DNA Methylation Influences miRNA Expression in Gonadotroph Pituitary Tumors

 ,
,
Abstract
:1. Introduction
2. Results
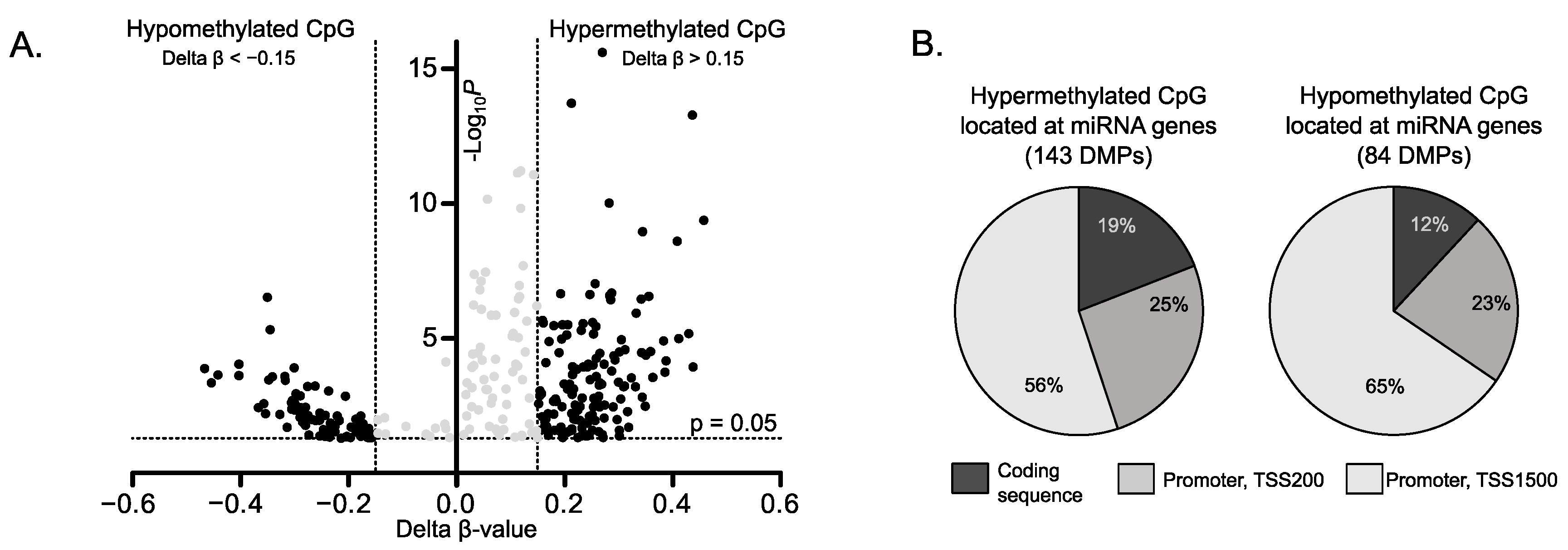
2.1. DNA Methylation Changes in miRNA Genes in Gonadotroph PitNETs
2.2. The Relationship between Impaired Methylation and Expression of miRNA Genes
2.3. Validating the Role of Selected Aberrantly Methylated miRNAs in a Large Patient Cohort
2.4. Predicting the mRNA Targets of Epigenetically Deregulated miRNAs
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Profiling Genome-Wide DNA Methylation with Microarray Technology
4.3. Assessment of miRNA Expression with Next Generation Sequencing (NGS)
4.4. Evaluation of DNA Methylation Pattern at Selected Genomic Regions
4.5. Determining miRNA Expression Level with qRT-PCR
4.6. miRNA Target-Prediction
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Greenman, Y.; Naftali, S. Non-functioning pituitary adenomas. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Hauser, B.M.; Lau, A.; Gupta, S.; Bi, W.L.; Dunn, I.F. The epigenomics of pituitary adenoma. Front. Endocrinol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, S.; Monzo, M.; Navarro, A. Epigenetic regulation mechanisms of microRNA expression. Biomol. Concepts 2017, 8, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Mao, Z.G.; Wang, X.; Du, Q.; Jian, M.; Zhu, D.; Xiao, Z.; Wang, H.J.; Zhu, Y.H. MicroRNAs and Target Genes in Pituitary Adenomas. Horm. Metab. Res. 2018, 50, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Syeda, Z.A.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory mechanism of microrna expression in cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kober, P.; Boresowicz, J.; Rusetska, N.; Maksymowicz, M.; Goryca, K.; Kunicki, J.; Bonicki, W.; Siedlecki, J.A.; Bujko, M. DNA methylation profiling in nonfunctioning pituitary adenomas. Mol. Cell. Endocrinol. 2018, 473, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. MirDIP 4.1 - Integrative database of human microRNA target predictions. Nucleic Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Nakayama, N.; Sakashita, G.; Nariai, Y.; Kato, H.; Sinmyozu, K.; Nakayama, J.I.; Kyo, S.; Urano, T.; Nakayama, K. Cancer-related transcription regulator protein NAC1 forms a protein complex with CARM1 for ovarian cancer progression. Oncotarget 2018, 9, 28408–28420. [Google Scholar] [CrossRef]
- Schult, D.; Hölsken, A.; Buchfelder, M.; Schlaffer, S.M.; Siegel, S.; Kreitschmann-Andermahr, I.; Fahlbusch, R.; Buslei, R. Expression pattern of neuronal intermediate filament α-internexin in anterior pituitary gland and related tumors. Pituitary 2015, 18, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Glaich, O.; Parikh, S.; Bell, R.E.; Mekahel, K.; Donyo, M.; Leader, Y.; Shayevitch, R.; Sheinboim, D.; Yannai, S.; Hollander, D.; et al. DNA methylation directs microRNA biogenesis in mammalian cells. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.-X.; Liu, Z.; Deng, F.; Wang, D.-D.; Li, X.-W.; Tian, T.; Zhang, J.; Tang, J.-H. MiR-145: A potential biomarker of cancer migration and invasion. Am. J. Transl. Res. 2019, 11, 6739–6753. [Google Scholar] [PubMed]
