Semi-Automated Heavy-Mineral Analysis by Raman Spectroscopy

Abstract
:1. Introduction
2. Methodology/Experimental
2.1. Instrumental Setup
2.1.1. Raman Spectrometry
2.1.2. Electron Microprobe Analysis (EMPA)
2.2. Sample Preparation
2.3. Raman Measurement Setup
- If TL gray value >= tTL:⚬ transparent grain
- If TL gray value < tTL and RL gray value < tRL:⚬ transparent grain
- If TL gray value < tTL and RL gray value > tRL:⚬ opaque grain
- If TL gray value < tTL:⚬ opaque grain
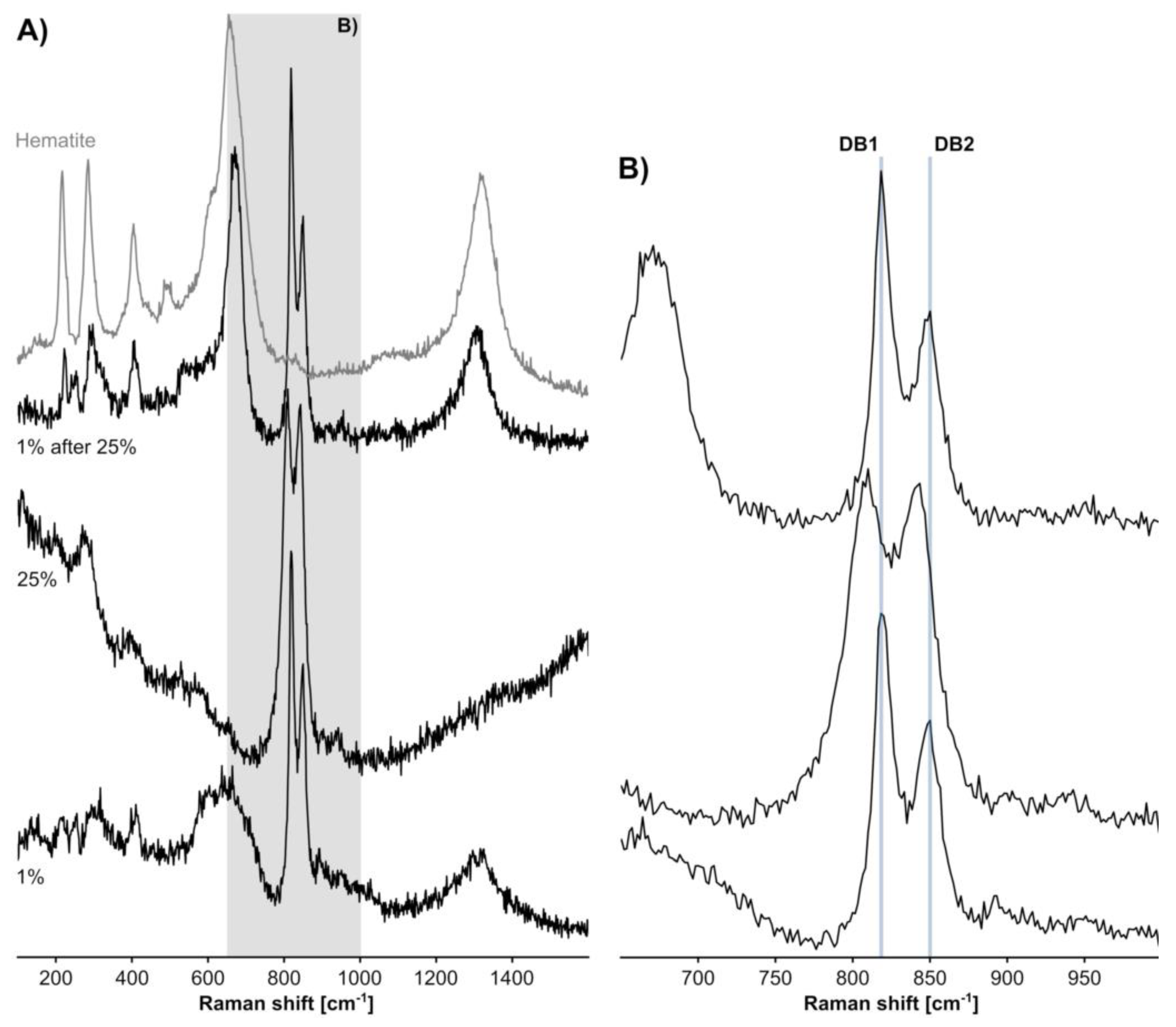
2.4. (Semi-) Automated Evaluation Routine
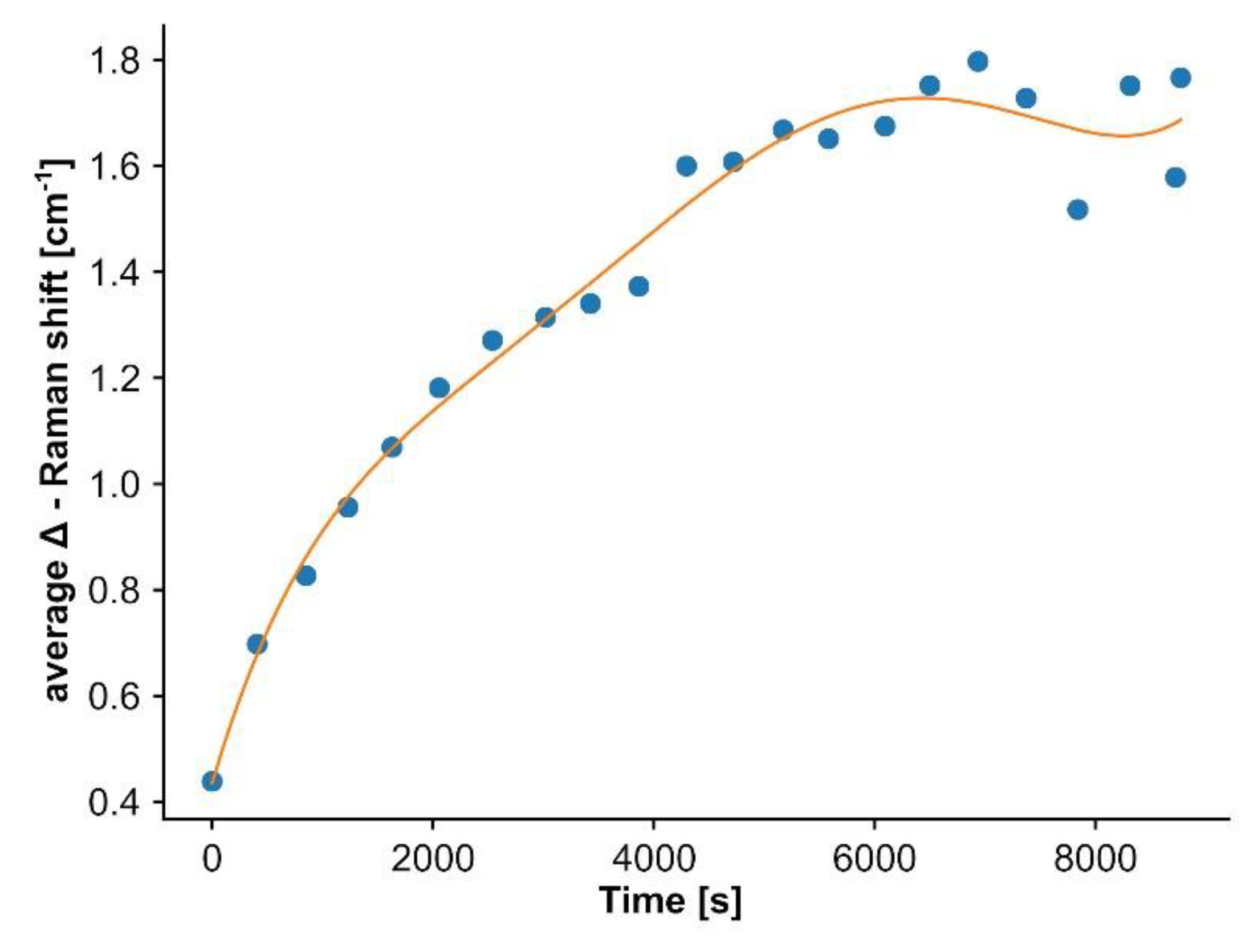
- Correcting for the temporal drift
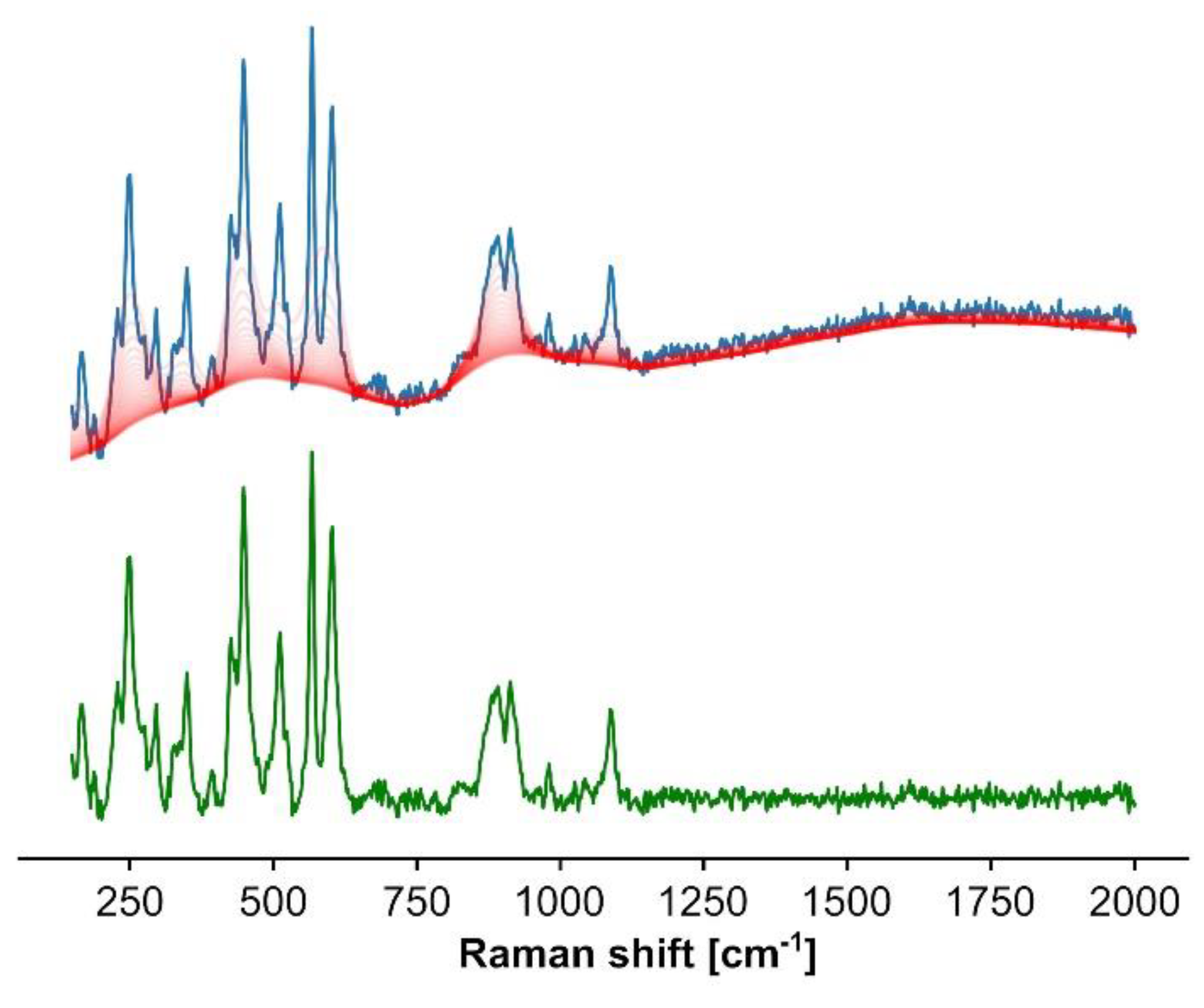
- Background removal
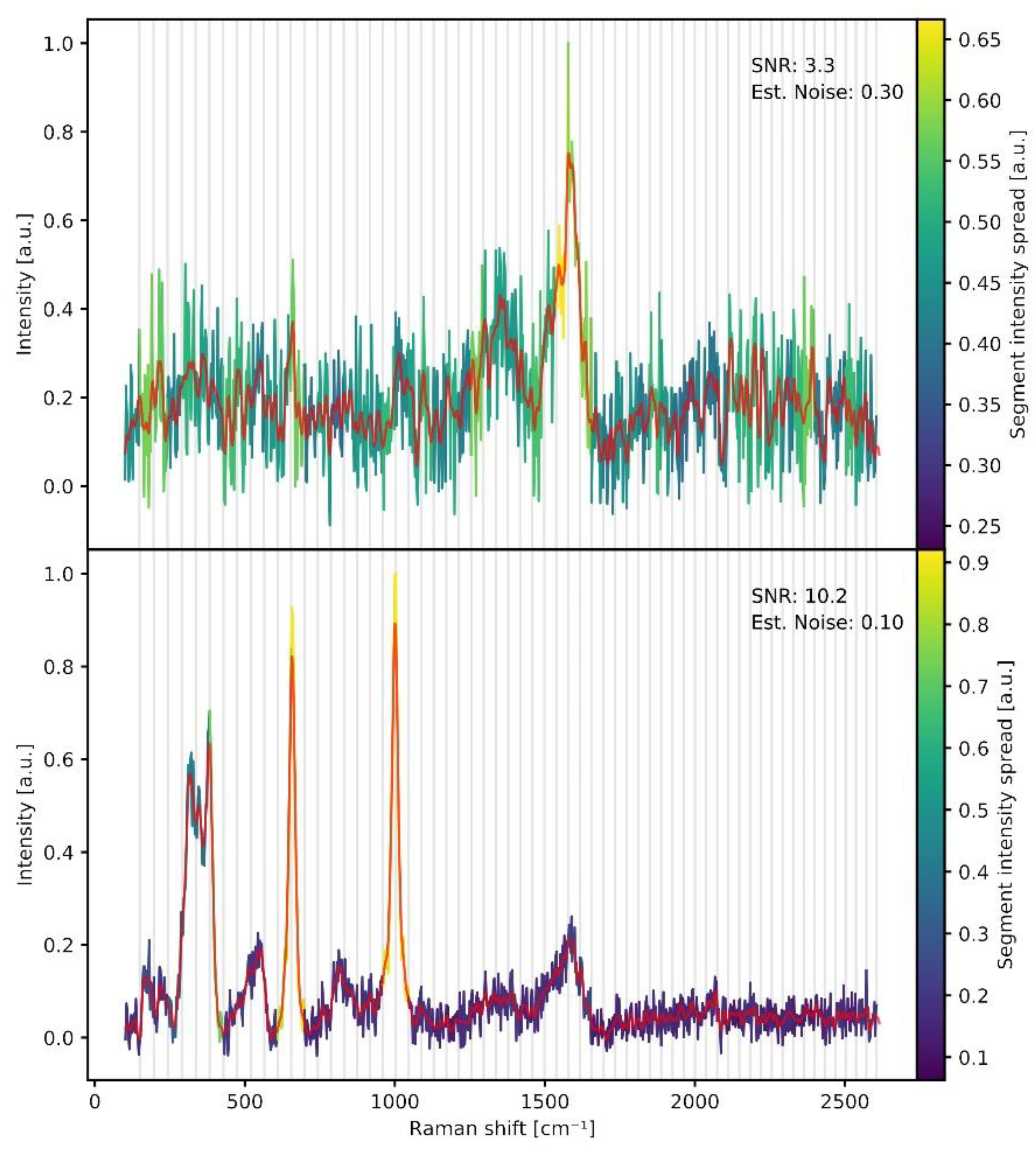
- Estimation of noise and exclusion of spectra below signal-to-noise (SNR) threshold
- Smoothing and scaling
- Correcting for embedding medium spectrum
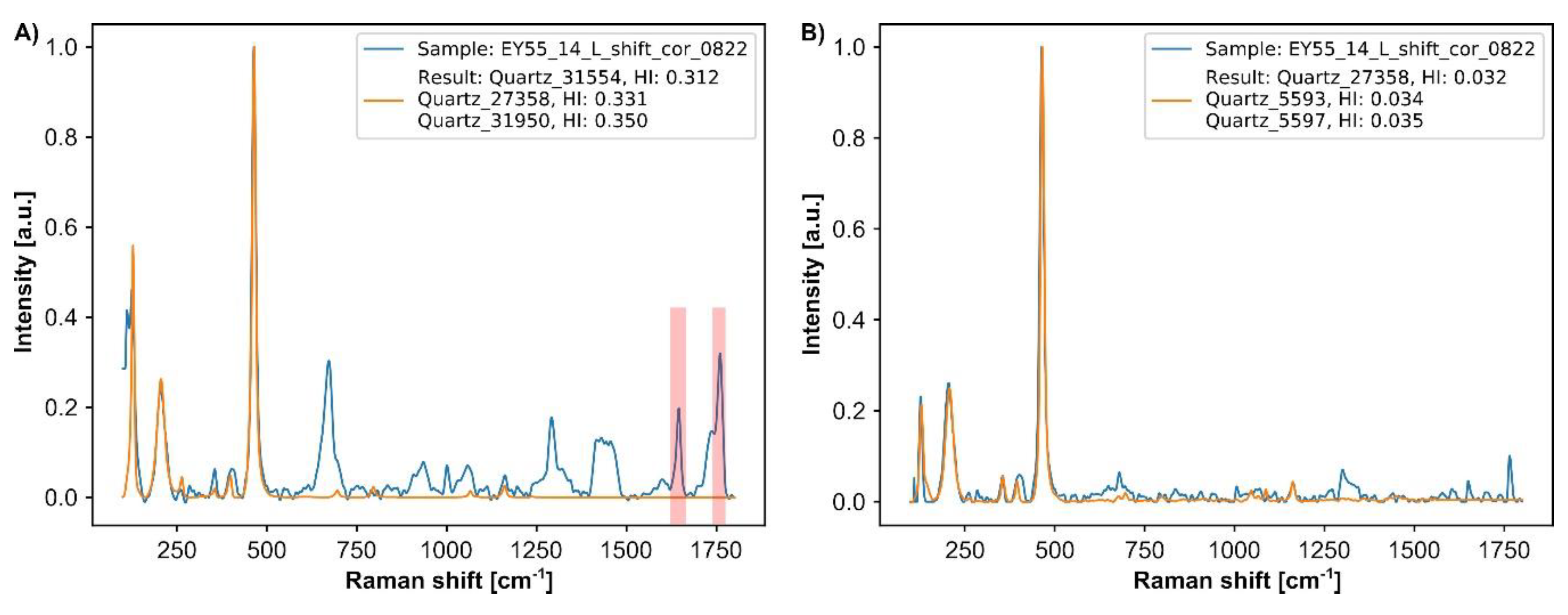
- Phase identification and compilation of HM assemblage
- Curve-fitting of interesting mineral groups (e.g., cpx, ol, grt) to semi-quantitatively assess chemical composition
3. Application (Case Study)
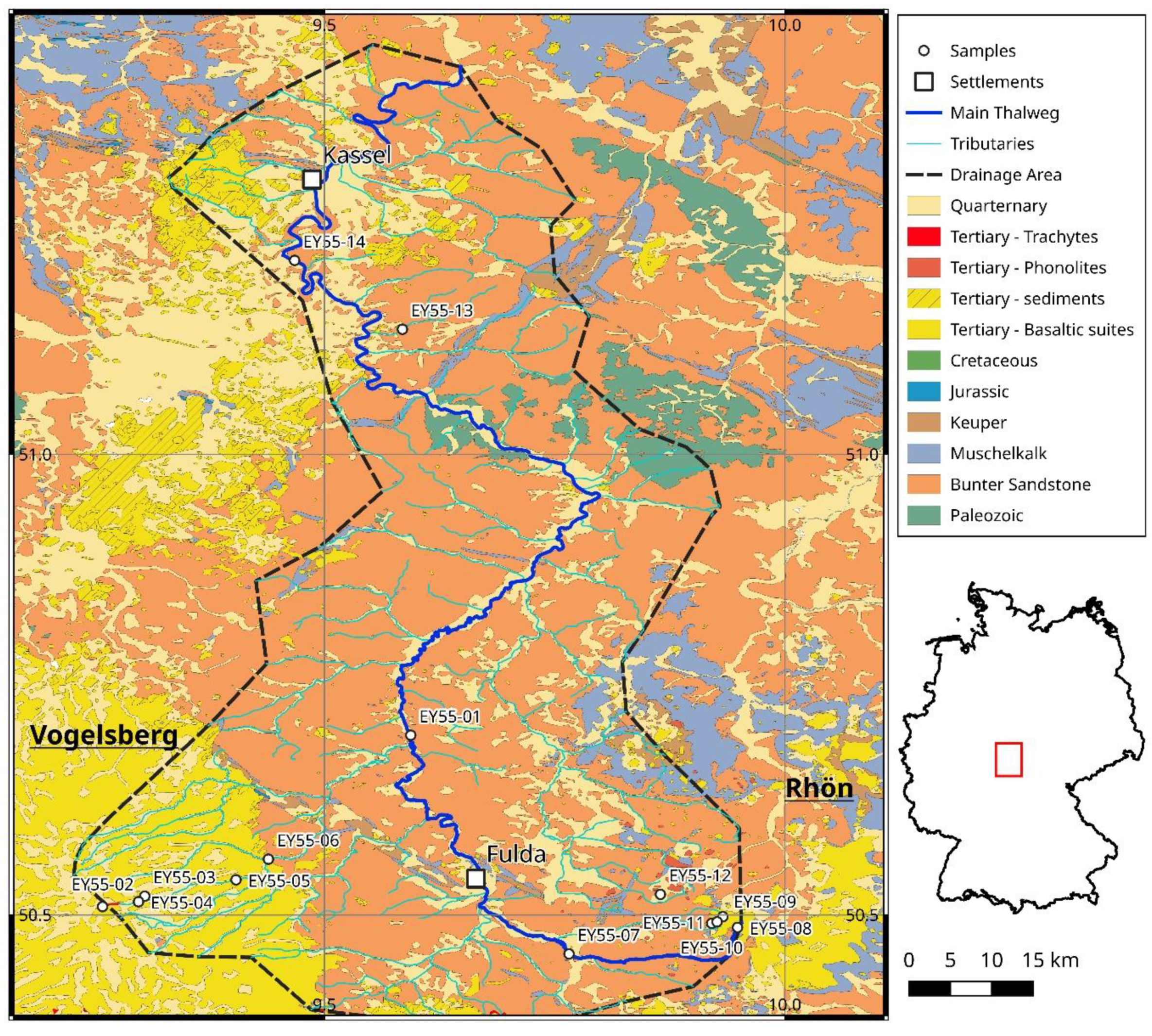
3.1. Samples
3.2. Results
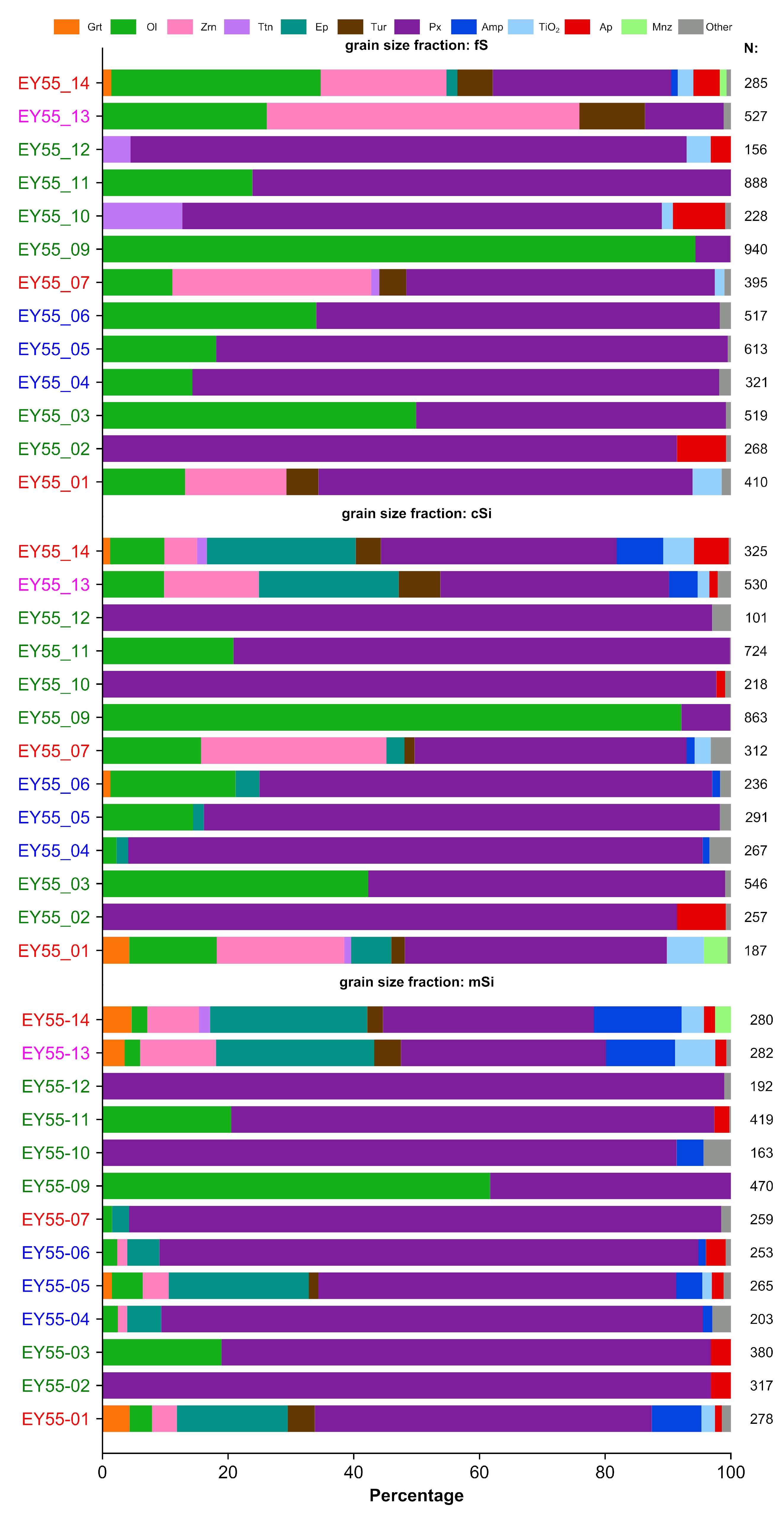
3.2.1. Heavy-Mineral Assemblages
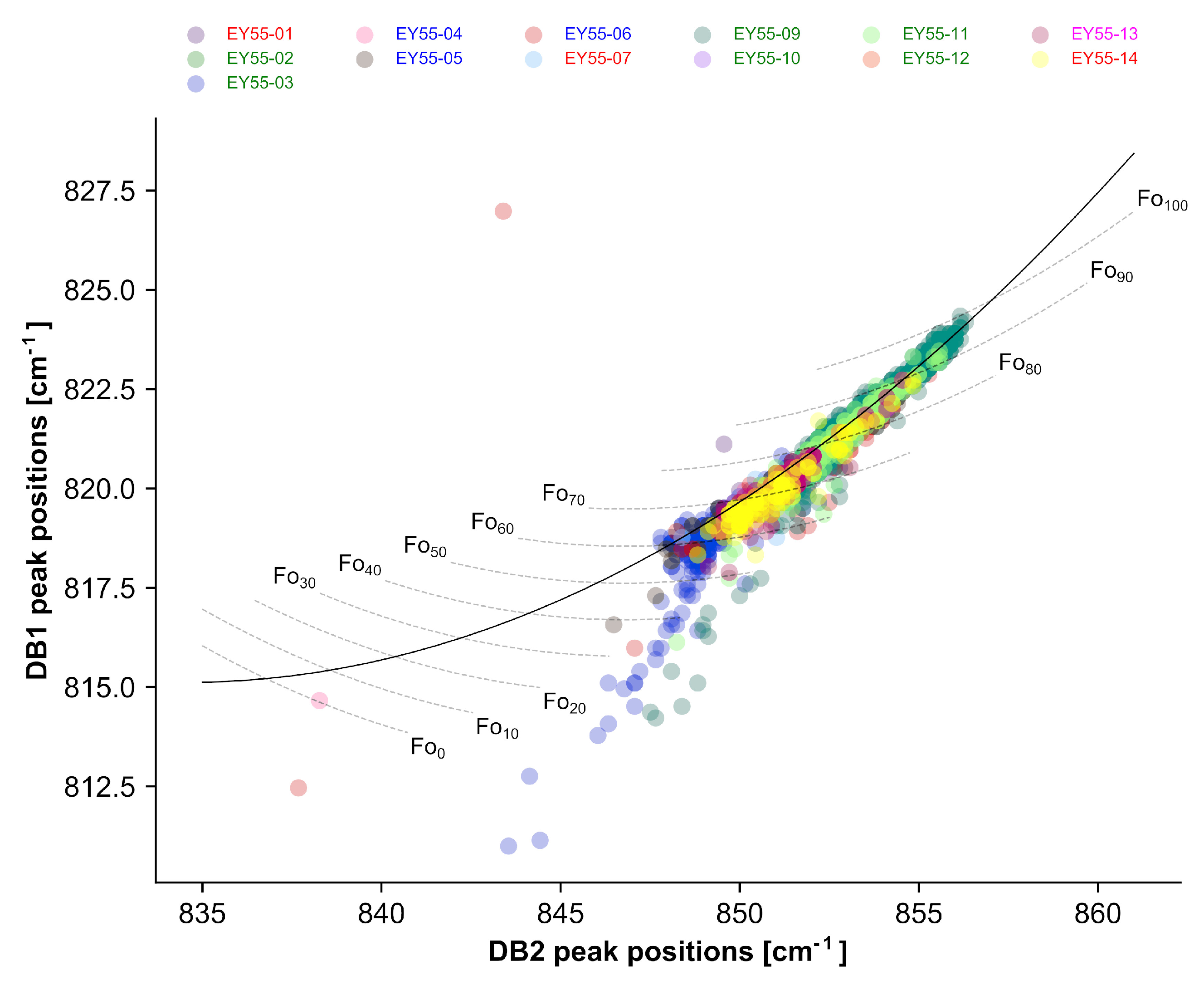
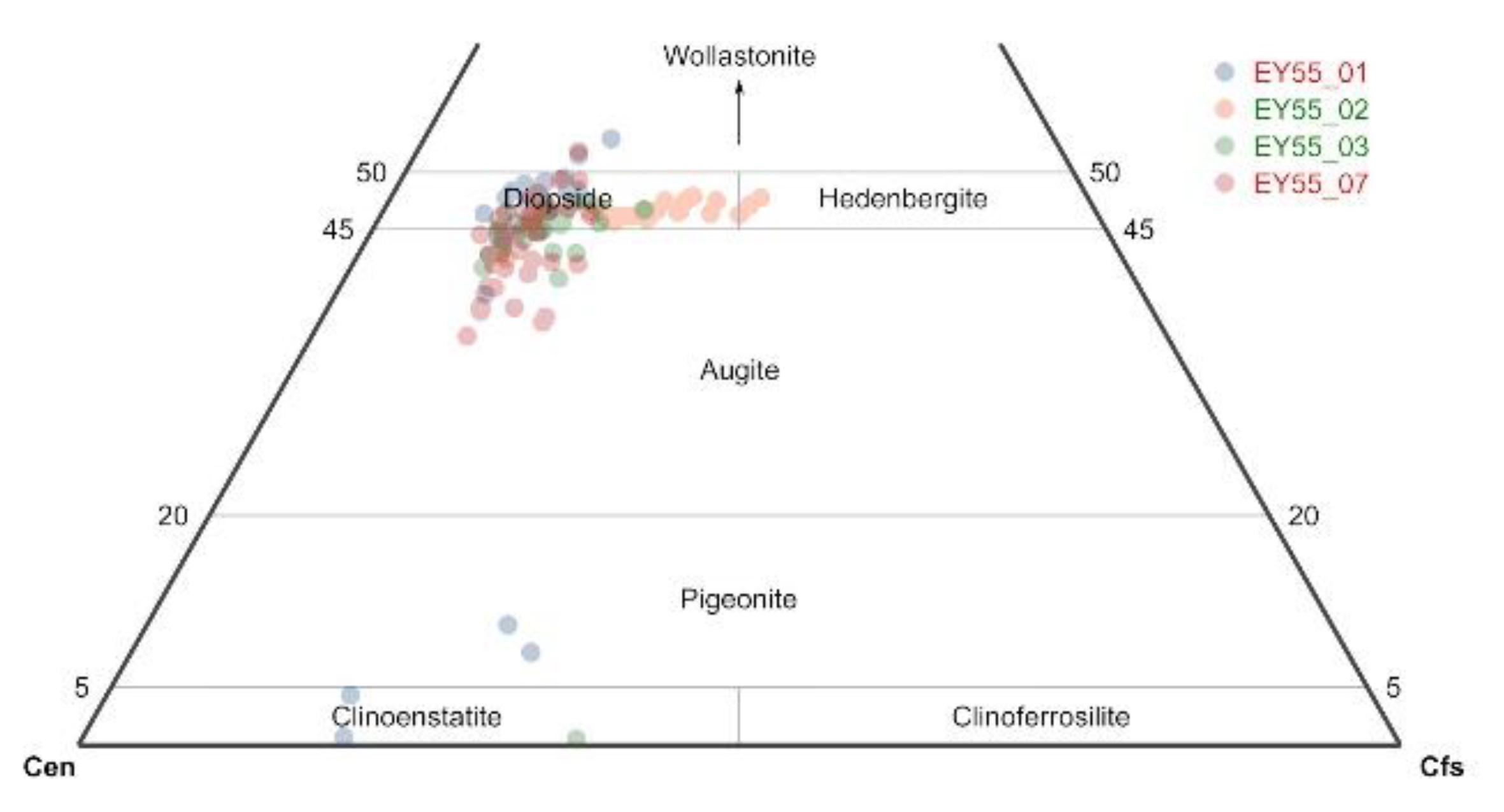
3.2.2. Semi-Quantitative Mineral Chemistry by Raman Spectroscopy: Olivine
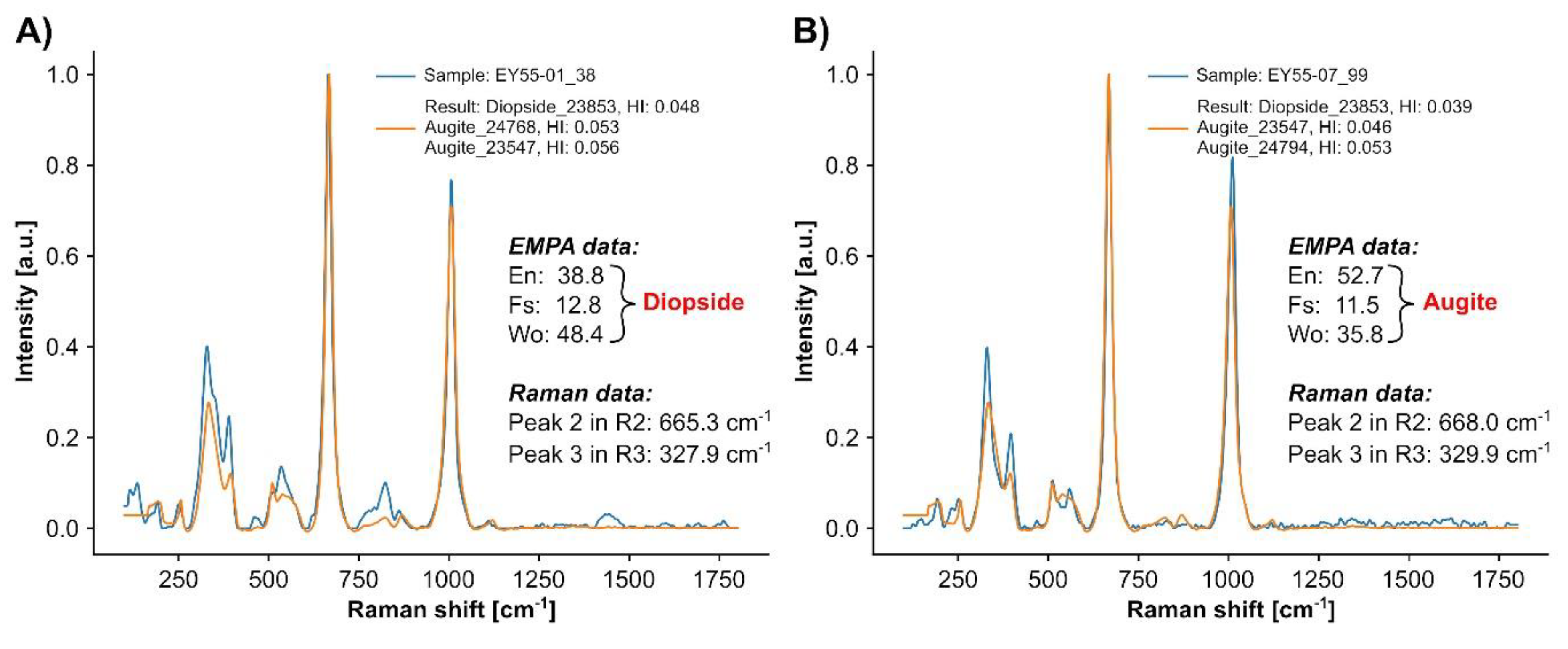
3.2.3. Semi-Quantitative Mineral Chemistry by Raman Spectroscopy: Pyroxene
4. Discussion
4.1. HM Assemblage
4.2. Olivine Composition
4.3. Pyroxene Composition
4.4. Remarks on the Methodology and Outlook
4.5. Potential Disadvantages
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weltje, G.J.; von Eynatten, H. Quantitative provenance analysis of sediments: Review and outlook. Sediment. Geol. 2004, 171, 1–11. [Google Scholar] [CrossRef]
