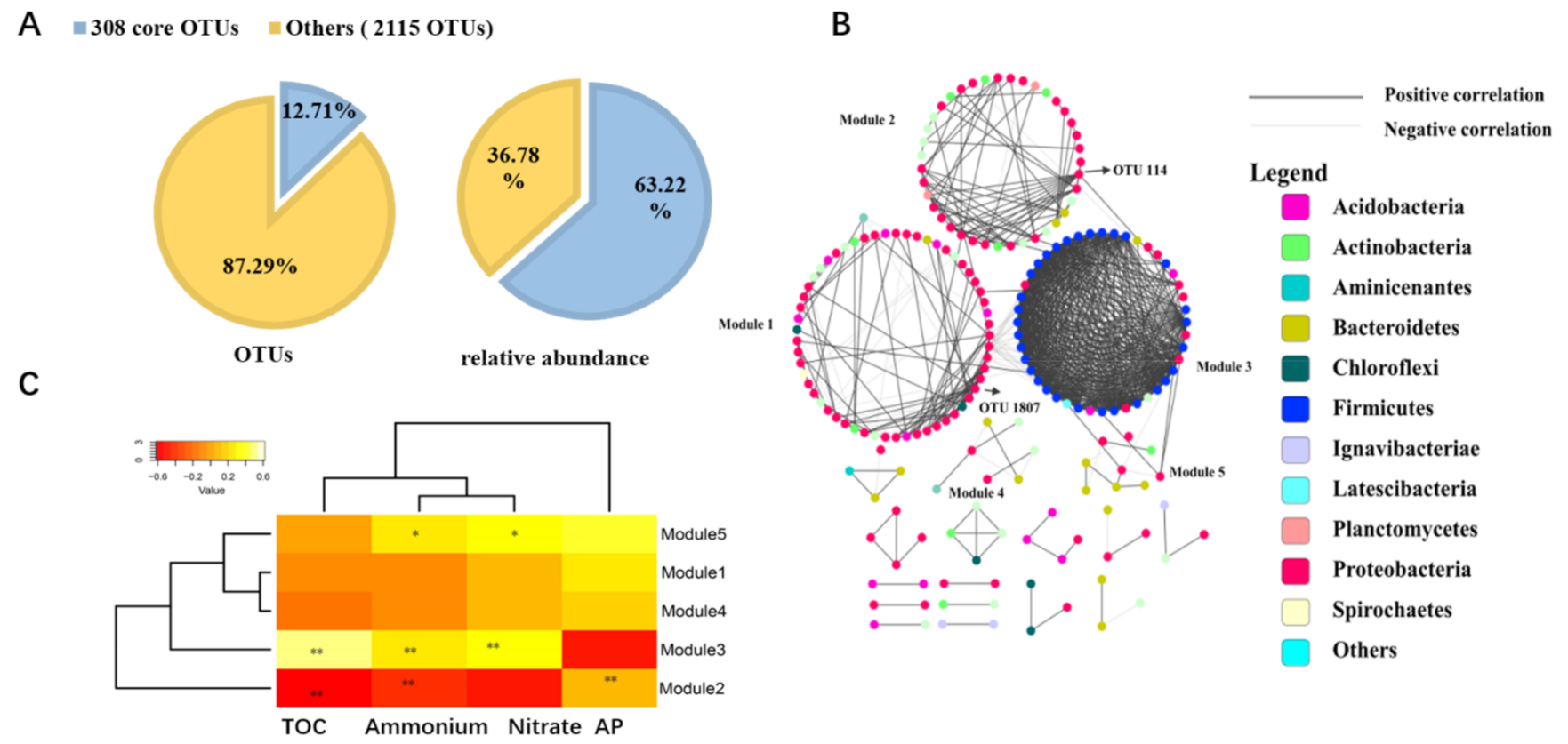
3.5. Core Microbial Community in Seagrass Microbial Rhizosphere
4. Discussion
4.1. Variations in the Taxonomical, Phylogenetical Diversity and Composition of Bacterial Communities
4.2. The Functional Structure of Microbial Communities in Seagrass Rhizosphere
4.3. The Core Microbial Community in Seagrass Rhizosphere across the Four Coral Reef Ecosystems
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, T.P.; Barnes, M.L.; Bellwood, D.R.; Cinner, J.E.; Cumming, G.S.; Jackson, J.B.C.; Kleypas, J.; van de Leemput, I.A.; Lough, J.M.; Morrison, T.H.; et al. Coral reefs in the Anthropocene. Nature 2017, 546, 82–90. [Google Scholar] [CrossRef]
- Andersson, A.J.; Gledhill, D. Ocean acidification and coral reefs: Effects on breakdown, dissolution, and net ecosystem calcification. Ann. Rev. Mar. Sci. 2013, 5, 321–348. [Google Scholar] [CrossRef]
- Hughes, T.P.; Baird, A.H.; Bellwood, D.R.; Card, M.; Connolly, S.R.; Folke, C.; Grosberg, R.; Hoegh-Guldberg, O.; Jackson, J.B.; Kleypas, J.; et al. Climate change, human impacts, and the resilience of coral reefs. Science 2003, 301, 929–933. [Google Scholar] [CrossRef] [Green Version]
- Sully, S.; Burkepile, D.E.; Donovan, M.K.; Hodgson, G.; van Woesik, R. A global analysis of coral bleaching over the past two decades. Nat. Commun. 2019, 10, 1264. [Google Scholar] [CrossRef] [Green Version]
- Unsworth, R.K.F.; Collier, C.J.; Henderson, G.M.; McKenzie, L.J. Tropical seagrass meadows modify seawater carbon chemistry: Implications for coral reefs impacted by ocean acidification. Environ. Res. Lett. 2012, 7, 024026. [Google Scholar] [CrossRef]
- Macreadie, P.I.; Anton, A.; Raven, J.A.; Beaumont, N.; Connolly, R.M.; Friess, D.A.; Kelleway, J.J.; Kennedy, H.; Kuwae, T.; Lavery, P.S.; et al. The future of Blue Carbon science. Nat. Commun. 2019, 10, 3998. [Google Scholar] [CrossRef] [Green Version]
- Macreadie, P.I.; Atwood, T.B.; Seymour, J.R.; Fontes, M.L.S.; Sanderman, J.; Nielsen, D.A.; Connolly, R.M. Vulnerability of seagrass blue carbon to microbial attack following exposure to warming and oxygen. Sci. Total Environ. 2019, 686, 264–275. [Google Scholar] [CrossRef]
- Lamb, J.B.; van de Water, J.A.; Bourne, D.G.; Altier, C.; Hein, M.Y.; Fiorenza, E.A.; Abu, N.; Jompa, J.; Harvell, C.D. Seagrass ecosystems reduce exposure to bacterial pathogens of humans, fishes, and invertebrates. Science 2017, 355, 731–733. [Google Scholar] [CrossRef]
