New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study


College of Resources and Civil Engineering, Northeastern University, Shenyang 110819, China
*
Author to whom correspondence should be addressed.
Minerals 2017, 7(12), 236; https://doi.org/10.3390/min7120236
Submission received: 31 October 2017
/
Revised: 19 November 2017
/
Accepted: 24 November 2017
/
Published: 27 November 2017
(This article belongs to the Special Issue Molecular Simulation of Mineral-Solution Interfaces)
Abstract
:A new understanding of the adsorption mechanism of oleate on cassiterite surfaces is presented by density functional theory (DFT) calculations. Various convergence tests were conducted to optimize the parameter settings for the rational simulation of cassiterite bulk unit cell and surface slabs. The calculated surface energies of four low-index cassiterite cleavage planes form an increasing sequence of (110) < (100) < (101) < (001), demonstrating (110) is the most thermodynamically stable surface of cassiterite. The interaction strengths of the oleate ion (OL−), OH−, and H2O on the SnO2 (110) face are in the order of H2O < OH− < OL−, which reveals that the OL− is able to replace the adsorbed H2O and OH− on the mineral surfaces. Mulliken population calculations and electron density difference analysis show that electrons transfer from the Sn atoms on the cassiterite (110) surface to the O atoms offered by carboxyl groups of oleate during the interaction. The populations of newly formed O1–Sn1 and O2–Sn2 bonds are 0.30 and 0.29, respectively, indicating that these two bonds are of a very low covalency. Density of states analysis reveals that the formation of an O1–Sn1 bond mainly results from the 5s and 5p orbitals of the Sn1 atom and the 2p orbital of the O1 atom.
1. Introduction
Cassiterite, SnO2, is the only economically viable mineral from which tin metal can be extracted. More than three-quarters of tin metal is produced from cassiterite. Cassiterite is usually separated by gravity concentration because of its high relative density [1]. However, the recovery of gravity separation can be as low as 60% to 70% when the cassiterite particle size is smaller than 40 μm [2,3]. Against such a background, froth flotation is usually used to rescue the fine and ultrafine cassiterite particles which are lost in gravity tailings [4,5,6,7].
One of the initially and most widely used collectors for cassiterite flotation is oleate [8,9,10,11]. Many works on the investigation of the interaction mechanisms between oleate and mineral surfaces by means of various testing methods have been reported and numerous valuable findings have been presented. Oliveira and Adamian [8] studied the separation of cassiterite and fluorite by flotation experiments using oleic acid as the collector, and found that cassiterite could be selectively separated from fluorite with a low oleate dosage at neutral pH. Xu and Qin [11] investigated the interaction of sodium oleate with cassiterite by zeta potential measurements and infrared spectrum studies, and the results demonstrated that oleate chemically absorbed on cassiterite surfaces by replacing the OH− ions at the mineral-collector interface. Quast [12] examined the adsorption of oleate on the micaceous hematite surfaces using zeta potential measurements, surface titration, and solution chemistry calculations, and found that the oleate ions competed with hydroxyl ions and adsorbed on the surface mainly by chemical bonding. Peck et al. [13] conducted a classic infrared study on the flotation of hematite using oleate as a collector. They noticed that the infrared radiation (IR) spectra of the ferric oleate was different from that of the chemisorbed oleate on hematite surfaces, which indicated that the chemisorption of oleate did not result in the formation of ferric oleate on the mineral surfaces.
Compared to these conventional measurement methods mentioned above, molecular modeling is a promising way to provide a deeper understanding of the interaction mechanism between reagents and mineral surfaces. Used in mineral processing, molecular modeling is able to predict the possible interactions between collectors and mineral surfaces, to calculate the adsorption energies, to confirm the favorable adsorption configurations, and to investigate the bonding mechanisms, which makes it pretty useful in investigating the adsorption of reagents on mineral surfaces. Rath et al. [14] have used density functional theory (DFT) calculations to investigate the adsorption of oleate on hematite, magnetite, and goethite. Rai et al. [15] have studied the interaction of various jadeite and spodumene cleavage planes with sodium oleate using a molecular dynamics (MD) simulation. Zhu et al. [16] have employed first-principles calculations to further investigate the adsorption of a novel collector, α-Bromolauric acid, on a quartz surface. Xu et al. [17] have employed an MD simulation to examine the adsorption of sodium oleate on spodumene anisotropic surface planes. Besides, it has been well established that molecular modeling is an efficient approach to design tailor made flotation reagents [18,19,20].
However, despite its extensive applications in the mineral processing field, little works have been done to study the adsorption of reagents on cassiterite using the molecular modeling method [21]. This work aims at investigating the adsorption and bonding mechanisms of oleate on cassiterite surfaces at an atomic level, and combines the results of micro-flotation tests, contact angle measurements, zeta potential analyses, and solution chemistry calculations [22] to provide new insights into the interaction between oleate and cassiterite. In order to acquire an optimum crystal structure, geometric optimization of cassiterite bulk cells was performed as a function of exchange-correlation functionals selections, the k-point set mesh and cutoff energy, respectively. Then, a series of calculations were done to obtain the most stable and rational surface structure for the following adsorption computations. Finally, the adsorption and bonding mechanisms of oleate on cassiterite were discussed in detail at an atomic level. China has the largest tin ore reserves in the world and most occur in hard-rock deposits, which means that the cassiterite in these ores needs flotation to beneficiate [10,23]. Under these circumstances, molecular modeling would play a great role in designing specific collectors for cassiterite flotation and predicting the possible interactions of reagents on mineral surfaces.