- Morton, A.C.; Hallsworth, C.R. Processes controlling the composition of heavy mineral assemblages in sandstones. Sediment. Geol. 1999, 124, 3–29. [Google Scholar] [CrossRef]
- Garzanti, E.; Andò, S.; Vezzoli, G. Grain-size dependence of sediment composition and environmental bias in provenance studies. Earth Planet. Sci. Lett. 2009, 277, 422–432. [Google Scholar] [CrossRef]
- Garzanti, E.; Andò, S.; Limonta, M.; Fielding, L.; Najman, Y. Diagenetic control on mineralogical suites in sand, silt, and mud (Cenozoic Nile Delta): Implications for provenance reconstructions. Earth Science Rev. 2018, 185, 122–139. [Google Scholar] [CrossRef]
- von Eynatten, H.; Tolosana-Delgado, R.; Karius, V.; Bachmann, K.; Caracciolo, L. Sediment generation in humid Mediterranean setting: Grain-size and source-rock control on sediment geochemistry and mineralogy (Sila Massif, Calabria). Sediment. Geol. 2016, 336, 68–80. [Google Scholar] [CrossRef]
- Mange, M.A.; Morton, A.C. Geochemistry of Heavy Minerals. In Heavy Minerals in Use, 1st ed.; Mange, M.A., Wright, D.T., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 345–391. [Google Scholar]
- von Eynatten, H.; Dunkl, I. Assessing the sediment factory: The role of single grain analysis. EarthScience Rev. 2012, 115, 97–120. [Google Scholar] [CrossRef]
- Morton, A.C.; Hallsworth, C. Identifying provenance-specific features of detrital heavy mineral assemblages in sandstones. Sediment. Geol. 1994, 90, 241–256. [Google Scholar] [CrossRef]
- Andò, S.; Garzanti, E. Raman spectroscopy in heavy-mineral studies. Geol. Soc. London Spec. Publ. 2013, 386, 395–412. [Google Scholar] [CrossRef]
- Mange, M.A.; Maurer, H.F.W. Schwerminerale in Farbe; Ferdinand Enke Verlag Stuttgart: Stuttgart, Germany, 1991. [Google Scholar]
- Fries, M.; Steele, A. Raman Spectroscopy and Confocal Raman Imaging in Mineralogy and Petrography. In Confocal Raman Microscopy; Dieing, T., Hollricher, O., Toporski, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; p. 289. [Google Scholar]