- Fraser, M.W.; Gleeson, D.B.; Grierson, P.F.; Laverock, B.; Kendrick, G.A. Metagenomic Evidence of Microbial Community Responsiveness to Phosphorus and Salinity Gradients in Seagrass Sediments. Front. Microbiol. 2018, 9, 1703. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-McCormick, V.; Kahlke, T.; Petrou, K.; Jeffries, T.; Ralph, P.J.; Seymour, J.R. Regional and Microenvironmental Scale Characterization of the Zostera muelleri Seagrass Microbiome. Front. Microbiol. 2019, 10, 1011. [Google Scholar] [CrossRef]
- Conte, C.; Rotini, A.; Manfra, L.; D’Andrea, M.M.; Winters, G.; Migliore, L. The Seagrass Holobiont: What We Know and What We Still Need to Disclose for Its Possible Use as an Ecological Indicator. Water 2021, 13, 406. [Google Scholar] [CrossRef]
- Garcias-Bonet, N.; Eguiluz, V.M.; Diaz-Rua, R.; Duarte, C.M. Host-association as major driver of microbiome structure and composition in Red Sea seagrass ecosystems. Environ. Microbiol. 2021, 23, 2021–2034. [Google Scholar] [CrossRef]
- Ugarelli, K.; Chakrabarti, S.; Laas, P.; Stingl, U. The Seagrass Holobiont and Its Microbiome. Microorganisms 2017, 5, 81. [Google Scholar] [CrossRef] [Green Version]
- Cúcio, C.; Overmars, L.; Engelen, A.H.; Muyzer, G. Metagenomic Analysis Shows the Presence of Bacteria Related to Free-Living Forms of Sulfur-Oxidizing Chemolithoautotrophic Symbionts in the Rhizosphere of the Seagrass Zostera marina. Front. Mar. Sci. 2018, 5, 171. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Cúcio, C.; Engelen, A.H.; Costa, R.; Muyzer, G. Rhizosphere Microbiomes of European Seagrasses Are Selected by the Plant, But Are Not Species Specific. Front. Microbiol. 2016, 7, 440. [Google Scholar] [CrossRef] [Green Version]
- Borum, J.; Pedersen, O.; Greve, T.M.; Frankovich, T.A.; Zieman, J.C.; Fourqurean, J.W.; Madden, C.J. The potential role of plant oxygen and sulphide dynamics in die-off events of the tropical seagrass, Thalassia testudinum. J. Ecol. 2005, 93, 148–158. [Google Scholar] [CrossRef]
- Pedersen, M.F.; Borum, J. Nitrogen Dynamics of Eelgrass Zostera-Marina during a Late Summer Period of High Growth and Low Nutrient Availability. Mar. Ecol. Prog. Ser. 1992, 80, 65–73. [Google Scholar] [CrossRef]
- Agawin, N.S.R.; Ferriol, P.; Sintes, E.; Moya, G. Temporal and spatial variability of in situ nitrogen fixation activities associated with the Mediterranean seagrass Posidonia oceanica meadows. Limnol. Oceanogr. 2017, 62, 2575–2592. [Google Scholar] [CrossRef]
- Pereg, L.L.; Lipkin, Y.; Sar, N. Different Niches of the Halophila-Stipulacea Seagrass Bed Harbor Distinct Populations of Nitrogen-Fixing Bacteria. Mar. Biol. 1994, 119, 327–333. [Google Scholar] [CrossRef]
