Higher Responsiveness to Rosuvastatin in Polygenic versus Monogenic Hypercholesterolemia: A Propensity Score Analysis

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Testing
2.2. Lipid Lowering Therapy
2.3. Statistical Analysis
3. Results
3.1. Molecular Testing
3.2. Patients’ Clinical Characteristics and Lipid Lowering Therapy Data
3.3. Influence of A Genetic Defect in FH on Response to LLT
3.4. Influence of A Genetic Defect in FH on Achieving the LDL-C Treatment Goals
4. Discussion
4.1. Limitations
4.2. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478. [Google Scholar] [CrossRef] [Green Version]
- Benn, M.; Watts, G.F.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Mutations causative of familial hypercholesterolaemia: Screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1. Eur. Hear. J. 2016, 37, 1384–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ference, B.A.; Yoo, W.; Alesh, I.; Mahajan, N.; Mirowska, K.K.; Mewada, A.; Kahn, J.; Afonso, L.; Williams, K.A.; Flack, J.M. Effect of long-term exposure to lower low-densilipoprotein cholesterol beginning early in life on the risk of coronary heart disease. A mendelian randomization analysis. Ration. Pharmacother. Cardiol. 2013, 9, 90–98. [Google Scholar] [CrossRef]
- Sharifi, M.; Futema, M.; Nair, D.; Humphries, S.E. Genetic Architecture of Familial Hypercholesterolaemia. Curr. Cardiol. Rep. 2017, 19, 44. [Google Scholar] [CrossRef] [PubMed]
- Humphries, S.E.; Cooper, J.; Seed, M.; Capps, N.; Durrington, P.; Jones, B.; McDowell, I.; Soran, H.; Neil, H. Simon Broome Familial Hyperlipidaemia Register Group Coronary heart disease mortality in treated familial hypercholesterolaemia: Update of the UK Simon Broome FH register. Atherosclerosis 2018, 274, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431. [Google Scholar] [CrossRef] [Green Version]
- Mikhailova, S.V.; Ivanoshchuk, D.E.; Timoshchenko, O.; Shakhtshneider, E.V. Genes Potentially Associated with Familial Hypercholesterolemia. Biomolecules 2019, 9, 807. [Google Scholar] [CrossRef] [Green Version]