- Hu, T.; Chong, Y.; Cai, B.; Liu, Y.; Lu, S.; Cowell, J.K. DNA methyltransferase 1–mediated CpG methylation of the miR-150-5p promoter contributes to fibroblast growth factor receptor 1– driven leukemogenesis. J. Biol. Chem. 2019, 294, 18122–18130. [Google Scholar] [CrossRef] [PubMed]
- Campos-Viguri, G.E.; Jiménez-Wences, H.; Peralta-Zaragoza, O.; Torres-Altamirano, G.; Soto-Flores, D.G.; Hernández-Sotelo, D.; Alarcón-Romero, L.D.C.; Jiménez-López, M.A.; Illades-Aguiar, B.; Fernández-Tilapa, G. MiR-23b as a potential tumor suppressor and its regulation by DNA methylation in cervical cancer. Infect. Agent. Cancer 2015, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lv, D.; Li, M.; Xuefei, Z.; Sun, G.; Bai, Y.; Dongmin, C. Hypermethylation of miRNA-589 promoter leads to upregulation of HDAC5 which promotes malignancy in non-small cell lung cancer. Int. J. Oncol. 2017, 50, 2079–2090. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, I.M.D.P.; Barros-Filho, M.C.; Dos Reis, M.B.; Beltrami, C.M.; Marchi, F.A.; Kuasne, H.; do Canto, L.M.; de Mello, J.B.H.; Abildgaard, C.; Pinto, C.A.L.; et al. Loss of DNA methylation is related to increased expression of miR-21 and miR-146b in papillary thyroid carcinoma. Clin. Epigenetics 2018, 10, 144. [Google Scholar] [CrossRef]
- Lynch, S.M.; O’Neill, K.M.; McKenna, M.M.; Walsh, C.P.; McKenna, D.J. Regulation of miR-200c and miR-141 by Methylation in Prostate Cancer. Prostate 2016, 76, 1146–1159. [Google Scholar] [CrossRef]
- Wong, N.W.; Chen, Y.; Chen, S.; Wang, X. OncomiR: An online resource for exploring pan-cancer microRNA dysregulation. Bioinformatics 2018, 34, 713–715. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Zhou, Y.; Zhang, Y.; Wang, T.; Wang, J. Role of MIR-489 in the proliferation and apoptosis of pancreatic carcinoma. J. B.U.ON. 2019, 24, 1574–1580. [Google Scholar]
- Liu, Q.; Yang, G.; Qian, Y. Loss of MicroRNA-489-3p promotes osteosarcoma metastasis by activating PAX3-MET pathway. Mol. Carcinog. 2017, 56, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Liu, H.; Hou, S.; Wu, L.; Yang, Z.; Shen, J.; Zhou, L.; Zheng, S.S.; Jiang, B. MiR-489 suppresses tumor growth and invasion by targeting HDAC7 in colorectal cancer. Clin. Transl. Oncol. 2018, 20, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; De La Rosa-Velázquez, I.A.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of miR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther. Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Han, S.; Zhang, K.; Chen, J.; Li, C.; Wang, R.; Chen, L. The roles of MicroRNA-141 in human cancers: From diagnosis to treatment. Cell. Physiol. Biochem. 2016, 38, 427–448. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.; Salvi, A.; Baiocchi, G.; Portolani, N.; De Petro, G. Functional Role of microRNA-23b-3p in Cancer Biology. MicroRNA 2018, 7, 156–166. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Zhang, Y.; Wang, D.; Gao, H.; Wang, L.; Gao, S. Prognostic role of microRNA-150 in various carcinomas: A meta-analysis. Onco. Targets. Ther. 2016, 9, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.M.; Jiang, X.S.; Li, H.Z.; Qian, L.X.; Du, M.Y.; Lu, Z.W.; Wu, J.; Tian, X.K.; Fei, Q.; He, X.; et al. MiR-184 Inhibits Tumor Invasion, Migration and Metastasis in Nasopharyngeal Carcinoma by Targeting Notch2. Cell. Physiol. Biochem. 2018, 49, 1564–1576. [Google Scholar] [CrossRef]