- Andò, S.; Vignola, P.; Garzanti, E. Raman counting: A new method to determine provenance of silt. Rend. Lincei 2011, 22, 327–347. [Google Scholar] [CrossRef]
- Korsakov, A.V.; Hutsebaut, D.; Theunissen, K.; Vandenabeele, P.; Stepanov, A.S. Raman mapping of coesite inclusions in garnet from the Kokchetav Massif (Northern Kazakhstan). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 68, 1046–1052. [Google Scholar] [CrossRef] [Green Version]
- Schönig, J.; Meinhold, G.; von Eynatten, H.; Lünsdorf, N.K. Tracing ultrahigh-pressure metamorphism at the catchment scale. Sci. Rep. 2018, 8, 2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönig, J.; von Eynatten, H.; Meinhold, G.; Lünsdorf, N.K. Diamond and coesite inclusions in detrital garnet of the Saxonian Erzgebirge, Germany. Geology 2019. [Google Scholar] [CrossRef]
- Vermeesch, P. How many grains are needed for a provenance study? Earth Planet. Sci. Lett. 2004, 224, 441–451. [Google Scholar] [CrossRef]
- Pullen, A.; Ibáñez-Mejía, M.; Gehrels, G.E.; Ibáñez-Mejía, J.C.; Pecha, M. What happens when n= 1000? Creating large-n geochronological datasets with LA-ICP-MS for geologic investigations. J. Anal. At. Spectrom. 2014, 29, 971–980. [Google Scholar] [CrossRef]
- Smith, D.C. The RAMANITA© method for non-destructive and in situ semi-quantitative chemical analysis of mineral solid-solutions by multidimensional calibration of Raman wavenumber shifts. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 61, 2299–2314. [Google Scholar] [CrossRef] [PubMed]
- Kuebler, K.E.; Jolliff, B.L.; Wang, A.; Haskin, L.A. Extracting olivine (Fo-Fa) compositions from Raman spectral peak positions. Geochim. Cosmochim. Acta 2006, 70, 6201–6222. [Google Scholar] [CrossRef]
- Wang, A.; Jolliff, B.L.; Haskin, L.A.; Kuebler, K.E.; Viskupic, K.M. Characterization and comparison of structural and compositional features of planetary quadrilateral pyroxenes by Raman spectroscopy. Am. Mineral. 2001, 86, 790–806. [Google Scholar] [CrossRef]