- Brodersen, K.E.; Siboni, N.; Nielsen, D.A.; Pernice, M.; Ralph, P.J.; Seymour, J.; Kuhl, M. Seagrass rhizosphere microenvironment alters plant-associated microbial community composition. Environ. Microbiol. 2018, 20, 2854–2864. [Google Scholar] [CrossRef]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Ling, J.; Lin, X.; Zhang, Y.; Zhou, W.; Yang, Q.; Lin, L.; Zeng, S.; Zhang, Y.; Wang, C.; Ahmad, M.; et al. Community Composition and Transcriptional Activity of Ammonia-Oxidizing Prokaryotes of Seagrass Thalassia hemprichii in Coral Reef Ecosystems. Front. Microbiol. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Cao, W.; Yu, K.; Wu, G.; Yang, J.; Su, X.; Wang, F. Evaluation of anthropogenic influences on the Luhuitou fringing reef via spatial and temporal analyses (from isotopic values). J. Geophys. Res. Ocean. 2017, 122, 4431–4443. [Google Scholar] [CrossRef]
- Yue, W.Z.; Sun, C.C.; Shi, P.; Engel, A.; Wang, Y.S.; He, W.H. Effect of temperature on the accumulation of marine biogenic gels in the surface microlayer near the outlet of nuclear power plants and adjacent areas in the Daya Bay, China. PLoS ONE 2018, 13, e0198735. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, S.Z.; Ge, B.Z.; Li, Y.X. Design optimization and uncertainty analysis of multi-energy complementary system for residential building in isolated area. Energy Convers. Manag. 2021, 241, 114310. [Google Scholar] [CrossRef]
- Lu, X.; Liu, Y.W.; Zhao, H.T.; Wang, Z.Y. Corrosion Behavior of Brass H62 in Harsh Marine Atmosphere in Nansha Islands, China. J. Mater. Eng. Perform. 2020, 29, 8156–8164. [Google Scholar] [CrossRef]
- Lakshmanan, V.; Ray, P.; Craven, K.D. Rhizosphere Sampling Protocols for Microbiome (16S/18S/ITS rRNA) Library Preparation and Enrichment for the Isolation of Drought Tolerance-Promoting Microbes. Methods Mol. Biol. 2017, 1631, 349–362. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellin, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Song, Z.; Zhang, H.; Liu, P.; Hu, X. Seagrass vegetation affect the vertical organization of microbial communities in sediment. Mar. Environ. Res. 2020, 162, 105174. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Zhang, T.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; et al. Comparative Analysis of the Gut Microbiota Composition between Captive and Wild Forest Musk Deer. Front. Microbiol. 2017, 8, 1705. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2020, 12, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Austral Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.H.; Yang, Y.F.; He, Z.L.; Luo, F.; Zhou, J.Z. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Zhong, J.X.; Yang, Y.F.; Scheuermann, R.H.; Zhou, J.Z. Application of random matrix theory to biological networks. Phys. Lett. A 2006, 357, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Li, X.D.; Zhao, Y.H.; Wang, L.; Zhang, C.Y. Soil microbial community structure in the rhizosphere of Robinia pseudoacacia L. seedlings exposed to elevated air temperature and cadmium-contaminated soils for 4 years. Sci. Total Environ. 2019, 650, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, Z.; Zhang, C.; Qu, X.; Chen, M.; Song, T.; Yuan, J. Seasonal Variation in the Rhizosphere and Non-Rhizosphere Microbial Community Structures and Functions of Camellia yuhsienensis Hu. Microorganisms 2020, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Zhang, X.; Zhang, Q.; Liu, F.; Zhang, J.; Gong, J. Seagrass (Zostera marina) Colonization Promotes the Accumulation of Diazotrophic Bacteria and Alters the Relative Abundances of Specific Bacterial Lineages Involved in Benthic Carbon and Sulfur Cycling. Appl. Environ. Microbiol. 2015, 81, 6901–6914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, S.; Bunk, B.; Sproer, C.; Rohde, M.; Klenk, H.P. Genome biology of a novel lineage of planctomycetes widespread in anoxic aquatic environments. Environ. Microbiol. 2018, 20, 2438–2455. [Google Scholar] [CrossRef]
- Zhang, X.L.; Zhang, Q.Q.; Yang, A.J.; Hou, L.J.; Zheng, Y.L.; Zhai, W.D.; Gong, J. Incorporation of Microbial Functional Traits in Biogeochemistry Models Provides Better Estimations of Benthic Denitrification and Anammox Rates in Coastal Oceans. J. Geophys. Res. Biogeosci. 2018, 123, 3331–3352. [Google Scholar] [CrossRef]
- Zhang, Y.; Ling, J.; Yang, Q.; Wen, C.; Yan, Q.; Sun, H.; Van Nostrand, J.D.; Shi, Z.; Zhou, J.; Dong, J. The functional gene composition and metabolic potential of coral-associated microbial communities. Sci. Rep. 2015, 5, 16191. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.F.; Ling, J.; Wang, Y.S.; Chen, B.; Zhang, Y.Y.; Dong, J.D. Cultivation-dependent analysis of the microbial diversity associated with the seagrass meadows in Xincun Bay, South China Sea. Ecotoxicology 2015, 24, 1540–1547. [Google Scholar] [CrossRef]
- Park, S.; Yoshizawa, S.; Kogure, K.; Yokota, A. Rubricoccus marinus gen. nov., sp nov., of the family ‘Rhodothermaceae’, isolated from seawater. Int. J. Syst. Evol. Microbiol. 2011, 61, 2069–2072. [Google Scholar] [CrossRef]