- Yang, J.; Lai, C.C.; Xian, Z.M.; Wang, W.Q.; Xu, B.L. Preliminary results indicate increased expression of MIR-184 in patients with renal carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6878–6887. [Google Scholar]
- Salem, M.; Shan, Y.; Bernaudo, S.; Peng, C. miR-590-3p targets cyclin G2 and FOXO3 to promote ovarian cancer cell proliferation, invasion, and spheroid formation. Int. J. Mol. Sci. 2019, 20, 1810. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Ye, L.; Feng, X.; Shi, R.; Sun, Z.; Li, Z.; Liu, T. MicroRNA-590-3p inhibits invasion and metastasis in triple-negative breast cancer by targeting slug. Am. J. Cancer Res. 2020, 10, 965–974. [Google Scholar]
- Leone, V.; Langella, C.; D’Angelo, D.; Mussnich, P.; Wierinckx, A.; Terracciano, L.; Raverot, G.; Lachuer, J.; Rotondi, S.; Jaffrain-Rea, M.-L.; et al. Mir-23b and miR-130b expression is downregulated in pituitary adenomas. Mol. Cell. Endocrinol. 2014, 390, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Hu, B.; Feng, Y.; Wang, Z.; Wang, X.; Zhu, D.; Zhu, Y.; Jiang, X.; Wang, H. Circoma1-mediated miR-145-5p suppresses tumor growth of nonfunctioning pituitary adenomas by targeting TPT1. J. Clin. Endocrinol. Metab. 2019, 104, 2419–2434. [Google Scholar] [CrossRef] [PubMed]
- Amaral, F.C.; Torres, N.; Saggioro, F.; Neder, L.; Machado, H.R.; Silva, W.A.; Moreira, A.C.; Castro, M. MicroRNAs differentially expressed in ACTH-secreting pituitary tumors. J. Clin. Endocrinol. Metab. 2009, 94, 320–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, T.; Faucz, F.R.; Azevedo, M.; Xekouki, P.; Iliopoulos, D.; Stratakis, C.A. Functional screen analysis reveals miR-26b and miR-128 as central regulators of pituitary somatomammotrophic tumor growth through activation of the PTEN-AKT pathway. Oncogene 2013, 32, 1651–1659. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Zhang, M.; Liu, H.F.; Wang, J.P. Effects of microRNA-21 targeting PITX2 on proliferation and apoptosis of pituitary tumor cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2995–3004. [Google Scholar]
- Cheunsuchon, P.; Zhou, Y.; Zhang, X.; Lee, H.; Chen, W.; Nakayama, Y.; Rice, K.A.; Tessa Hedley-Whyte, E.; Swearingen, B.; Klibanski, A. Silencing of the imprinted DLK1-MEG3 locus in human clinically nonfunctioning pituitary adenomas. Am. J. Pathol. 2011, 179, 2120–2130. [Google Scholar] [CrossRef]
- Mao, Z.G.; He, D.S.; Zhou, J.; Yao, B.; Xiao, W.W.; Chen, C.H.; Zhu, Y.H.; Wang, H.J. Differential expression of microRNAs in GH-secreting pituitary adenomas. Diagn. Pathol. 2010, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Chen, L.; Hu, X.; Tang, J.; He, L.; Hu, J.; Fei, F.; Wang, Q. Next-generation sequencing of microRNAs reveals a unique expression pattern in different types of pituitary adenomas. Endocr. J. 2019, 66, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Vila-Casadesús, M.; Gironella, M.; Lozano, J.J. MiRComb: An R package to analyse miRNA-mRNA interactions. Examples across five digestive cancers. PLoS ONE 2016, 11, 1–18. [Google Scholar] [CrossRef]