- Bersani, D.; Andò, S.; Vignola, P.; Moltifiori, G.; Marino, I.G.; Lottici, P.P.; Diella, V. Micro-Raman spectroscopy as a routine tool for garnet analysis. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2009, 73, 484–491. [Google Scholar] [CrossRef]
- Leissner, L.; Schlüter, J.; Horn, I.; Mihailova, B. Exploring the potential of Raman spectroscopy for crystallochemical analyses of complex hydrous silicates: I. Amphiboles. Am. Mineral. 2015, 100, 2682–2694. [Google Scholar] [CrossRef]
- Lenz, C.; Nasdala, L.; Talla, D.; Hauzenberger, C.; Seitz, R.; Kolitsch, U. Laser-induced REE3+ photoluminescence of selected accessory minerals - An ‘advantageous artefact’ in Raman spectroscopy. Chem. Geol. 2015, 415, 1–16. [Google Scholar] [CrossRef]
- Nasdala, L.; Wenzel, M.; Vavra, G.; Irmer, G.; Wenzel, T.; Kober, B. Metamictisation of natural zircon: Accumulaton versus thermal annealing of radioactivity-induced damage. Contrib. Mineral. Petrol. 2001, 141, 125–144. [Google Scholar] [CrossRef]
- Hanchar, J.M.; Ruschel, K.; Nasdala, L.; Škoda, R.; Finger, F.; Kronz, A.; Többens, D.M.; Möller, A. A Raman spectroscopic study on the structural disorder of monazite–(Ce). Mineral. Petrol. 2012, 105, 41–55. [Google Scholar]
- Heller, B.M.; Lünsdorf, N.K.; Dunkl, I.; Molnár, F.; von Eynatten, H. Estimation of radiation damage in titanites using Raman spectroscopy. Am. Mineral. 2019, 104, 857–868. [Google Scholar] [CrossRef]
- Mezger, K.; Krogstad, E.J. Interpretation of discordant U-Pb zircon ages: An evaluation. J. Metamorph. Geol. 1997, 15, 127–140. [Google Scholar] [CrossRef]
- Zack, T.; Moraes, R.; Kronz, A. Temperature dependence of Zr in rutile: Empirical calibration of a rutile thermometer. Contrib. Mineral. Petrol. 2004, 148, 471–488. [Google Scholar] [CrossRef]