- Hendricks-Sturrup, R.M.; Clark-LoCascio, J.; Lu, C. A Global Review on the Utility of Genetic Testing for Familial Hypercholesterolemia. J. Pers. Med. 2020, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; A Cooper, J.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; Whittall, R.A.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of Variant Selection for the LDL Cholesterol Genetic Risk Score in the Diagnosis of the Polygenic Form of Clinical Familial Hypercholesterolemia and Replication in Samples from 6 Countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef]
- E Humphries, S.; A Whittall, R.; Hubbart, C.S.; Maplebeck, S.; A Cooper, J.; Soutar, A.K.; Naoumova, R.; Thompson, G.R.; Seed, M.; Durrington, P.N.; et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: Relation to plasma lipid levels and coronary heart disease risk. J. Med. Genet. 2006, 43, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Masana, L.; Zamora, A.; Plana, N.; Comas-Cufí, M.; García-Gil, M.; Lluch, R.M.; Ponjoan, A.; Alves, L.; Elosua, R.; Marrugat, J.; et al. Incidence of Cardiovascular Disease in Patients with Familial Hypercholesterolemia Phenotype: Analysis of 5 Years Follow-Up of Real-World Data from More than 1.5 Million Patients. J. Clin. Med. 2019, 8, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrov, I.S.; Postadzhiyan, A.S.; Tokmakova, M.P.; Kitova, L.G.; Tsonev, S.N.; Addison, J.; Petkova, R.T.; Lachev, V.I. Management of High and Very High-Risk Subjects with Familial Hypercholesterolemia: Results from an Observational Study in Bulgaria. Folia Medica 2018, 60, 389–396. [Google Scholar] [CrossRef]
- Hrechanina, O.; Isayeva, G.; Kolesnikova, O.; Isakova, Y. Relations Between Familial Hypercholesterolemia and Early Ischemic Heart Disease: An Analysis of Medical Documentation Data. Serbian J. Exp. Clin. Res. [CrossRef]
- Sharifi, M.; Higginson, E.; Bos, S.; Gallivan, A.; Harvey, D.; Li, K.W.; Abeysekera, A.; Haddon, A.; Ashby, H.; Shipman, K.; et al. Greater preclinical atherosclerosis in treated monogenic familial hypercholesterolemia vs. polygenic hypercholesterolemia. Atherosclerosis 2017, 263, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, P.C.J.D.L.; Morgan, A.C.; Jannes, C.E.; Turolla, L.; Krieger, J.E.; Santos, R.D.; Pereira, A.D.C. Presence and type of low density lipoprotein receptor (LDLR) mutation influences the lipid profile and response to lipid-lowering therapy in Brazilian patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2014, 233, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Mickiewicz, A.; Chmara, M.; Futema, M.; Fijałkowski, M.; Chlebus, K.; Gałąska, R.; Bandurski, T.; Pajkowski, M.; Żuk, M.; Wasąg, B.; et al. Efficacy of clinical diagnostic criteria for familial hypercholesterolemia genetic testing in Poland. Atherosclerosis 2016, 249, 52–58. [Google Scholar] [CrossRef]
- E Heath, K.; E Humphries, S.; Middleton-Price, H.; Boxer, M. A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur. J. Hum. Genet. 2001, 9, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Chmara, M.; Kubalska, J.; Bednarska-Makaruk, M.; Węgrzyn, A.; Pronicka, E.; Wehr, A.; Rynkiewicz, A.; Limon, J. Molecular Characterization of Polish Patients With Familial Hypercholesterolemia: Novel And Recurrent LDLR Gene Mutations. Atheroscler. Suppl. 2008, 9, 111. [Google Scholar] [CrossRef]
- Schuster, H.; Rauh, G.; Muller, S.; Keller, C.; Wolfram, G.; Zöllner, N. Allele-specific and asymmetric polymerase chain reaction amplification in combination: A one step polymerase chain reaction protocol for rapid diagnosis of familial defective apolipoprotein B-100. Anal. Biochem. 1992, 204, 22–25. [Google Scholar] [CrossRef]
- Iacocca, M.A.; Chora, J.R.; Carrié, A.; Freiberger, T.; Leigh, S.E.; Defesche, J.C.; Kurtz, C.L.; Distefano, M.; Santos, R.D.; Humphries, S.E.; et al. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum. Mutat. 2018, 39, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Bourbon, M.; Williams, M.; Humphries, S.E. Clinical utility of the polygenic LDL-C SNP score in familial hypercholesterolemia. Atherosclerosis 2018, 277, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Hear. J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooldridge, J.M. Econometric Analysis of Cross Section and Panel Data; MIT press: Cambridge, MA, USA, 2010. [Google Scholar]
- Szymański, F.M.; Barylski, M.; Cybulska, B.; Wożakowska-Kapłon, B.; Krasiński, Z.; Mamcarz, A.; Widecka, K.; Płatek, A.E.; Dudek, D.; Mickiewicz, A.; et al. Recommendation for the management of dyslipidemia in Poland—Third Declaration of Sopot. Interdisciplinary Expert Position Statement endorsed by the Polish Cardiac Society Working Group on Cardiovascular Pharmacotherapy. Cardiol. J. 2018, 25, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; A Ference, B.