- Hu, G.; Zhao, X.; Wang, C.; Geng, Y.; Zhao, J.; Xu, J.; Zuo, B.; Zhao, C.; Wang, C.; Zhang, X. MicroRNA-145 attenuates TNF-α-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 2017, 8, e3140. [Google Scholar] [CrossRef]
- Gao, P.; Xing, A.Y.; Zhou, G.Y.; Zhang, T.G.; Zhang, J.P.; Gao, C.; Li, H.; Shi, D.B. The molecular mechanism of microRNA-145 to suppress invasion-metastasis cascade in gastric cancer. Oncogene 2013, 32, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cheng, Y.; Ren, X.; Hori, T.; Huber-Keener, K.J.; Zhang, L.; Yap, K.L.; Liu, D.; Shantz, L.; Qin, Z.H.; et al. Dysfunction of nucleus accumbens-1 activates cellular senescence and inhibits tumor cell proliferation and oncogenesis. Cancer Res. 2012, 72, 4262–4275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Wang, J.; Wang, G.; Jin, Y.; Luo, X.; Xia, X.; Gong, J.; Hu, J. P55PIK transcriptionally activated by MZF1 promotes colorectal cancer cell proliferation. BioMed Res. Int. 2013, 2013, 868131. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Rhein, M.; Hagemeier, L.; Klintschar, M.; Muschler, M.; Bleich, S.; Frieling, H. DNA methylation results depend on DNA integrity- role of post mortem interval. Front. Genet. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helen, M.M. Deleterious effects of formalin- fixation and delays to fixation on RNA and miRNA-Seq profiles. Sci. Rep. 2019, 9, 6980. [Google Scholar]
- Ferreira, P.G.; Muñoz-Aguirre, M.; Reverter, F.; Sá Godinho, C.P.; Sousa, A.; Amadoz, A.; Sodaei, R.; Hidalgo, M.R.; Pervouchine, D.; Carbonell-Caballero, J.; et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Kober, P.; Boresowicz, J.; Rusetska, N.; Maksymowicz, M.; Paziewska, A.; Dąbrowska, M.; Kunicki, J.; Bonicki, W.; Ostrowski, J.; Siedlecki, J.A.; et al. The role of aberrant DNA methylation in misregulation of gene expression in gonadotroph nonfunctioning pituitary tumors. Cancers 2019, 11, 1650. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, 1–10. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. MiRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| HM450K CpG Site | CpG Location | miRNA | Discovery Group Spearman R; adj. p-Value | Validation Group Spearman R; p-Value |
|---|---|---|---|---|
| cg23121785 | TSS1500 | hsa-miR-184 | −0.577; adj. p = 0.0244 | −0.524; p < 0.0001 |
| cg01310120 | TSS200 | hsa-miR-145-5p | −0.608; adj. p = 0.0163 | −0.312; p = 0.0061 |
| cg23917868 | TSS200 | hsa-miR-145-5p | −0.672; adj. p = 0.0122 | −0.448; p < 0.0001 |
| cg02515217 | TSS200 | hsa-miR-21-5p | −0,796; adj. p = 0.0011 | −0.336; p = 0.0030 |
| Putative Onco-miRNAs Based on Identification of Target mRNAs in NCG 6.0 | |||||
|---|---|---|---|---|---|
| miRNA | DNA Methylation/Expression Results | Target Gene Prediction | |||
