
Behavior of Chemokine Receptor 6 (CXCR6) in Complex with CXCL16 Soluble form Chemokine by Molecular Dynamic Simulations: General Protein‒Ligand Interaction Model and 3D-QSAR Studies of Synthetic Antagonists



Abstract
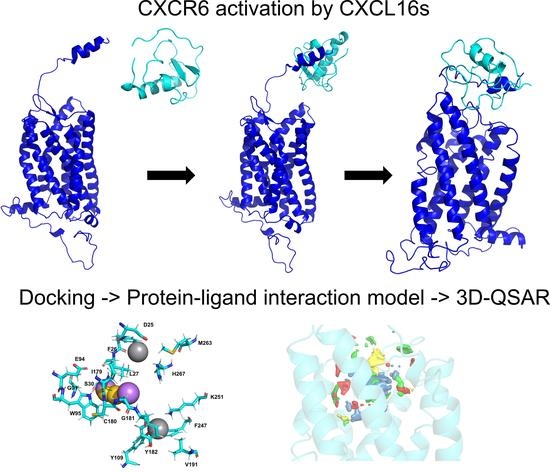
:
1. Introduction
2. Materials and Methods
2.1. Protein Structures, Homology Modeling of CXCR6 and CXCL16s, and Relaxation by All Atom Molecular Dynamics (MD) Simulations
2.2. Protein‒Protein Docking
2.3. Coarse-Grained Molecular Dynamics (CG-MD) Simulations
2.4. Ligand Docking, General Protein–Ligand Interaction Model, and Receptor-Based 3D-QSAR Model
2.5. Manipulations of Complexes and Figures
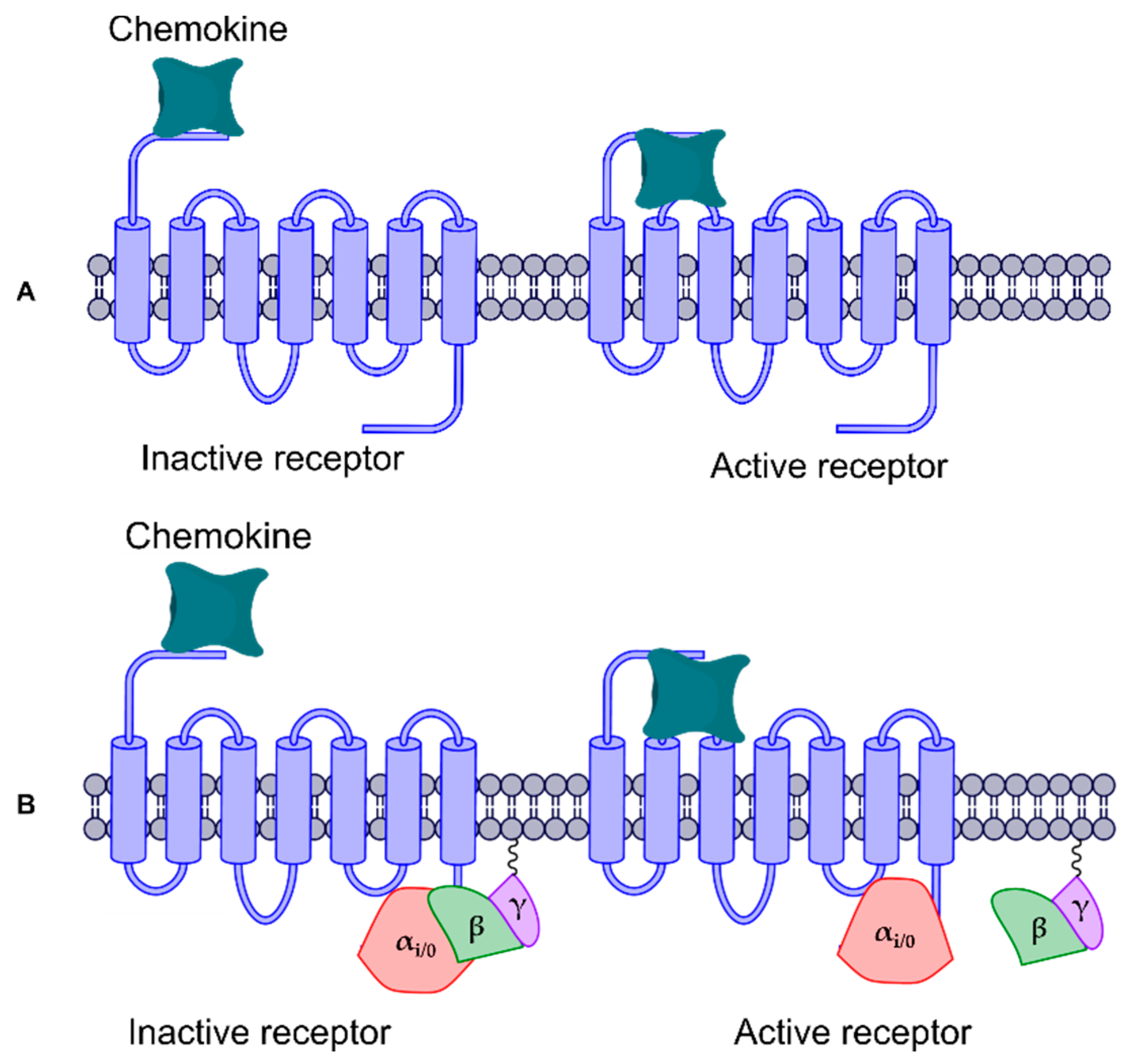
3. Results and Discussion
3.1. Protein Structures and Homology Modeling
3.2. Relaxation of CXCR6 and CXCL16s
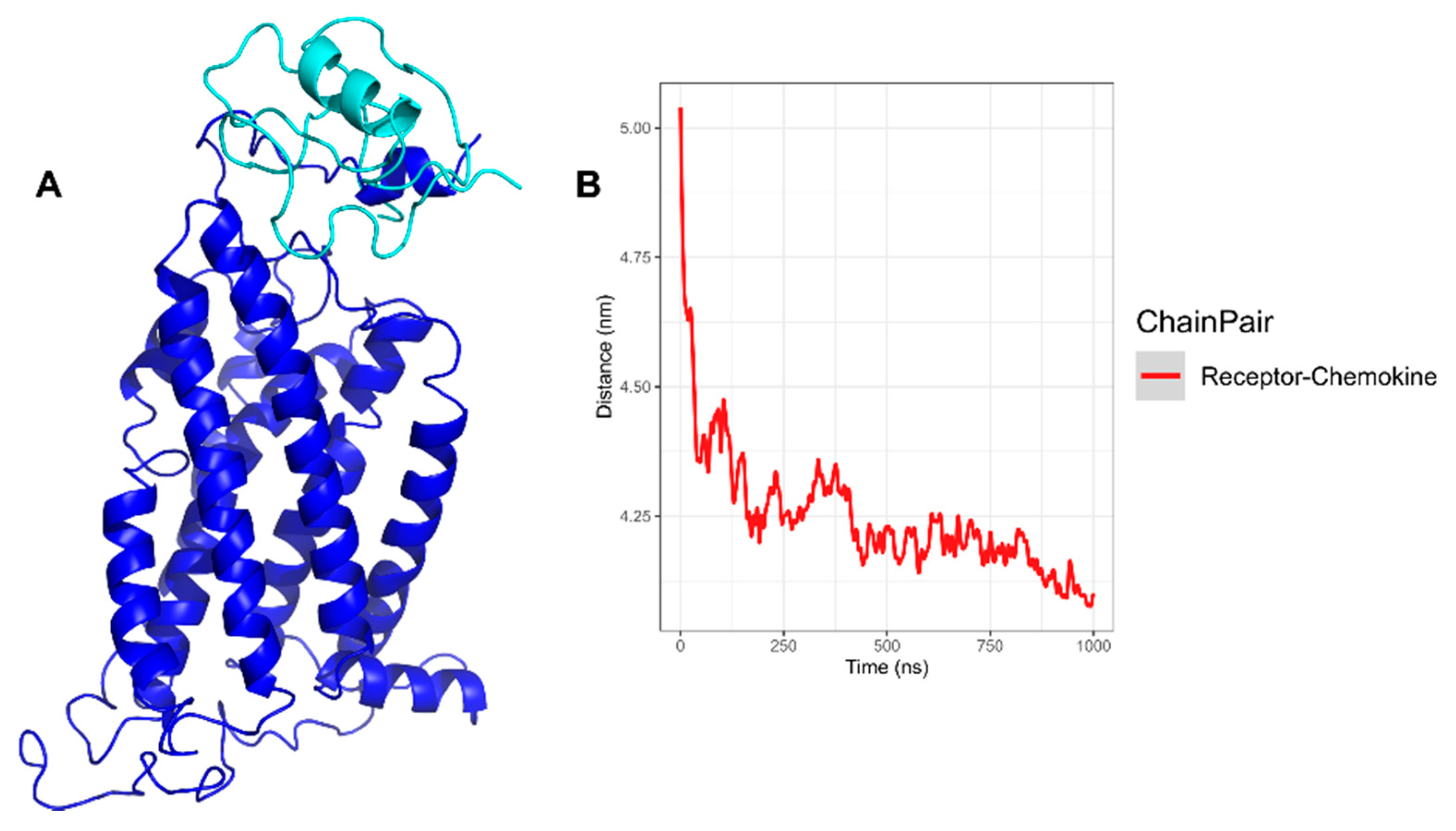
3.3. CXCR6/Chemokine Complex Building and CG-MD Simulations
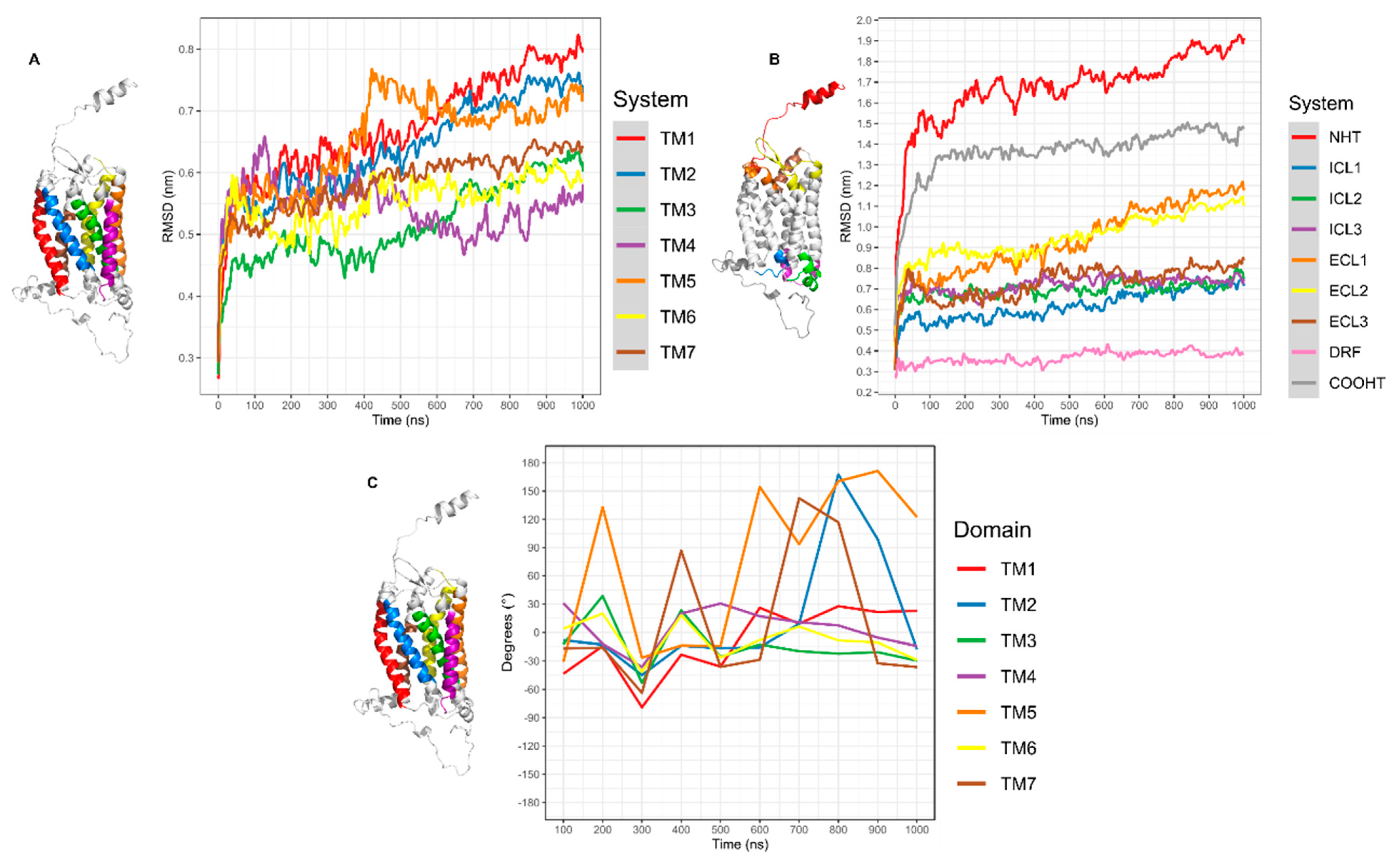
3.3.1. CXCR6 CG-MD Simulations
3.3.2. CXCR6‒CXCL16s CG-MD Simulations
3.4. General Protein‒Ligand Interaction Model and Receptor-Based 3D-QSAR Model of CXCR6 Antagonists
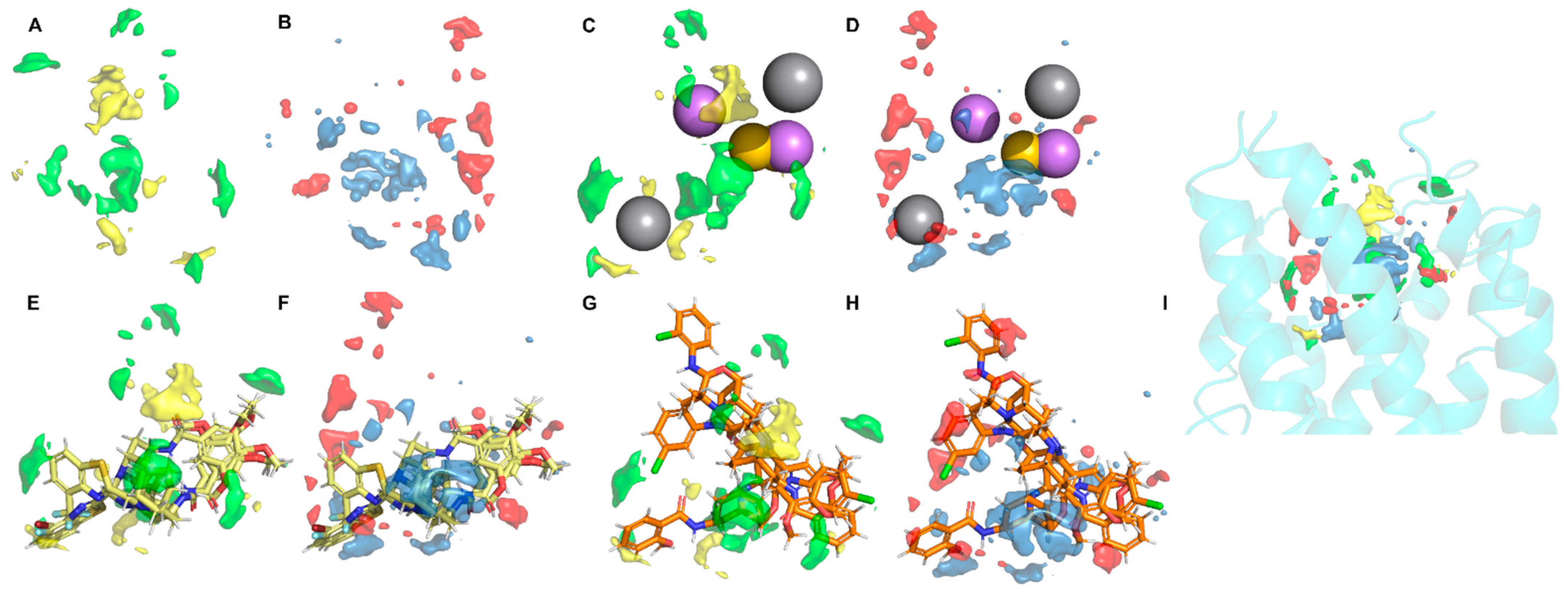
3.4.1. General Protein‒Ligand Interaction Model Based on pIC50
3.4.2. Receptor-Based 3D-QSAR Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Chea, S.; Possot, C.; Perchet, T.; Petit, M.; Cumano, A.; Golub, R. CXCR6 expression is important for retention and circulation of ILC precursors. Mediat. Inflamm. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Wang, X.; Wang, J.; Zu, L.; Cheng, G.; Hao, M.; Sun, X.; Xue, Y.; Lu, J.; Wang, J. CXCL16/CXCR6 chemokine signaling mediates breast cancer progression by pERK1/2-dependent mechanisms. Oncotarget 2015, 6, 14165–14178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, A.; Stabile, H.; Gismondi, A.; Santoni, A.; Bernardini, G. Chemokine regulation of innate lymphoid cell tissue