- Ugarelli, K.; Laas, P.; Stingl, U. The Microbial Communities of Leaves and Roots Associated with Turtle Grass (Thalassia testudinum) and Manatee Grass (Syringodium filliforme) are Distinct from Seawater and Sediment Communities, but Are Similar between Species and Sampling Sites. Microorganisms 2019, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Brodersen, K.E.; Nielsen, D.A.; Ralph, P.J.; Kuhl, M. Oxic microshield and local pH enhancement protects Zostera muelleri from sediment derived hydrogen sulphide. New Phytol. 2015, 205, 1264–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, B.C.; Bougoure, J.; Ryan, M.H.; Bennett, W.W.; Colmer, T.D.; Joyce, N.K.; Olsen, Y.S.; Kendrick, G.A. Oxygen loss from seagrass roots coincides with colonisation of sulphide-oxidising cable bacteria and reduces sulphide stress. ISME J. 2018, 13, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Kuypers, M.M. Microbiology: A division of labour combined. Nature 2015, 528, 487–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radecker, N.; Pogoreutz, C.; Voolstra, C.R.; Wiedenmann, J.; Wild, C. Nitrogen cycling in corals: The key to understanding holobiont functioning? Trends Microbiol. 2015, 23, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Welsh, D.T. Nitrogen fixation in seagrass meadows: Regulation, plant-bacteria interactions and significance to primary productivity. Ecol. Lett. 2000, 3, 58–71. [Google Scholar] [CrossRef]
- van Kessel, M.A.; Speth, D.R.; Albertsen, M.; Nielsen, P.H.; Op den Camp, H.J.; Kartal, B.; Jetten, M.S.; Lucker, S. Complete nitrification by a single microorganism. Nature 2015, 528, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Daims, H.; Lebedeva, E.V.; Pjevac, P.; Han, P.; Herbold, C.; Albertsen, M.; Jehmlich, N.; Palatinszky, M.; Vierheilig, J.; Bulaev, A.; et al. Complete nitrification by Nitrospira bacteria. Nature 2015, 528, 504–509. [Google Scholar] [CrossRef]
- Schlesner, H. Blastopirellula. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–13. [Google Scholar] [CrossRef]
- More, K.D.; Giosan, L.; Grice, K.; Coolen, M.J.L. Holocene paleodepositional changes reflected in the sedimentary microbiome of the Black Sea. Geobiology 2019, 17, 436–448. [Google Scholar] [CrossRef]
- Martin, B.C.; Alarcon, M.S.; Gleeson, D.; Middleton, J.A.; Fraser, M.W.; Ryan, M.H.; Holmer, M.; Kilminster, K. Root microbiomes as indicators of seagrass health. FEMS Microbiol. Ecol. 2019, 96, fiz201. [Google Scholar] [CrossRef]
- Moriarty, D.J.W.; Pollard, P.C. Diel Variation of Bacterial Productivity in Seagrass (Zostera-Capricorni) Beds Measured by Rate of Thymidine Incorporation into DNA. Mar. Biol. 1982, 72, 165–173. [Google Scholar] [CrossRef]