- Triebold, S.; Luvizotto, G.L.; Tolosana-Delgado, R.; Zack, T.; von Eynatten, H. Discrimination of TiO2 polymorphs in sedimentary and metamorphic rocks. Contrib. Mineral. Petrol. 2011, 161, 581–596. [Google Scholar] [CrossRef]
- Fleet, W.F. Petrological Notes on the Old Red Sandstone of the West Midlands. Geol. Mag. 1926, 63, 505–516. [Google Scholar] [CrossRef]
- Moore, D.M.; Reynolds, R.C. X-Ray Diffraction and the Identification and Analysis of Clay Minerals, 2nd ed.; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Tributh, H.; Lagaly, G. Aufbereitung und Identifizierung von Boden- und Lagerstättentonen. 1. GIT-Fachzeitschrift für das Lab. 1986, 30, 524–529. [Google Scholar]
- de Faria, D.L.A.; Lopes, F.N. Heated goethite and natural hematite: Can Raman spectroscopy be used to differentiate them? Vib. Spectrosc. 2007, 45, 117–121. [Google Scholar] [CrossRef]
- Hanesch, M. Raman spectroscopy of iron oxides and (oxy)hydroxides at low laser power and possible applications in environmental magnetic studies. Geophys. J. Int. 2009, 177, 941–948. [Google Scholar] [CrossRef]
- Besl, P.J.; McKay, N.D. A Method for Registration of 3-D shapes. IEEE Trans. Pattern Anal. Mach. Intell. 1992, 14, 239–256. [Google Scholar] [CrossRef]
- Dörfer, T.; Bocklitz, T.; Tarcea, N.; Schmitt, M.; Popp, J. Checking and improving calibration of raman spectra using chemometric approaches. Zeitschrift fur Phys. Chemie 2011, 225, 753–764. [Google Scholar] [CrossRef]
- Popp, J.; Tarcea, N. Raman data analysis. In EMU Notes in Mineralogy, Volume 12—Applications of Raman Spectroscopy to Earth Sciences and Cultural Heritage; Dubessy, J., Caumon, M.-C., Rull, F., Eds.; European Mineralogical Union & Mineralogical Society of Great Britain & Ireland: London, UK, 2012; p. 504. [Google Scholar]
- McCreery, R. McCreery Group—National Institute for Nanotechnology, University of Alberta, 2015. Available online: https://www.chem.ualberta.ca/~mccreery/ramanmaterials.html (accessed on 24 May 2019).