distribution and function. Cytokine Growth Factor Rev. 2018, 42, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yin, L.; Dai, Y. Modification of SR-PSOX functions by multi-point mutations of basic amino acid residues. Biochimie 2013, 95, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Scholz, F.; Schulte, A.; Adamski, F.; Hundhausen, C.; Mittag, J.; Schwarz, A.; Kruse, M.L.; Proksch, E.; Ludwig, A. Constitutive expression and regulated release of the transmembrane chemokine CXCL16 in human and murine skin. J. Investig. Dermatol. 2007, 127, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Koenen, A.; Babendreyer, A.; Schumacher, J.; Pasqualon, T.; Schwarz, N.; Seifert, A.; Deupi, X.; Ludwig, A.; Dreymueller, D. The DRF motif of CXCR6 as chemokine receptor adaptation to adhesion. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, A.; Hundhausen, C.; Lambert, M.; Broadway, N.; Andrews, R.; Bickett, D.; Leesnitzer, M.; Becherer, J. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef]
- Reiss, K.; Ludwig, A.; Saftig, P. Breaking up the tie: Disintegrin-like metalloproteinases as regulators of cell migration in inflammation and invasion. Pharmacol. Ther. 2006, 111, 985–1006. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Weber, C. Transmembrane chemokines: Versatile “special agents” in vascular inflammation. Thromb. Haemost. 2007, 97, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Gough, P.J.; Garton, K.J.; Wille, P.T.; Rychlewski, M.; Dempsey, P.J.; Raines, E.W. A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand 16. J. Immunol. 2004, 172, 3678–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Unutmaz, D.; KewalRamani, V.N.; Littman, D.R. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 1997, 388, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Loetscher, M.; Amara, A.; Oberlin, E.; Brass, N.; Legler, D.F.; Loetscher, P.; D’Apuzzo, M.; Meese, E.; Rousset, D.; Virelizier, J.L.; et al. TYMSTR, a putative chemokine receptor selectively expressed in activated T cells, exhibits HIV-1 coreceptor function. Curr. Biol. 1997, 7, 652–660. [Google Scholar] [CrossRef] [Green Version]