- Holmer, M.; Duarte, C.M.; Boschker, H.T.S.; Barron, C. Carbon cycling and bacterial carbon sources in pristine and impacted Mediterranean seagrass sediments. Aquat. Microb. Ecol. 2004, 36, 227–237. [Google Scholar] [CrossRef] [Green Version]
- Sogin, E.; Michellod, D.; Gruber-Vodicka, H.; Bourceau, P.; Geier, B.; Meier, D.; Seidel, M.; Hach, P.F.; Procaccini, G.; Dubilier, N.; et al. Seagrass excretes sugars to their rhizosphere making them the sweet spots in the sea. bioRxiv 2019, 797522. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DYH | SYT | XST | XSH | XSC | XSS | NXH | NXT | |
|---|---|---|---|---|---|---|---|---|---|
| Latitude (° E) | 22.54 | 18.2 | 16.84 | 16.84 | 16.84 | 16.84 | 9.71 | 10.22 | |
| Longtitude (° N) | 114.45 | 119.47 | 112.34 | 112.34 | 112.34 | 112.34 | 114.29 | 114.2 | |
| Water | pH | 8.14 (0) | 8.15 (0.01) | 8.24 (0.01) | 8.24 (0.03) | 8.24 (0.03) | 8.24 (0.01) | 8.15 (0.01) | 8.21 (0.01) |
| Salinity | 28.37 (0.07) | 24.7 (0.3) | 34.52 (0.05) | 34.52 (0.15) | 34.57 (0.08) | 34.52 (0.05) | 34.31 (0) | 34.54 (0.09) | |
| DO (mg/L) | 5.82 (0.05) | 6.7 (0.2) | 9.47 (0.17) | 9.47 (0.51) | 9.64 (0.59) | 9.47 (0.17) | 7.64 (0.03) | 8.36 (0.13) | |
| Nitrate (mg/L) | 0.153 (0.004) | 0.044 (0.002) | 0.049 (0.002) | 0.049 (0.005) | 0.046 (0.001) | 0.049 (0.002) | 0.06 (0.001) | 0.035 (0) | |
| Nitrite (mg/L) | 0.001 (0) | 0.025 (0.001) | 0.016 (0) | 0.016 (0.001) | 0.016 (0.001) | 0.016 (0) | 0.001 (0) | 0.001 (0) | |
| Ammonium (mg/L) | 0.069 (0) | 0.121 (0.002) | 0.094 (0.002) | 0.094 (0.005) | 0.091 (0.006) | 0.094 (0.002) | 0.048 (0.001) | 0.032 (0.001) | |
| Phosphate (mg/L) | 0.007 (0.001) | 0.015 (0) | 0.01 (0.001) | 0.01 (0.002) | 0.01 (0.001) | 0.01 (0.001) | 0.006 (0) | 0.006 (0) | |
| Sediment | Ammonium (mg/kg) | 3.02 (0.01) | 5.97 (0.02) | 4.37 (0.01) | 2.91 (0.03) | 1.45 (0.01) | 4.78 (0.02) | 6.39 (0.03) | 5.48 (0.12) |
| Nitrate (mg/kg) | 22.3 (2.63) | 24.4 (3.68) | 44.0 (2.10) | 62.4 (3.26) | 69.4 (4.06) | 44.0 (2.24) | 32.60 (1.12) | 45.60 (1.56) | |
| AP (mg/kg) | 18.0 (2.01) | 15.0 (1.28) | 14.0 (3.06) | 11.0 (1.85) | 15.0 (2.30) | 15.0 (1.60) | 18 (2.45) | 17 (3.01) | |
| TOC (%) | 18.4% (1.89) | 32.5% (2.36) | 43.4% (1.03) | 33.2% (2.64) | 40.3% (3.12) | 21.8% (3.56) | 36.80 (2.63) | 42.06 (3.42) |
| Microbial Communities | Microbial Communities | Phylogenetic Composition (PD) | Taxonomic Alpha Diversity | |||
|---|---|---|---|---|---|---|
| Richness | Shannon | Simpson | ||||
| Species | p | p | p | p | ||
| XSC | XST | 0.9420 | 0.6700 | 0.5250 | 0.3240 | |
| XSC | XSH | 0.4550 | 0.3870 | 0.7380 | 0.7380 | |
| XSC | XSS | 0.5250 | 0.6700 | 0.3240 | 0.5250 | |
| XST | XSH | 0.0802 | 0.9690 | 0.0810 | 0.0330 | |