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Hear. J. 2019, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Masana, L.; Plana, N.; Pérez-Calahorra, S.; Ibarretxe, D.; Lamiquiz-Moneo, I.; Pedro-Botet, J.; Suarez-Tembra, M.; Valdivielso, P.; Ortega, E.; De Miguel-Etayo, P. How many familial hypercholesterolemia patients are eligible for PCSK9 inhibition? Atherosclerosis 2017, 262, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Gouni-Berthold, I. The efficacy of anti-PCSK9 antibodies: Results from recent trials. Atheroscler. Suppl. 2017, 30, 9–18. [Google Scholar] [CrossRef]
- E Kosmas, C.; Estrella, A.M.; Sourlas, A.; Silverio, D.; Hilario, E.; Montan, P.D.; Guzman, E. Inclisiran: A New Promising Agent in the Management of Hypercholesterolemia. Diseases 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Iacocca, M.A.; Ban, M.R.; A Hegele, R. Efficacy of Evolocumab in Monogenic vs Polygenic Hypercholesterolemia. CJC Open 2019, 1, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Sijbrands, E.; Lombardi, M.P.; Westendorp, R.G.; Leuven, J.A.G.; Meinders, A.; Van Der Laarse, A.; Frants, R.R.; Havekes, L.M.; Smelt, A.H. Similar response to simvastatin in patients heterozygous for familial hypercholesterolemia with mRNA negative and mRNA positive mutations. Atherosclerosis 1998, 136, 247–254. [Google Scholar] [CrossRef]
- Chaves, F.J.; Real, J.T.; García-García, A.B.; Civera, M.; Armengod, M.E.; Ascaso, J.F.; Carmena, R. Genetic Diagnosis of Familial Hypercholesterolemia in a South European Outbreed Population: Influence of Low-Density Lipoprotein (LDL) Receptor Gene Mutations on Treatment Response to Simvastatin in Total, LDL, and High-Density Lipoprotein Cholesterol. J. Clin. Endocrinol. Metab. 2001, 86, 4926–4932. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Raslova, K.; Vohnout, B.; Bláha, V.; Satny, M.; Kyselak, O.; Vaclova, M.; Urbanek, R.; Maskova, J.; Soska, V.; et al. Real-life LDL-C treatment goals achievement in patients with heterozygous familial hypercholesterolemia in the Czech Republic and Slovakia: Results of the PLANET registry. Atherosclerosis 2018, 277, 355–361. [Google Scholar] [CrossRef] [PubMed]