- Petit, S.J.; Chayen, N.E.; Pease, J.E. Site-directed mutagenesis of the chemokine receptor CXCR6 suggests a novel paradigm for interactions with the ligand CXCL16. Eur. J. Immunol. 2008, 38, 2337–2350. [Google Scholar] [CrossRef]
- Hydes, T.; Noll, A.; Salinas-Riester, G.; Abuhilal, M.; Armstrong, T.; Hamady, Z.; Primrose, J.; Takhar, A.; Walter, L.; Khakoo, S.I. IL-12 and IL-15 induce the expression of CXCR6 and CD49a on peripheral natural killer cells. Immun. Inflamm. Dis. 2018, 6, 34–46. [Google Scholar] [CrossRef]
- Booth, V.; Keizer, D.W.; Kamphuis, M.B.; Clark-Lewis, I.; Sykes, B.D. The CXCR3 binding chemokine IP-10/CXCL10: Structure and receptor interactions. Biochemistry 2002, 41, 10418–10425. [Google Scholar] [CrossRef]
- Benredjem, B.; Girard, M.; Rhainds, D.; St.-Onge, G.; Heveker, N. Mutational analysis of atypical chemokine receptor 3 (ACKR3/CXCR7) interaction with its chemokine ligands CXCL11 and CXCL12. J. Biol. Chem. 2017, 292, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.; Huma, Z.E.; Lane, J.R.; Liu, X.; Bridgford, J.L.; Payne, R.J.; Canals, M.; Stone, M.J. Evaluation and extension of the two-site, two-step model for binding and activation of the chemokine receptor CCR1. J. Biol. Chem. 2019, 294, 3464–3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera-Durán, G.; Romo-Mancillas, A. Computational study of C-X-C chemokine receptor (CXCR)3 binding with its natural agonists chemokine (C-X-C Motif) ligand (CXCL)9, 10 and 11 and with synthetic antagonists: Insights of receptor activation towards drug design for vitiligo. Molecules 2020, 25, 4413. [Google Scholar] [CrossRef]