| Methylation status in tumor | DNA methylation/expression relationship | Putative target mRNA | Correlation (Spearman R, non-adjusted p-value) | Confidence of target prediction with MirDIP | |
| hsa-miR-134-5p | Hypomethylated | Positive | SCN9A | −0.531, 0.0080 | Very high |
| hsa-miR-145-5p | Hypermethylated | Negative | MAP2K4 | −0.539, 0.0071 | Very high |
| CDH2 | −0.515, 0.0100 | Very high | |||
| hsa-miR-150-5p | Hypermethylated | Negative | CACNA2D1 | −0.514, 0.0102 | Very high |
| hsa-miR-21-5p | Hypermethylated | Negative | RNF111 | −0.558, 0.0053 | Very high |
| hsa-miR-23b-3p | Hypermethylated | Negative | CCDC6 | −0.505, 0.0116 | Very high |
| Putative onco-miRNAs based on literature-based interpretation of role of target mRNA | |||||
| miRNA | DNA Methylation/Expression Results | Target Gene Prediction | |||
| Methylation status in tumor | DNA methylation/expression relationship | Putative target mRNA | Correlation (Spearman R, non-adjusted p-value) | Confidence of target prediction with MirDIP | |
| hsa-miR-23b-3p | Hypermethylated | Negative | NACC1 | −0.510, 0.0107 | Very high |
| PIK3R3 | −0.504, 0.0117 | Very high | |||
| hsa-miR-134-5p | Hypomethylated | Positive | INA | −0.5103, 0.0107 | Very high |
| Characteristic | Genome-Wide DNA Methylation/Whole Transcriptome Profiling | DNA Pyrosequencing/qRT-PCR |
|---|---|---|
| PitNET patients (number of patients) | 15 | 80 |
| Age (years) | ||
| Range | 36–73 | 34–80 |
| Median | 57 | 61 |
| Gender (number of patients) | ||
| Male | 10 | 46 |
| Female | 5 | 34 |
| Histopathology (number of patients) | ||
| Gonadotroph PA | 15 | 74 |
| Null-cell/gonadotroph PA * | 0 | 6 |
| Clinical classification (number of patients) | ||
| Invasive NFPA | 5 | 49 |
| Non-invasive NFPA | 10 | 25 |
| Unknown | - | 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boresowicz, J.; Kober, P.; Rusetska, N.; Maksymowicz, M.; Paziewska, A.; Dąbrowska, M.; Zeber-Lubecka, N.; Kunicki, J.; Bonicki, W.; Ostrowski, J.; et al. DNA Methylation Influences miRNA Expression in Gonadotroph Pituitary Tumors. Life 2020, 10, 59. https://doi.org/10.3390/life10050059
Boresowicz J, Kober P, Rusetska N, Maksymowicz M, Paziewska A, Dąbrowska M, Zeber-Lubecka N, Kunicki J, Bonicki W, Ostrowski J, et al. DNA Methylation Influences miRNA Expression in Gonadotroph Pituitary Tumors. Life. 2020; 10(5):59. https://doi.org/10.3390/life10050059
Chicago/Turabian StyleBoresowicz, Joanna, Paulina Kober, Natalia Rusetska, Maria Maksymowicz, Agnieszka Paziewska, Michalina Dąbrowska, Natalia Zeber-Lubecka, Jacek Kunicki, Wiesław Bonicki, Jerzy Ostrowski, and et al. 2020. "DNA Methylation Influences miRNA Expression in Gonadotroph Pituitary Tumors" Life 10, no. 5: 59. https://doi.org/10.3390/life10050059
APA StyleBoresowicz, J., Kober, P., Rusetska, N., Maksymowicz, M., Paziewska, A., Dąbrowska, M., Zeber-Lubecka, N., Kunicki, J., Bonicki, W., Ostrowski, J., Siedlecki, J. A., & Bujko, M. (2020). DNA Methylation Influences miRNA Expression in Gonadotroph Pituitary Tumors. Life, 10(5), 59. https://doi.org/10.3390/life10050059