- Ryan, C.G.; Clayton, E.; Griffin, W.L.; Sie, S.H.; Cousens, D.R. SNIP, a statistics-sensitive background treatment for the quantitative analysis of PIXE spectra in geoscience applications. Nucl. Inst. Methods Phys. Res. B 1988, 34, 396–402. [Google Scholar] [CrossRef]
- Morháč, M.; Matoušek, V. Peak clipping algorithms for background estimation in spectroscopic data. Appl. Spectrosc. 2008, 62, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.J. On the Use of Windows for Harmonic Analysis with the Discrete Fourier Transform. Proc. IEEE 1978, 66, 51–83. [Google Scholar] [CrossRef]
- McCreery, R.L. Raman Spectroscopy for Chemical Analysis. In Volume 157 in Chemical Analysis—A Series of Monographs on Analytical Chemistry and Its Applications; Winefordner, J.D., Ed.; Wiley-Interscience: Hoboken, NJ, USA, 2000. [Google Scholar] [Green Version]
- Lafuente, B.; Downs, T.R.; Yang, H.; Stone, N. The power of databases: the RRUFF project. In Highlights in Mineralogical Crystallography; Armbruster, T., Danisi, R.M., Eds.; De Greuyter: Berlin, Germany, 2015; pp. 1–30. [Google Scholar]
- Rodriguez, J.D.; Westenberger, B.J.; Buhse, L.F.; Kauffman, J.F. Quantitative evaluation of the sensitivity of library-based Raman spectral correlation methods. Anal. Chem. 2011, 83, 4061–4067. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Osadchy, M.; Ashton, L.; Foster, M.; Solomon, C.J.; Gibson, S.J. Deep convolutional neural networks for Raman spectrum recognition: A unified solution. Analyst 2017, 142, 4067–4074. [Google Scholar] [CrossRef]
- Nasdala, L.; Smith, D.C.; Kaindl, R.; Ziemann, M.A. Raman spectroscopy: Analytical perspectives in mineralogical research. EMU Notes Mineral. 2004, 6, 281–343. [Google Scholar]
- Lünsdorf, N.K.; Lünsdorf, J.O. Evaluating Raman spectra of carbonaceous matter by automated, iterative curve-fitting. Int. J. Coal Geol. 2016, 160–161, 51–62. [Google Scholar] [CrossRef]
- Sindowski, K.-H. Schüttungsrichtungen und Mineral-Provinzen im westdeutschen Buntsandstein. Geol. Jahrb. 1957, 73, 277–294. [Google Scholar]
- Okrajek, A. Sedimentpetrographische Untersuchung toniger und sandiger Lagen des Mittleren Buntsandsteins in Bohrungen und Tagesaufschlüssen Süd-Niedersachsens. Beiträge zur Mineral. und Petrogr. 1965, 11, 507–534. [Google Scholar]
- Heim, D. Petrographische Beitrage zur Paläogeographie des Buntsandsteins. Notizblatt des Hess. Landesamtes für Bodenforsch. zu Wiesbad. 1966, 94, 235–258. [Google Scholar]
- Brüning, U. Stratigraphie und Lithofazies des Unteren Buntsandsteins in Südniedersachsen und Nordhessen. Geol. Jahrb. 1986, 90, 125. [Google Scholar]
- Pryor, W.A. Petrology of the Weissliegendes sandstones in the Harz and Werra-Fulda areas, Germany. Geol. Rundsch. 1971, 60, 524–552. [Google Scholar] [CrossRef]
- Scheffer, F.; Meyer, B.; Kalk, E. Mineraluntersuchungen am Würm-Löß südniedersächsischer Lößfluren als Voraussetzung für die Mineralanalyse verschiedener Lößbodentypen. Chem. Erde. 1958, 19, 338–360. [Google Scholar]
- Bogaard, P.J.F.; Wörner, G. Petrogenesis of Basanitic to Tholeiitic Volcanic Rocks from the Miocene Vogelsberg, Central Germany. J. Petrol. 2003, 44, 569–602. [Google Scholar] [CrossRef] [Green Version]
- Reischmann, T.; Schraft, A. Der Vogelsberg - Geotope im größten Vulkangebiet Mitteleuropas; Hessisches Landesamt für Umwelt und Geologie: Wiesbaden, Germany, 2009. [Google Scholar]
- Ficke, B. Petrologische Untersuchungen an tertiären basaltischen bis phonolitschen Vulkaniten der Rhön. Mineral. Petrol. 1961, 7, 337–436. [Google Scholar] [CrossRef]
- Chopelas, A. Single crystal Raman spectra of forsterite, fayalite, and monticellite. Am. Mineral. 1991, 76, 1101–1109. [Google Scholar]
- Huang, E.; Chen, C.H.; Huang, T.; Lin, E.H.; Xu, J.A. Raman spectroscopic characteristics of Mg-Fe-Ca pyroxenes. Am. Mineral. 2000, 85, 473–479. [Google Scholar] [CrossRef]
- Tribaudino, M.; Montovani, L.; Bersani, D.; Lottici, P. Raman spectroscopy of (Ca,Mg)MgSi2O6 clinopyroxenes. Am. Mineral. 2012, 97, 1339–1347. [Google Scholar] [CrossRef]
- Mursky, G.A.; Thompson, R.M. A specific gravity index for minerals. Can. Mineral. 1958, 6, 273–287. [Google Scholar]
- Poldervaart, A. Correlation of physical properties and chemical composition in the plagioclase, olivine and orthopyroxene series. Am. Mineral. 1950, 35, 1067–1079. [Google Scholar]
- Komar, P.D.; Reimers, C.E. Grain shape effects on settling rates. J. Geol. 1978, 86, 193–209. [Google Scholar] [CrossRef]
- Le Roux, J.P. Settling velocity of ellipsoidal grains as related to shape entropy. Sediment. Geol. 1996, 101, 15–20. [Google Scholar] [CrossRef]
- Garzanti, E.; Andò, S.; Vezzoli, G. Settling equivalence of detrital minerals and grain-size dependence of sediment composition. Earth Planet. Sci. Lett. 2008, 273, 138–151. [Google Scholar] [CrossRef]
- Cornwall, C.; Bandfield, J.L.; Titus, T.N.; Schreiber, B.C.; Montgomery, D.R. Physical abrasion of mafic minerals and basalt grains: Application to martian aeolian deposits. Icarus 2015, 256, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.L.; Milnes, A.R.; Eggleton, R.A. Weathering of Basalt: Formation of Iddingsite. Clays Clay Miner. 1987, 35, 418–428. [Google Scholar] [CrossRef]
- Gialanella, S.; Girardi, F.; Ischia, G.; Lonardelli, I.; Mattarelli, M.; Montagna, M. On the goethite to hematite phase transformation. J. Therm. Anal. Calorim. 2010, 102, 867–873. [Google Scholar] [CrossRef]
- Pine, A.S.; Tannenwald, P.E. Temperature dependence of Raman linewidth and shift in α-quartz. Phys. Rev. 1969, 178, 1424–1430. [Google Scholar] [CrossRef]
- Franz, L.; Seifert, W.; Kramer, W. Thermal evolution of the mantle underneath the Mid-German Crystalline Rise: evidence from mantle xenoliths from the Rhone area (Central Germany). Mineral. Petrol. 1997, 61, 1–25. [Google Scholar] [CrossRef]
- Brantley, S.L.; Chen, Y. Chemical weathering rates of pyroxenes and amphiboles. In Chemical Weathering Rates of Silicate Minerals; White, A.F., Brantley, S.L., Eds.; Mineralogical Society of America: Washington, DC, USA, 1995; p. 583. [Google Scholar]