- Neumann, A.; Engel, V.; Mahardhika, A.B.; Schoeder, C.T.; Namasivayam, V.; Kieć-Kononowicz, K.; Müller, C.E. Computational investigations on the binding mode of ligands for the cannabinoid-activated G protein-coupled receptor GPR18. Biomolecules 2020, 10, 686. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Foley, J.F.; Zhang, H.H.; Hurt, D.E.; Richards, J.L.; Smith, C.S.; Liao, F.; Farber, J.M. Selectivity in the use of G i/o proteins is determined by the DRF motif in CXCR6 and is cell-type specific. Mol. Pharmacol. 2015, 88, 894–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, T.; Ernst, O.P.; Palczewski, K.; Hofmann, K.P. Activation of rhodopsin: New insights from structural and biochemical studies. Trends Biochem. Sci. 2001, 26, 318–324. [Google Scholar] [CrossRef]
- Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W.; Choe, H.-W.; Park, J.H.; Heck, M.; Ernst, O.P. A G protein-coupled receptor at work: The rhodopsin model. Trends Biochem. Sci. 2009, 34, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Deupi, X. Relevance of rhodopsin studies for GPCR activation. Biochim. Biophys. Acta Bioenergy 2014, 1837, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, K.N.; Pfeffer, J.; Dutta, A.; Klein-Seetharaman, J. Vibrational resonance, allostery, and activation in rhodopsin-like G protein-coupled receptors. Sci. Rep. 2016, 6, 37290. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Hu, H.; Ramachandran, S.; Erickson, J.W.; Cerione, R.A.; Skiniotis, G. Structures of the rhodopsin-transducin complex: Insights into G-protein activation. Mol. Cell 2019, 75, 781–790.e3. [Google Scholar] [CrossRef]
- Gooden, M.J.M.; Wiersma, V.R.; Boerma, A.; Leffers, N.; Boezen, H.M.; Ten Hoor, K.A.; Hollema, H.; Walenkamp, A.M.E.; Daemen, T.; Nijman, H.W.; et al. Elevated serum CXCL16 is an independent predictor of poor survival in ovarian cancer and may reflect pro-metastatic ADAM protease activity. Br. J. Cancer 2014, 110, 1535–1544. [Google Scholar] [CrossRef]