- De Isla, L.P.; Alonso, R.; Watts, G.F.; Mata, N.; Cerezo, A.S.; Muñiz, O.; Fuentes, F.; Díaz, J.L.D.; De Andrés, R.; Zambón, D.; et al. Attainment of LDL-Cholesterol Treatment Goals in Patients With Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 1278–1285. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, É.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Hear. J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.-H.; Gaudette, É.; Goldman, D.P. PCSK9 Inhibitors Show Value for Patients and the US Health Care System. Value Heal. 2017, 20, 1270–1278. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, M.; Futema, M.; Nair, N.-K.C.; Humphries, S.E. Polygenic Hypercholesterolemia and Cardiovascular Disease Risk. Curr. Cardiol. Rep. 2019, 21, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.A.; Pedersen, T.R.; Gaciong, Z.A.; Ceska, R.; Ezhov, M.V.; Connolly, D.L.; Jukema, J.W.; Toth, K.; Tikkanen, M.J.; Im, K.; et al. Effect of the PCSK9 Inhibitor Evolocumab on Total Cardiovascular Events in Patients With Cardiovascular Disease: A Prespecified Analysis From the FOURIER Trial. JAMA Cardiol. 2019, 4, 613–619. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Parameter | Polygenic Hypercholesterolemia n = 65 | Monogenic Hypercholesterolemia n = 47 | p Value |
|---|---|---|---|
| Age, years * | 54.37 ± 12.54 | 50.57 ± 13.49 | 0.134 |
| Female | 42 (64.62%) | 28 (59.57%) | 0.586 |
| Prevention Primary | 48 (73.85%) | 31 (65.96%) | 0.366 |
| Prevention Secondary | 17 (26.15%) | 16 (34.04%) | 0.366 |
| CVD | 15 (23.08%) | 15 (31.91%) | 0.297 |
| CAD | 13 (20.00%) | 13 (27.66%) | 0.343 |
| CAD age, years * | 49.45 ± 10.96 | 47.30 ± 3.56 | 0.548 |
| MI | 21 (32.31%) | 8 (17.02%) | 0.311 |
| MI age, years * | 48.70 ± 11.25 | 47.38 ± 5.40 | 0.748 |
| PCI | 7 (10.77%) | 9 (19.15%) | 0.211 |
| PCI age, years * | 46.57 ± 12.16 | 46.78 ± 3.35 | 0.966 |
| CABG | 4 (6.15%) | 3 (6.38%) | 0.961 |
| CABG age, years * | 48.00 ± 5.20 | 51.5 ± 0.71 | 0.364 |
| Stroke/TIA | 3 (4.62%) | 2 (4.26%) | 0.748 |
| Smoking | 11 (16.92%) | 7 (14.89%) | 0.783 |
| HA | 22 (33.85%) | 23 (48.94%) | 0.169 |
| DM | 6 (9.23%) | 2 (4.26%) | 0.303 |
| BMI (kg/m2)* | 26.57 ± 4.11 | 26.24 ± 4.62 | 0.720 |
| FH Definite Probable Possible | 2 (3.08%) 29 (44.62%) 34 (52.31%) | 24 (51.06%) 21 (44.68%) 2 (4.26%) | <0.001 0.995 <0.001 |