- Dubessy, J.; Caumon, M.-C.; Rull, F.; Sharma, S. Instrumentation in Raman spectroscopy: elementary theory and practice. In EMU Notes in Mineralogy—Volume 12 Applications of Raman spectroscopy to Earth sciences and cultural heritage; Dubessy, J., Caumon, M.-C., Rull, F., Eds.; European Mineralogical Union & Mineralogical Society of Great Britain & Ireland: London, UK, 2012; pp. 83–172. [Google Scholar]
- Cover, T.M.; Hart, P.E. Nearest Neighbor Pattern Classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Mason, L.; Bartlett, P.; Baxter, J.; Frean, M. Boosting Algorithms as Gradient Descent. In Advances in Neural Information Processing Systems 12; MIT Press: Cambridge, MA, USA, 2000; p. 1098. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 297, 273–297. [Google Scholar] [CrossRef]
- Drucker, H.; Burges, C.J.C.; Kaufman, L.; Smola, A.; Vapnik, V.; Long, W.; Nj, B. Support Vector Regression Machines. In Advances in Neural Information Processing Systems 9; MIT Press: Cambridge, MA, USA, 1996; p. 1118. [Google Scholar]
- Totten, M.W.; Hanan, M.A. Heavy minerals in shale. In Heavy Minerals in Use; Mange, M.A., Wright, D.T., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; p. 1283. [Google Scholar]
- Andò, S.; Garzanti, E.; Padoan, M.; Limonta, M. Corrosion of heavy minerals during weathering and diagenesis: A catalog for optical analysis. Sediment. Geol. 2012, 280, 165–178. [Google Scholar] [CrossRef]
- Kohn, M.J. “Thermoba-Raman-try”: Calibration of spectroscopic barometers and thermometers for mineral inclusions. Earth Planet. Sci. Lett. 2014, 388, 187–196. [Google Scholar] [CrossRef]
- Zoubir, A. Raman Imaging; Springer: Berlin/Heidelberg, Germany, 2012; Volume 168. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | gsf: | fS (63–125 µm) | cSi (30–63 µm) | mSi (10–30 µm) |
|---|---|---|---|---|
| Laser wavelength (nm) | transparent | 532 | ||
| opaque | ||||
| Laser power (% of 100 mW) | transparent | 25 | 25 | 10 |
| opaque | 1 | 1 | 1 | |
| Laser polarization | transparent | circular (lambda/4 retarder plate) | ||
| opaque | ||||
| Spectral grating (gr/mm) | transparent | 1800 | ||
| opaque | ||||
| Spectrometer position (cm−1) | transparent | 1000 | ||
| opaque | ||||
| Spectral range (cm−1) | transparent | 110–780 | ||
| opaque | ||||
| Slit size (µm) | transparent | 100 | ||
| opaque | ||||
| Hole size (µm) | transparent | 100 | ||
| opaque | ||||
| Objective | transparent | 50x, 0.5 NA, LWD | 100x, 0.8 NA, LWD | |
| opaque | ||||
| Maximum exposure time (s) | transparent | 30 | 120 | |
| opaque | 300 | |||
| Number of accumulations | transparent | 1 | ||
| opaque | 2 | |||
| Maximum intensity cts | transparent | 5000 | ||
| opaque | 10,000 | |||
| tTL (gray value 0–255) | 10 | |||
| tRL (gray value 0–255) | 130 | |||
| Sample Code | N | E | Type | Note |
|---|---|---|---|---|
| EY55-01 | 50.69526 | 9.59377 | recent sediment | Fulda river, north of VVC |
| EY55-02 | 50.50903 | 9.25902 | Trachyte | Hard rock with weathering crust, cm-sized Fsp phenocrysts |
| EY55-03 | 50.52040 | 9.30481 | Basalt | Hard rock with weathering crust, mm-sized olivine phenocrysts |
| EY55-04 | 50.51437 | 9.29809 | recent sediment | Fulda tributary on VVC |
| EY55-05 | 50.53819 | 9.40401 | recent sediment | Fulda tributary on VVC |
| EY55-06 | 50.56057 | 9.43914 | recent sediment | Fulda tributary on VVC |
| EY55-07 | 50.45762 | 9.76565 | recent sediment | Fulda river, south of VVC |
| EY55-08 | 50.48646 | 9.94867 | recent sediment | near Fulda river spring, limestone gravel, not used for analysis |
| EY55-09 | 50.49809 | 9.93273 | Basalt | Hard rock, peridotite xenoliths |
| EY55-10 | 50.49091 | 9.92062 | Trachyte | Hard rock with weathering crust, fine grained |
| EY55-11 | 50.49251 | 9.92645 | Basalt | Hard rock with weathering crust, fine grained, few phenocrysts |
| EY55-12 | 50.52239 | 9.86492 | Phonolite | Hard rock, fresh |
| EY55-13 | 51.13599 | 9.58469 | recent sediment | Fulda tributary draining only Bunter sandstone formations |
| EY55-14 | 51.21082 | 9.46757 | recent sediment | Fulda river, most distal sample |
| Grain Size Fraction | Sample | trns | trns n.n. | opq | opq n.n. | TS | NS | GH | MH | NH | HR | Grt | Ol | Zrn | Ttn | Ep | Tur | Px | Amp | TiO2 | Ap | Mnz | Other | n | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| fine sand | EY55-01 | 849 | 722 | 260 | 218 | 1109 | 169 | 478 | 135 | 110 | 66 | 0.00 | 13.17 | 16.10 | 0.00 | 0.00 | 5.12 | 59.51 | 0.00 | 4.63 | 0.00 | 0.00 | 1.46 | 410 | Ttn, Amp, Spl, Brt, Ap |
| EY55-02 | 459 | 411 | 581 | 572 | 1040 | 57 | 319 | 34 | 58 | 78 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 91.42 | 0.00 | 0.00 | 7.84 | 0.00 | 0.75 | 268 | Ol, TiO2, | |
| EY55-03 | 868 | 725 | 109 | 97 | 977 | 155 | 531 | 68 | 127 | 73 | 0.00 | 49.90 | 0.00 | 0.00 | 0.00 | 0.00 | 49.33 | 0.00 | 0.00 | 0.00 | 0.00 | 0.77 | 519 | Ap | |
| EY55-04 | 845 | 601 | 350 | 314 | 1195 | 280 | 338 | 158 | 105 | 56 | 0.00 | 14.33 | 0.00 | 0.00 | 0.00 | 0.00 | 83.80 | 0.00 | 0.00 | 0.00 | 0.00 | 1.87 | 321 | Zrn, Ttn, Ep, Ap, Mnz | |
| EY55-05 | 1013 | 776 | 162 | 147 | 1175 | 252 | 625 | 66 | 85 | 81 | 0.00 | 18.11 | 0.00 | 0.00 | 0.00 | 0.00 | 81.40 | 0.00 | 0.00 | 0.00 | 0.00 | 0.49 | 613 | Ttn, TiO2, Spl | |
| EY55-06 | 829 | 675 | 371 | 329 | 1200 | 196 | 529 | 77 | 70 | 78 | 0.00 | 34.04 | 0.00 | 0.00 | 0.00 | 0.00 | 64.22 | 0.00 | 0.00 | 0.00 | 0.00 | 1.74 | 517 | Zrn, Ep, Spl, Ap | |
| EY55-07 | 562 | 516 | 441 | 417 | 1003 | 70 | 415 | 56 | 45 | 80 | 0.00 | 11.14 | 31.65 | 1.27 | 0.00 | 4.30 | 49.11 | 0.00 | 1.52 | 0.00 | 0.00 | 1.01 | 395 | Ep, Amp, Spl, Mnz | |
| EY55-09 | 1012 | 985 | 4 | 3 | 1016 | 28 | 941 | 16 | 28 | 96 | 0.00 | 94.36 | 0.00 | 0.00 | 0.00 | 0.00 | 5.53 | 0.00 | 0.00 | 0.00 | 0.00 | 0.11 | 940 | Ap | |
| EY55-10 | 617 | 566 | 398 | 361 | 1015 | 88 | 232 | 102 | 232 | 41 | 0.00 | 0.00 | 0.00 | 12.72 | 0.00 | 0.00 | 76.32 | 0.00 | 1.75 | 8.33 | 0.00 | 0.88 | 228 | Ol, Zrn, Ep, Amp | |
| EY55-11 | 1028 | 991 | 74 | 74 | 1102 | 37 | 888 | 93 | 10 | 90 | 0.00 | 23.87 | 0.00 | 0.00 | 0.00 | 0.00 | 76.13 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 888 | Titano-Mag | |