- Hu, W.; Liu, Y.; Zhou, W.; Si, L.; Ren, L. CXCL16 and CXCR6 are coexpressed in human lung cancer In Vivo and mediate the invasion of lung cancer cell lines In Vitro. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Li, J.; Ley, K. Lymphocyte migration into atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, Q.; Zhen, J.; Xu, Y.; Sun, S. Simvastatin ameliorates renal lipidosis through the suppression of renal CXCL16 expression in mice with adriamycin-induced nephropathy. Int. J. Clin. Exp. Pathol. 2015, 8, 15696–15707. [Google Scholar] [PubMed]
- Fallahi, P.; Corrado, A.; Di Domenicantonio, A.; Frenzilli, G.; Antonelli, A.; Martina Ferrari, S. CXCR3, CXCR5, CXCR6, and CXCR7 in diabetes. Curr. Drug Targets 2016, 17, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Li, Y.; Li, H.; Lyu, M.; Zhang, D.; Fu, R.; Guan, Y.; Wang, S.; Sun, B.; Dou, X.; et al. Increased plasma sCXCL16 levels may have a relationship with Th1/Th2 imbalance in primary immune thrombocytopenia. Cytokine 2017, 99, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Elmaci, İ.; Altinoz, M.; Sari, R. Immune pathobiology of schwannomas: A concise review. J. Neurol. Surg. Part A Cent. Eur. Neurosurg. 2018, 79, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zhen, X.; Xiong, B.; Wang, B.; Zhang, W.; Zhou, W. CXCR6 is expressed in human prostate cancer in vivo and is involved in the in vitro invasion of PC3 and LNCap cells. Cancer Sci. 2008, 99, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Peddibhotla, S.; Hershberger, P.M.; Jason Kirby, R.; Sugarman, E.; Maloney, P.R.; Hampton Sessions, E.; Divlianska, D.; Morfa, C.J.; Terry, D.; Pinkerton, A.B.; et al. Discovery of small molecule antagonists of chemokine receptor CXCR6 that arrest tumor growth in SK-HEP-1 mouse xenografts as a model of hepatocellular carcinoma. Bioorg. Med. Chem. Lett. 2020, 30, 126899. [Google Scholar] [CrossRef] [PubMed]