2. Simulation Details
In this work, the CASTEP module in Materials Studio 6.0 was used to carry out the geometry optimization, energy calculations, and properties analysis.
2.1. Cassiterite Crystal Cell Optimization
The dependence of the rationality of cassiterite unit-cell on exchange-correlation functionals selection, the k-point set mesh and cutoff energy, was tested. Afterwards, the calculated crystal lattice parameters were compared with experimental values to determine the validity of the computed cassiterite crystal cell. After the convergence tests, the XRD pattern of the computed cassiterite unit-cell was compared with that of the experimental cassiterite sample to guarantee the rationality of the simulation work. All the convergence criteria during the entire simulation work were set as shown in Table 1. All the optimal parameters confirmed in the bulk cell geometry optimization remained constant in all the subsequent simulation jobs, unless with additional specifications.
2.2. Calculation of the Surface Energy
All the surface structures simulated in this work were obtained by cleaving the optimized cassiterite bulk cell. Afterwards, the surface energy for these surface slabs was calculated by the following formula:
where Eslab is the total energy of the calculated surface structure, and Euc is that of the optimized unit cell. N and n represent the atom numbers in the surface slabs and bulk unit cell, respectively. A is the total surface area of the surface slabs.
Esurf = [Eslab − (N/n) · Euc]/A
As mentioned in the literature [16], surface energy is a function of the number of the atoms in the slab and the vacuum depth. Thus, surface energy convergence tests were performed as a function of the surface slab thickness and the vacuum depth to obtain accurate cassiterite surface slabs.
2.3. Computation of Adsorption Energy
The total adsorption energy is regarded as a measure of affinity of different adsorbates on cassiterite surfaces. Usually, the adsorption energy of a spontaneous chemical reaction is subtractive, which indicates a negative adsorption energy, meaning that the adsorbate can adsorb on the surface favorably. The expression used for the adsorption energy calculation is shown as below:
where Ecomplex, Eadsorbate, and Emineral refer to the total energy of the surface-adsorbate complex, adsorbate, and cassiterite surface slab after optimization, respectively. It is worth noting that the pure calculated adsorption energy cannot represent the real energy change during the reagent–mineral interaction [15], but it can be a good measure of the relative adsorption strength between different adsorbates and mineral surfaces. A more negative adsorption energy implies a stronger binding [24].
ΔEads = Ecomplex − (Eadsorbate + Emineral)
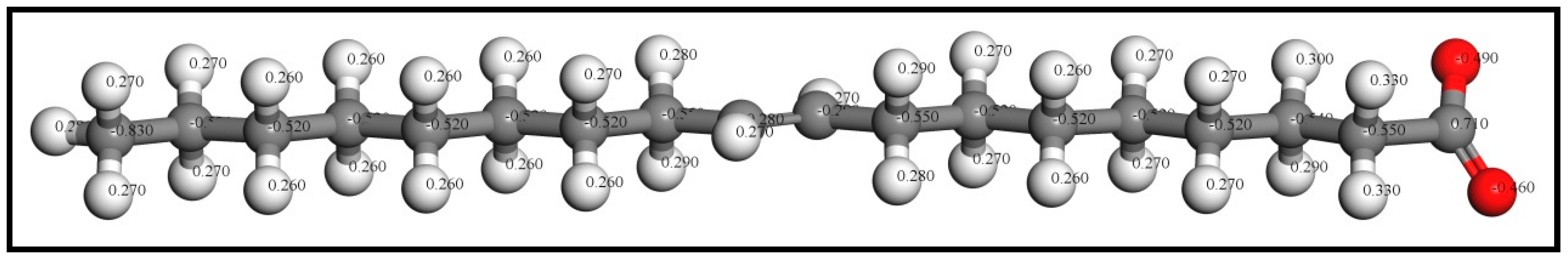
Based on the results of the flotation tests and solution chemistry calculations [22], the oleate ion (OL−) generated by ionization is the effective component which will bind with the cassiterite surfaces. All the adsorbates, namely, OL−, OH−, and H2O, were firstly placed in a 20 Å × 20 Å × 40 Å lattice cell for optimization calculations and the k-point set was changed to a gamma point only. After that, the optimized ions were separately docked on the cassiterite surfaces for the following adsorption calculation. The optimized oleate ion structure with its partial Mulliken charges is presented in Figure 1.
3. Results and Discussion
3.1. Cassiterite Bulk Cell Optimization
The first and most important thing is to build a rational cassiterite bulk cell, which is the first guarantee that all the simulation work is carried out correctly. In this work, convergence tests were conducted to evaluate the dependences of the cassiterite crystal rationality on the exchange-correlation functionals selection, the k-point set mesh and cutoff energy.