| Family history of hypercholesterolemia in adults aged >18 years defined as LDL-C > 4.9 mmol/L (190 mg/dL) | 22 (33.85%) | 39 (82.98%) | <0.001 |
| Family history of hypercholesterolemia in children defined as LDL-C >4.0 mmol/L (155 mg/dL) | 6 (9.23%) | 12 (25.53%) | 0.021 |
| Family history of premature CAD (in men below age 55, in women below 60 years) | 49 (75.38%) | 35 (74.47%) | 0.912 |
| Corneal arcus <45 y | 1 (1.54%) | 5 (10.64%) | 0.046 |
| Tendinous xanthomata | 0 | 1 (2.13%) | 0.420 |
| Parameter | Polygenic Hypercholesterolemia N = 65 | Monogenic Hypercholesterolemia N = 47 | p Value |
|---|---|---|---|
| Lipid profile parameters before and after treatment | |||
| TC (mmol/L) * baseline after treatment | 8.6 ± 1.2 5.0 ± 0.9 | 10.0 ± 1.8 5.7 ± 1.2 | <0.001 <0.001 |
| LDL-C (mmol/L) * baseline after treatment | 6.2 ± 1.2 2.9 ± 0.7 | 7.6 ± 1.5 3.8 ± 1.1 | <0.001 <0.001 |
| non HDL-C (mmol/L) * baseline after treatment | 7.0 ± 1.4 3.5 ± 0.9 | 8.3 ± 1.7 4.3 ± 1.1 | <0.001 <0.001 |
| TG (mmol/L) * baseline after treatment | 1.7 ± 0.8 1.3 ± 0.6 | 1.6 ± 0.6 1.1 ± 0.5 | 0.431 0.136 |
| HDL-C (mmol/L) * baseline after treatment | 1.6 ± 0.4 1.5 ± 0.4 | 1.6 ± 0.5 1.5 ± 0.4 | 0.499 0.421 |
| Lipid lowering therapy | |||
| Rosuvastatin 5–10 mg daily | 22 (34%) | 9 (19%) | 0.134 |
| Rosuvastatin 15–20 mg daily | 20 (31%) | 22 (47%) | |
| Rosuvastatin 30–40 mg daily | 23 (36%) | 16 (34%) | |
| Ezetimibe use | 15 (23%) | 20 (43%) | 0.029 |
| High intensity rosuvastatin therapy (20–40 mg daily) | 41 (63%) | 37 (79%) | 0.075 |
| High intensity rosuvastatin in combination with ezetimibe | 10 (24%) | 14 (38%) | 0.199 |
| Statin intolerance | 7 (11%) | 5 (11%) | 0.617 |
| SAMS | 4 (6%) | 4 (9%) | 0.451 |
| LDL-C treatment goals | |||
| LDL-C < 2.5 mmol/L | 25 (38%) | 6 (13%) | 0.003 |
| LDL-C < 2.5 mmol/L achieved in primary prevention | 19/48 (40%) | 5/31 (16%) | 0.027 |
| LDL-C < 1.8 mmol/L in secondary prevention | 1/17 (6%) | 0/16 (0%) | 0.515 |
| LDL-C goal achieved | 20 (31%) | 5 (11%) | 0.012 |
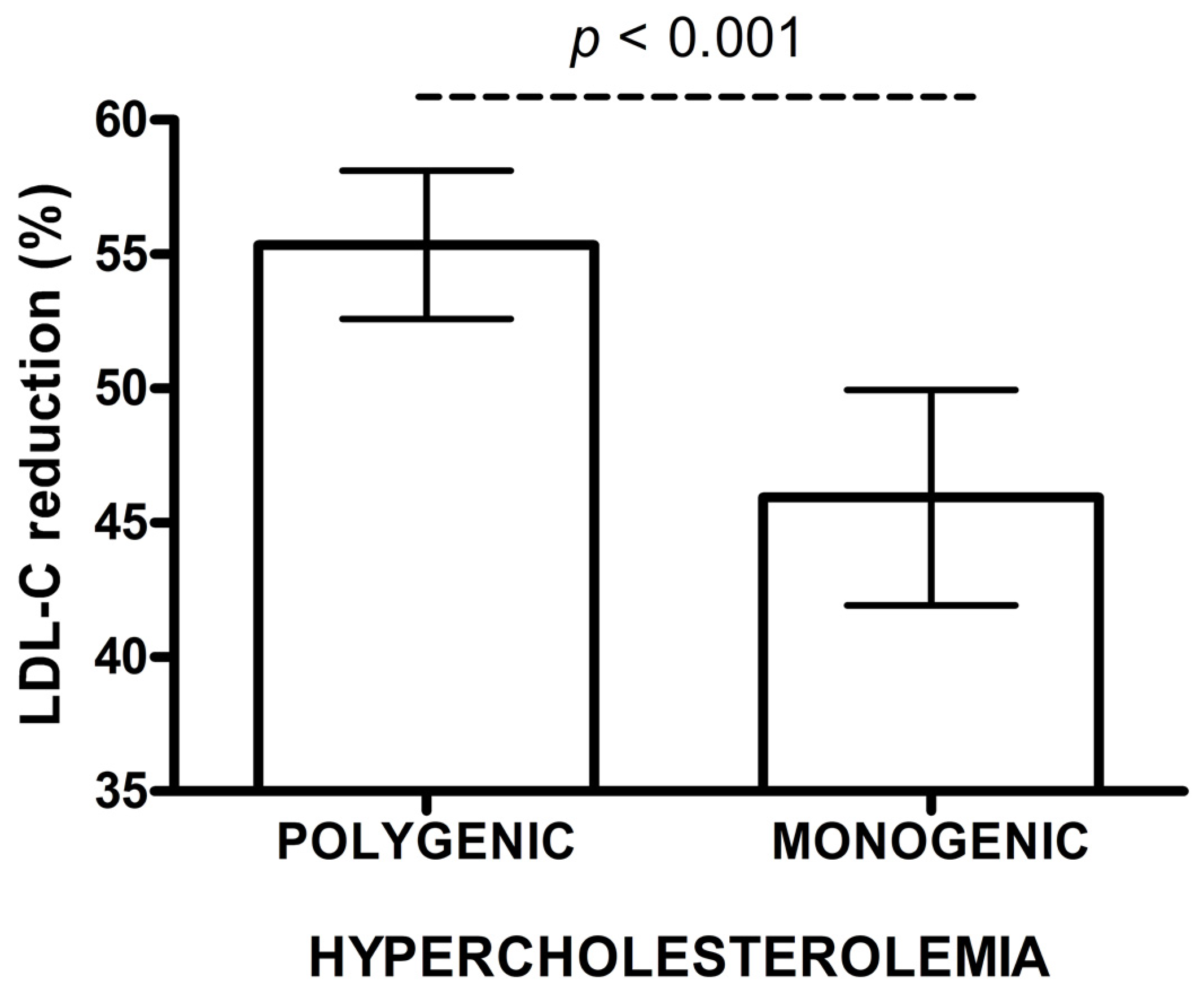
| A. LDL-C reduction (%) | ||||
| Inverse Probability Weighted Regression Adjustment (IPWRA) | ||||
| Group | Mean,% * | p value | 95% CI | |
| lower | upper | |||
| Monogenic | 45.9 | <0.001 | 42.0 | 49.8 |