- Floudas, C.A.; Fung, H.K.; McAllister, S.R.; Mönnigmann, M.; Rajgaria, R. Advances in protein structure prediction and de novo protein design: A review. Chem. Eng. Sci. 2006, 61, 966–988. [Google Scholar] [CrossRef]
- De Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsutake, A.; Nagai, T.; Takano, H. Relaxation mode analysis for simulations of biomolecules. Seibutsu Butsuri 2013, 1637, 164102. [Google Scholar] [CrossRef] [Green Version]
- Dämgen, M.A.; Biggin, P.C. Computational methods to examine conformational changes and ligand-binding properties: Examples in neurobiology. Neurosci. Lett. 2019, 700, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Thelen, M. New insights in chemokine signaling. F1000 Res. 2018, 7, 95. [Google Scholar] [CrossRef]
- Wasilko, D.J.; Johnson, Z.L.; Ammirati, M.; Che, Y.; Griffor, M.C.; Han, S.; Wu, H. Structural basis for chemokine receptor CCR6 activation by the endogenous protein ligand CCL20. Nat. Commun. 2020, 11, 3031. [Google Scholar] [CrossRef] [PubMed]
- Medina-Ruiz, D.; Erreguin-Luna, B.; Luna-Vázquez, F.J.; Romo-Mancillas, A.; Rojas-Molina, A.; Ibarra-Alvarado, C. Vasodilation elicited by isoxsuprine, identified by high-throughput virtual screening of compound libraries, involves activation of the NO/cGMP and H2S/KATP pathways and blockade of α1-adrenoceptors and calcium channels. Molecules 2019, 24, 987. [Google Scholar] [CrossRef] [Green Version]
- The Uniprot Consortium Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yang, J.; Jang, R.; Zhang, Y. GPCR-I-TASSER: A hybrid approach to g protein-coupled receptor structure modeling and the application to the human genome. Structure 2015, 23, 1538–1549. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.I.; Hunter, H.N.; Tack, B.F.; Vogel, H.J. Human macrophage inflammatory protein 3α: Protein and peptide nuclear magnetic resonance solution structures, dimerization, dynamics, and anti-infective properties. Antimicrob. Agents Chemother. 