| EY55-12 | 766 | 666 | 262 | 255 | 1028 | 107 | 203 | 245 | 218 | 30 | 0.00 | 0.00 | 0.00 | 4.49 | 0.00 | 0.00 | 88.46 | 0.00 | 3.85 | 3.21 | 0.00 | 0.00 | 156 | Mag, Feldspathoides | |
| EY55-13 | 622 | 600 | 378 | 378 | 1000 | 22 | 552 | 37 | 11 | 92 | 0.00 | 26.19 | 49.72 | 0.00 | 0.00 | 10.44 | 12.52 | 0.00 | 0.00 | 0.00 | 0.00 | 1.14 | 527 | Gr, Amp, Ttn, Ant | |
| EY55-14 | 728 | 706 | 272 | 258 | 1000 | 36 | 668 | 18 | 20 | 95 | 1.40 | 33.33 | 20.00 | 0.00 | 1.75 | 5.61 | 28.42 | 1.05 | 2.46 | 4.21 | 1.05 | 0.70 | 285 | Ttn, Opx, Cld, Mica, Brt | |
| coarse silt | EY55-01 | 652 | 544 | 378 | 315 | 1030 | 171 | 350 | 112 | 82 | 64 | 4.28 | 13.90 | 20.32 | 1.07 | 6.42 | 2.14 | 41.71 | 0.00 | 5.88 | 0.00 | 3.74 | 0.53 | 187 | Sil, St, Ap |
| EY55-02 | 397 | 348 | 722 | 722 | 1119 | 49 | 290 | 33 | 25 | 83 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 91.44 | 0.00 | 0.00 | 7.78 | 0.00 | 0.78 | 257 | Ol | |
| EY55-03 | 767 | 671 | 293 | 278 | 1060 | 111 | 561 | 33 | 77 | 84 | 0.00 | 42.31 | 0.00 | 0.00 | 0.00 | 0.00 | 56.78 | 0.00 | 0.00 | 0.00 | 0.00 | 0.92 | 546 | Ap | |
| EY55-04 | 745 | 535 | 256 | 236 | 1001 | 230 | 281 | 140 | 114 | 53 | 0.00 | 2.25 | 0.00 | 0.00 | 1.87 | 0.00 | 91.39 | 1.12 | 0.00 | 0.00 | 0.00 | 3.37 | 267 | Grt, Zrn, Ttn, TiO2, Ap | |
| EY55-05 | 745 | 507 | 290 | 274 | 1035 | 254 | 305 | 94 | 108 | 60 | 0.00 | 14.43 | 0.00 | 0.00 | 1.72 | 0.00 | 82.13 | 0.00 | 0.00 | 0.00 | 0.00 | 1.72 | 291 | Zrn, Amp, Ap | |
| EY55-06 | 655 | 445 | 354 | 315 | 1000 | 249 | 250 | 81 | 114 | 56 | 1.27 | 19.92 | 0.00 | 0.00 | 3.81 | 0.00 | 72.03 | 1.27 | 0.00 | 0.00 | 0.00 | 1.69 | 236 | Ttn, Tur, TiO2, Ap | |
| EY55-07 | 551 | 467 | 449 | 391 | 1000 | 142 | 321 | 80 | 66 | 69 | 0.00 | 15.71 | 29.49 | 0.00 | 2.88 | 1.60 | 43.27 | 1.28 | 2.56 | 0.00 | 0.00 | 3.21 | 312 | Grt, Ttn, Cld, Ap, Mnz | |
| EY55-09 | 954 | 933 | 47 | 46 | 1002 | 22 | 866 | 31 | 36 | 93 | 0.00 | 92.12 | 0.00 | 0.00 | 0.00 | 0.00 | 7.76 | 0.00 | 0.00 | 0.00 | 0.00 | 0.12 | 863 | Chr | |
| EY55-10 | 908 | 603 | 92 | 90 | 1000 | 307 | 284 | 136 | 183 | 47 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 97.71 | 0.00 | 0.00 | 1.38 | 0.00 | 0.92 | 218 | Ttn, Titano-Mag | |
| EY55-11 | 934 | 910 | 82 | 81 | 1016 | 25 | 729 | 137 | 44 | 80 | 0.00 | 20.86 | 0.00 | 0.00 | 0.00 | 0.00 | 79.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.14 | 724 | Ap | |
| EY55-12 | 931 | 714 | 81 | 81 | 1012 | 216 | 232 | 254 | 228 | 32 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 97.03 | 0.00 | 0.00 | 0.00 | 0.00 | 2.97 | 101 | Ttn, Ap, Mnz, Zeo | |
| EY55-13 | 667 | 617 | 333 | 333 | 1000 | 50 | 554 | 41 | 22 | 90 | 0.00 | 9.81 | 15.09 | 0 | 22.26 | 6.60 | 36.42 | 4.53 | 1.89 | 1.32 | 0.00 | 2.08 | 530 | Grt, Ttn, Sil, St, Mnz | |
| EY55-14 | 778 | 716 | 222 | 218 | 1000 | 66 | 648 | 44 | 24 | 91 | 1.23 | 8.62 | 5.23 | 1.54 | 23.69 | 4.00 | 37.54 | 7.38 | 4.92 | 5.54 | 0.00 | 0.31 | 325 | St, Cld, Chm, Sd, Mnz, | |
| medium silt | EY55-01 | 492 | 441 | 208 | 168 | 700 | 91 | 290 | 113 | 38 | 66 | 4.32 | 3.60 | 3.96 | 0.00 | 17.63 | 4.32 | 53.60 | 7.91 | 2.16 | 1.08 | 0.00 | 1.44 | 278 | Ttn, And, St, Spl, Mnz |
| EY55-02 | 391 | 386 | 265 | 265 | 656 | 5 | 323 | 42 | 20 | 84 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 96.85 | 0.00 | 0.00 | 3.15 | 0.00 | 0.00 | 317 | ||
| EY55-03 | 545 | 503 | 161 | 161 | 706 | 42 | 387 | 53 | 63 | 77 | 0.00 | 18.95 | 0.00 | 0.00 | 0.00 | 0.00 | 77.89 | 0.00 | 0.00 | 3.16 | 0.00 | 0.00 | 380 | ||
| EY55-04 | 486 | 378 | 214 | 214 | 700 | 108 | 214 | 97 | 67 | 57 | 0.00 | 2.46 | 1.48 | 0.00 | 5.42 | 0.00 | 86.21 | 1.48 | 0.00 | 0.00 | 0.00 | 2.96 | 203 | Grt, Chm, TiO2, Ap, Mnz | |
| EY55-05 | 457 | 412 | 248 | 248 | 705 | 45 | 289 | 84 | 39 | 70 | 1.51 | 4.91 | 4.15 | 0.00 | 22.26 | 1.51 | 56.98 | 4.15 | 1.51 | 1.89 | 0.00 | 1.13 | 265 | Ttn, Ky, St, Mnz | |
| EY55-06 | 448 | 418 | 164 | 163 | 612 | 31 | 260 | 112 | 46 | 62 | 0.00 | 2.37 | 1.58 | 0.00 | 5.14 | 0.00 | 85.77 | 1.19 | 0.00 | 3.16 | 0.00 | 0.79 | 253 | Tur, TiO2 | |
| EY55-07 | 435 | 397 | 271 | 269 | 706 | 40 | 264 | 81 | 50 | 67 | 0.00 | 1.54 | 0.00 | 0.00 | 2.70 | 0.00 | 94.21 | 0.00 | 0.00 | 0.00 | 0.00 | 1.54 | 259 | Grt, Ttn, Amp, Ap | |
| EY55-09 | 1185 | 1131 | 15 | 15 | 1200 | 54 | 511 | 297 | 323 | 45 | 0.00 | 61.70 | 0.00 | 0.00 | 0.00 | 0.00 | 38.30 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 470 | ||
| EY55-10 | 481 | 394 | 221 | 207 | 702 | 101 | 178 | 107 | 109 | 45 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 93.25 | 3.68 | 0.00 | 0.00 | 0.00 | 3.07 | 163 | Ol, Ep, Rt, Ap, Mnz, Titano-Mag | |
| EY55-11 | 613 | 600 | 105 | 96 | 718 | 22 | 465 | 80 | 55 | 78 | 0.00 | 20.53 | 0.00 | 0.00 | 0.00 | 0.00 | 76.85 | 0.00 | 0.00 | 2.39 | 0.00 | 0.24 | 419 | Ant, Titano-Mag | |
| EY55-12 | 638 | 559 | 84 | 72 | 722 | 91 | 229 | 177 | 133 | 42 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 98.96 | 0.00 | 0.00 | 0.00 | 0.00 | 1.04 | 192 | Ol, Ap, Mag, Lorenzenite | |
| EY55-13 | 488 | 455 | 217 | 216 | 705 | 34 | 366 | 58 | 31 | 80 | 3.55 | 2.48 | 12.06 | 0.00 | 25.18 | 4.26 | 32.62 | 10.99 | 6.38 | 1.77 | 0.00 | 0.71 | 282 | Ttn, And | |
| EY55-14 | 496 | 449 | 207 | 204 | 703 | 50 | 385 | 34 | 30 | 86 | 4.64 | 2.50 | 8.21 | 1.79 | 25.00 | 2.50 | 33.57 | 13.93 | 3.57 | 1.79 | 2.50 | 0.00 | 280 | Sil, And, St, Chm, Sd |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lünsdorf, N.K.; Kalies, J.; Ahlers, P.; Dunkl, I.; von Eynatten, H. Semi-Automated Heavy-Mineral Analysis by Raman Spectroscopy. Minerals 2019, 9, 385. https://doi.org/10.3390/min9070385
Lünsdorf NK, Kalies J, Ahlers P, Dunkl I, von Eynatten H. Semi-Automated Heavy-Mineral Analysis by Raman Spectroscopy. Minerals. 2019; 9(7):385. https://doi.org/10.3390/min9070385
Chicago/Turabian StyleLünsdorf, Nils Keno, Jannick Kalies, Patrick Ahlers, István Dunkl, and Hilmar von Eynatten. 2019. "Semi-Automated Heavy-Mineral Analysis by Raman Spectroscopy" Minerals 9, no. 7: 385. https://doi.org/10.3390/min9070385
APA StyleLünsdorf, N. K., Kalies, J., Ahlers, P., Dunkl, I., & von Eynatten, H. (2019). Semi-Automated Heavy-Mineral Analysis by Raman Spectroscopy. Minerals, 9(7), 385. https://doi.org/10.3390/min9070385