| Polygenic | 55.4 | 52.7 | 58.1 | |
| Ancova | ||||
| Group | Mean,% * | p value | 95% CI | |
| lower | upper | |||
| Monogenic | 41.5 | <0.001 | 35.4 | 47.6 |
| Polygenic | 51.6 | 46.6 | 56.6 | |
| Linear Regression Model | ||||
| Variable | F(3,107) = 8.23 p < 0.001 | |||
| Coefficient | p value | 95% CI | ||
| lower | upper | |||
| Constant | 27.41 | <0.001 | 15.61 | 39.20 |
| Monogenic/Polygenic | −10.13 | <0.001 | −15.72 | −4.54 |
| LDL baseline | 0.11 | <0.001 | 0.06 | 0.16 |
| Diabetes melitus | −9.38 | 0.052 | −18.84 | 0.08 |
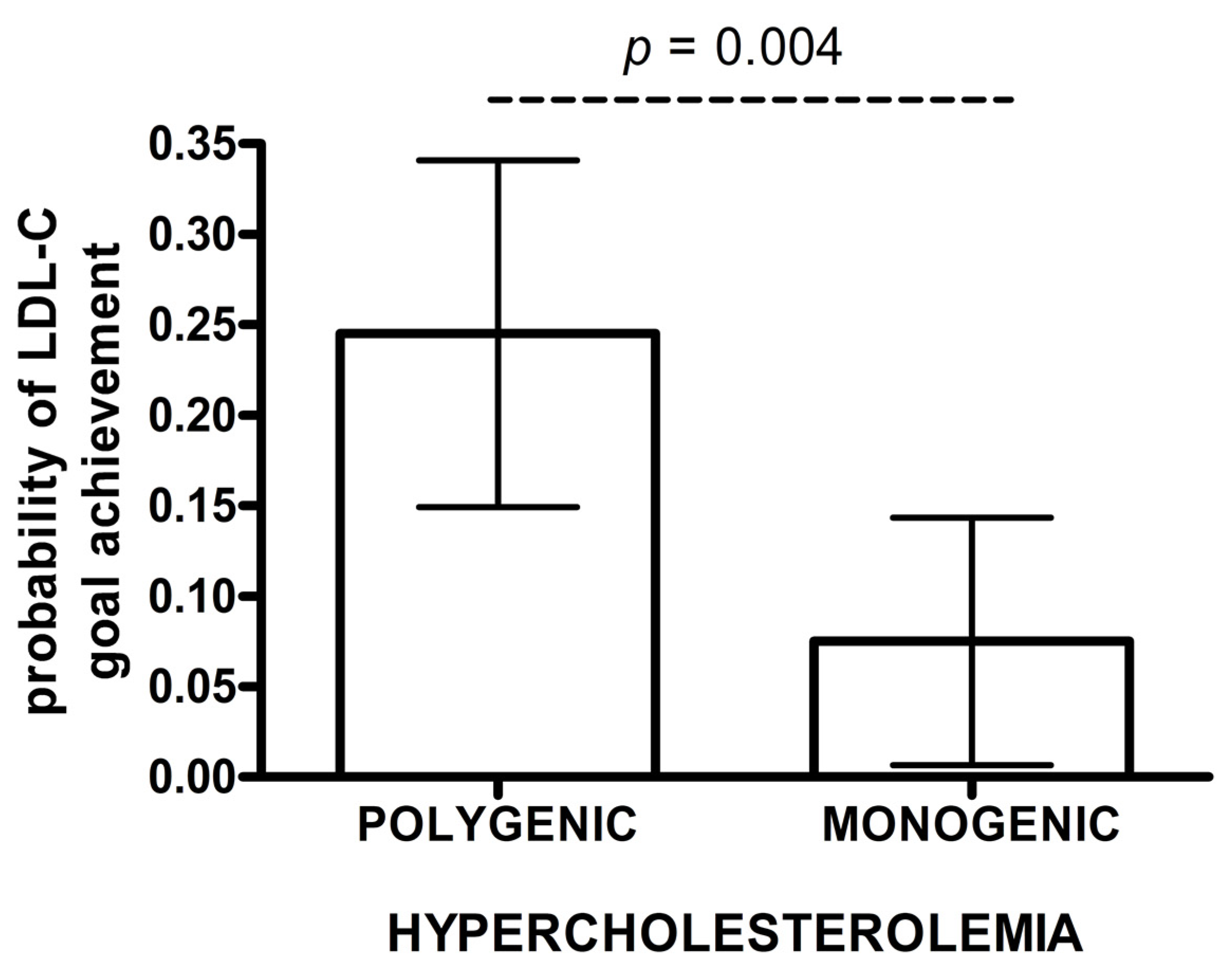
| B. LDL-C Goal Achievement | ||||
| Inverse Probability Weighted Regression Adjustment (IPWRA) | ||||
| Group | Probability of goal achievement | p value | 95% CI | |
| lower | upper | |||
| Monogenic | 0.075 | 0.004 | 0.008 | 0.142 |
| Polygenic | 0.245 | 0.151 | 0.339 | |
| Risk Ratio Polygenic vs. Monogenic = 3.28 | 1.23 | 8.72 | ||
| Logistic Regression | ||||
| Variable | LR Chi2 (2) = 16.90 p < 0.001 | |||
| Odds ratio | p value | 95% CI | ||
| lower | upper | |||
| Constant | 0.625 | 0.103 | 0.355 | 1.100 |
| Monogenic/Polygenic | 0.281 | 0.023 | 0.094 | 0.840 |
| CVD | 0.087 | 0.020 | 0.011 | 0.686 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mickiewicz, A.; Futema, M.; Ćwiklinska, A.; Kuchta, A.; Jankowski, M.; Kaszubowski, M.; Chmara, M.; Wasąg, B.; Fijałkowski, M.; Jaguszewski, M.; et al. Higher Responsiveness to Rosuvastatin in Polygenic versus Monogenic Hypercholesterolemia: A Propensity Score Analysis. Life 2020, 10, 73. https://doi.org/10.3390/life10050073
Mickiewicz A, Futema M, Ćwiklinska A, Kuchta A, Jankowski M, Kaszubowski M, Chmara M, Wasąg B, Fijałkowski M, Jaguszewski M, et al. Higher Responsiveness to Rosuvastatin in Polygenic versus Monogenic Hypercholesterolemia: A Propensity Score Analysis. Life. 2020; 10(5):73. https://doi.org/10.3390/life10050073
Chicago/Turabian StyleMickiewicz, Agnieszka, Marta Futema, Agnieszka Ćwiklinska, Agnieszka Kuchta, Maciej Jankowski, Mariusz Kaszubowski, Magdalena Chmara, Bartosz Wasąg, Marcin Fijałkowski, Miłosz Jaguszewski, and et al. 2020. "Higher Responsiveness to Rosuvastatin in Polygenic versus Monogenic Hypercholesterolemia: A Propensity Score Analysis" Life 10, no. 5: 73. https://doi.org/10.3390/life10050073