2008, 52, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins Struct. Funct. Bioinform. 2017, 85, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and its limits in rigid body protein-protein docking. Structure 2020, 28, 1071–1081.e3. [Google Scholar] [CrossRef]
- Xia, B.; Vajda, S.; Kozakov, D. Accounting for pairwise distance restraints in FFT-based protein-protein docking. Bioinformatics 2016, 32, 3342–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yershova, A.; Jain, S.; LaValle, S.M.; Mitchell, J.C. Generating uniform incremental grids on SO (3) using the hopf fibration. Int. J. Rob. Res. 2010, 29, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. Ligplot: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ingólfsson, H.I.; Cheng, X.; Lee, J.; Marrink, S.J.; Im, W. CHARMM-gui martini maker for coarse-grained simulations with the martini force field. J. Chem. Theory Comput. 2015, 11, 4486–4494. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-3; Maestro, Schrödinger, LLC: New York, NY, USA, 2019.
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 12 August 2020).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock4 with GPUs and gradient-based local search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. Pharmer: Efficient and exact pharmacophore search. J. Chem. Inf. Model. 2011, 51, 1307–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.P.; Richards, G.; de la Iglesia, B.; Rayward-Smith, V.J. Clustering rules: A comparison of partitioning and hierarchical clustering algorithms. J. Math. Model. Algorithms 2006, 5, 475–504. [Google Scholar] [CrossRef]
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions. R Package Version 2.1.0. Available online: https://cran.r-project.org/package=cluster (accessed on 12 August 2020).