| XST | XSS | 0.2180 | 0.1060 | 0.9870 | 0.9870 | |
| XSS | XSH | 0.0240 | 0.0330 | 0.0330 | 0.0810 | |
| Location | ||||||
| H. ovalis | XSH | DYH | 0.9500 | 0.3700 | 0.0200 | 0.0200 |
| XSH | CGX | 0.2300 | 0.3700 | 0.3700 | 0.3700 | |
| DYH | CGX | 0.1300 | 0.0200 | 0.3700 | 0.3700 | |
| T. hemprichii | ||||||
| XST | SYT | 0.5490 | 0.5500 | 0.3700 | 0.8960 | |
| XST | NXT | 0.5490 | 0.3000 | 0.0370 | 0.0650 | |
| SYT | NXT | 0.0930 | 0.0300 | 0.0200 | 0.1730 | |
| Microbial Community | Microbial Community | Delta Unifrac | P Unifrac | Delta Bray | P Bray | Delta Euclidean | P Euclidean | Delta Sorensen | P Sorensen | |
|---|---|---|---|---|---|---|---|---|---|---|
| Species | ||||||||||
| XSC | XST | 0.184 | 0.087 | 0.294 | 0.016 | 1.260 | 0.156 | 0.099 | 0.293 | |
| XSC | XSH | 0.177 | 0.084 | 0.308 | 0.109 | 1.590 | 0.294 | 0.152 | 0.282 | |
| XSC | XSS | 0.049 | 0.100 | 0.088 | 0.100 | 1.040 | 0.500 | 0.061 | 0.300 | |
| XST | XSH | 0.219 | 0.025 * | 0.366 | 0.015 * | 1.429 | 0.017 * | 0.136 | 0.124 | |
| XST | XSS | 0.176 | 0.035 * | 0.284 | 0.017 * | 1.073 | 0.009 ** | 0.083 | 0.016 * | |
| XSH | XSS | 0.169 | 0.011 * | 0.299 | 0.012 * | 1.403 | 0.037 * | 0.135 | 0.109 | |
| Location | ||||||||||
| H. ovalis | XSH | NSH | 0.039 | 0.100 | 0.088 | 0.100 | 1.024 | 0.100 | 0.111 | 0.100 |
| XSH | DYH | 0.035 | 0.100 | 0.125 | 0.100 | 1.024 | 0.100 | 0.114 | 0.100 | |
| NSH | DYH | 0.033 | 0.100 | 0.122 | 0.100 | 0.667 | 0.100 | 0.042 | 0.100 | |
| T. hemprichii | XST | NST | 0.070 | 0.100 | 0.110 | 0.100 | 20.104 | 0.100 | 0.130 | 0.100 |
| XST | SYT | 0.064 | 0.100 | 0.112 | 0.100 | 19.395 | 0.100 | 0.132 | 0.100 | |
| NST | SYT | 0.026 | 0.100 | 0.105 | 0.100 | 18.762 | 0.100 | 0.121 | 0.100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, J.; Zhou, W.; Yang, Q.; Yin, J.; Zhang, J.; Peng, Q.; Huang, X.; Zhang, Y.; Dong, J. Spatial and Species Variations of Bacterial Community Structure and Putative Function in Seagrass Rhizosphere Sediment. Life 2021, 11, 852. https://doi.org/10.3390/life11080852
Ling J, Zhou W, Yang Q, Yin J, Zhang J, Peng Q, Huang X, Zhang Y, Dong J. Spatial and Species Variations of Bacterial Community Structure and Putative Function in Seagrass Rhizosphere Sediment. Life. 2021; 11(8):852. https://doi.org/10.3390/life11080852
Chicago/Turabian StyleLing, Juan, Weiguo Zhou, Qingsong Yang, Jianping Yin, Jian Zhang, Qiuying Peng, Xiaofang Huang, Yuhang Zhang, and Junde Dong. 2021. "Spatial and Species Variations of Bacterial Community Structure and Putative Function in Seagrass Rhizosphere Sediment" Life 11, no. 8: 852. https://doi.org/10.3390/life11080852