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2011, 17, 201–208. [Google Scholar] [CrossRef]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Naceiri Mrabti, N.; Elhallaoui, M. QSAR study and molecular docking of benzimidazole derivatives as potent activators of AMP-activated protein kinase. J. Taibah Univ. Sci. 2017, 11, 18–39. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L. The PyMOL Molecular Graphics System; Version 2.0; Schrödinger Inc.: New York, NY, USA, 2015. [Google Scholar]
- Hillisch, A.; Pineda, L.F.; Hilgenfeld, R. Utility of homology models in the drug discovery process. Drug Discov. Today 2004, 9, 659–669. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, P.; Chaturvedi, S.; Swastika Pal, S.; Jain, N.; Mishra, A.K. Improvising 5-HT 7 R homology model for design of high affinity ligands: Model validation with docking, embrace minimization, MM-GBSA, and molecular dynamic simulations. J. Biomol. Struct. Dyn. 2018, 36, 2475–2494. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- States, D.J.; Gish, W. QGB: Combined use of sequence similarity and codon bias for coding region identification. J. Comput. Biol. 1994, 1, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobolev, O.V.; Afonine, P.V.; Moriarty, N.W.; Hekkelman, M.L.; Joosten, R.P.; Perrakis, A.; Adams, P.D. A global ramachandran score identifies protein structures with unlikely stereochemistry. Structure 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Mattioli, I.; Gong, J.-H.; Loetscher, P. Structure-function relationship between the human chemokine receptor CXCR3 and its ligands. J. Biol. Chem. 2003, 278, 289–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | C-Score | TM-Score | Ramachandran Favored | * z-Score |
|---|---|---|---|---|
| CXCR6-M1 | −0.46 | 0.65 ± 0.13 | 85.29% | −3.03 ± 0.38 |
| CXCR6 | - | - | 84.71% | −3.08 ± 0.37 |
| CXCR6 ** | - | - | 91.76% | −2.05 ± 0.39 |
| CXCL16s-M1 | −1.51 | 0.53 ± 0.15 | 67.57% | −6.24 ± 0.71 |
| CXCL16s-C1 | - | - | 89.19% | −2.50 ± 0.81 |
| TM1 | TM2 | TM3 * | TM4 | TM5 | TM6 | TM7 |
|---|---|---|---|---|---|---|
| 33–59 | 69–89 | 104–128 | 144–164 | 188–215 | 232–259 | 276–293 |
| Pharmacophoric Features | Residues Around 5 Å |
|---|---|
| HAc | L27, S30, G91, E94, W95, I179, C180, G181, Y182 |
| HDn | D25, F26, L27, Y109, Y182, V191, F247, K251, M263, H267 |
| Hph | L27, S30, W95, I179, C180 |
| Models | q2 | R2 | F Test | Test Group |
|---|---|---|---|---|
| Model 7 | 0.6397 | 0.8731 | 72.8899 | 18, 74, 38, 2, 72, 80, 28, 52, 16, 15, 12, 73, 70, 26, 47, 13, 32, 54, 65, 62, 59, 79, 56 |
| Model 1 | 0.6329 | 0.8639 | 67.2740 | 33, 30, 39, 22, 61, 28, 36, 53, 9, 66, 56, 59, 24, 48, 72, 18, 2, 52, 13, 41, 62, 46, 75 |
| Model 4 | 0.4906 | 0.8718 | 72.0680 | 27, 50, 25, 9, 2, 23, 14, 67, 41, 70, 33, 28, 39, 79, 66, 36, 13, 1, 71, 55, 82, 3, 76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilera-Durán, G.; Romo-Mancillas, A. Behavior of Chemokine Receptor 6 (CXCR6) in Complex with CXCL16 Soluble form Chemokine by Molecular Dynamic Simulations: General Protein‒Ligand Interaction Model and 3D-QSAR Studies of Synthetic Antagonists. Life 2021, 11, 346. https://doi.org/10.3390/life11040346
Aguilera-Durán G, Romo-Mancillas A. Behavior of Chemokine Receptor 6 (CXCR6) in Complex with CXCL16 Soluble form Chemokine by Molecular Dynamic Simulations: General Protein‒Ligand Interaction Model and 3D-QSAR Studies of Synthetic Antagonists. Life. 2021; 11(4):346. https://doi.org/10.3390/life11040346
Chicago/Turabian StyleAguilera-Durán, Giovanny, and Antonio Romo-Mancillas. 2021. "Behavior of Chemokine Receptor 6 (CXCR6) in Complex with CXCL16 Soluble form Chemokine by Molecular Dynamic Simulations: General Protein‒Ligand Interaction Model and 3D-QSAR Studies of Synthetic Antagonists" Life 11, no. 4: 346. https://doi.org/10.3390/life11040346