Various exchange-correlation functionals have been raised to describe the exchange and correlation potential, but only one can be the best for a certain structure. Generalised gradient approximation (GGA) and local-density approximation (LDA) are the two favorite types at present. However, because of having inherent problems [25,26,27], LDA is not applicable to metal oxides. Thus, in this work, the five most common GGA functionals were used to optimize the cassiterite unit-cell, and the computed lattice constants were contrasted with those observed in X-ray diffraction to determine the suitable functional. Under the condition of cutoff energy 340 eV and k-point set mesh 3 × 3 × 4, the calculation tests were carried out using GGA functionals Wu-Cohen (WC), Perdew-Burke-Ernzerhof (PBE), Perdew-Burke-Ernzerhof (RPBE), Perdew-Wang’s 1991 (PW91) and Perdew-Burke-Ernzerhof solids (PBESOL), respectively. The experimental and computed lattice parameters are listed in Table 2.
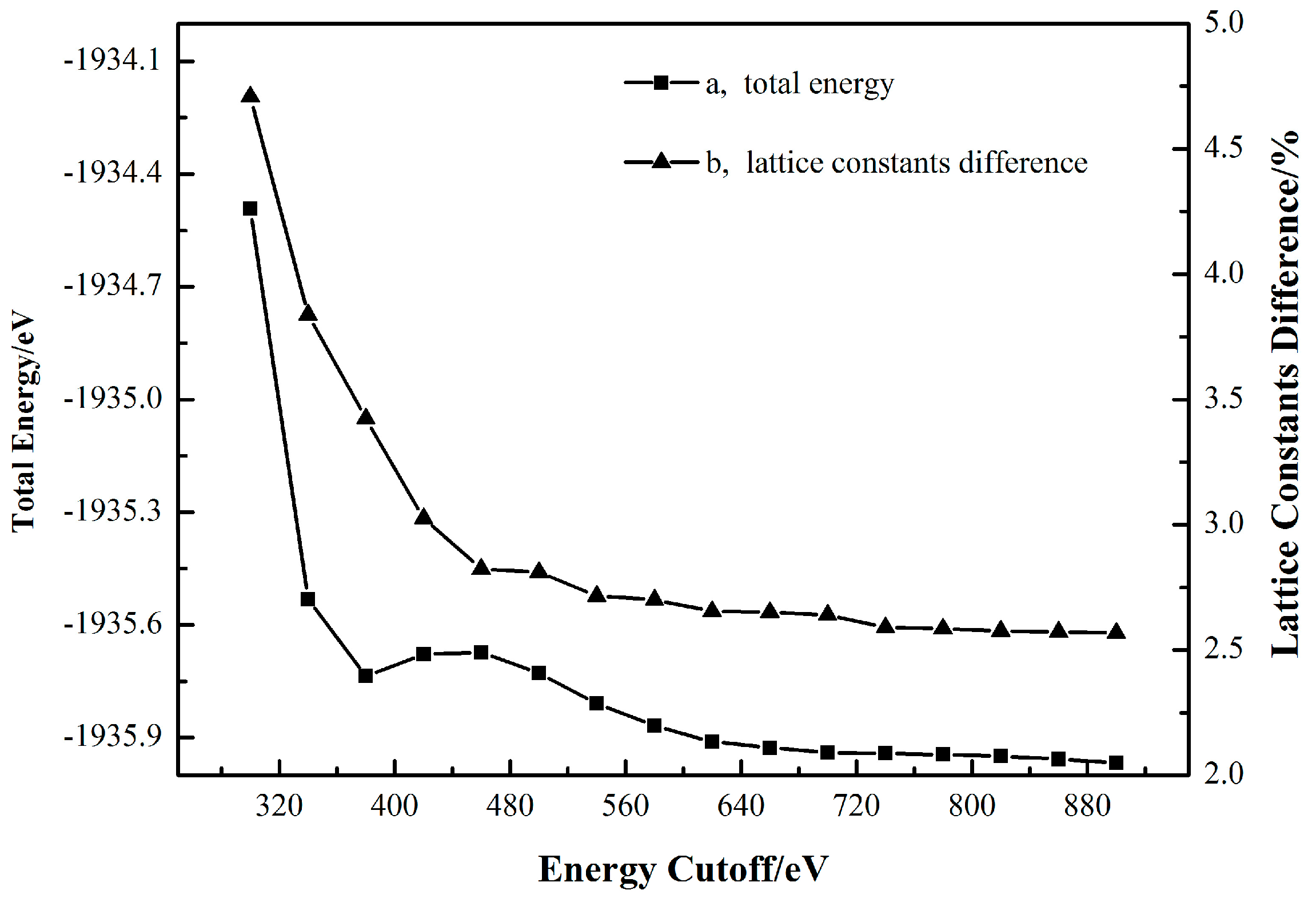
Table 2 shows that the use all these five GGA functionals can generate excellent computational cassiterite bulk cells, and none of the lattice constant differences exceed 5% [14]. Besides, when the functional is set as GGA-WC, a relatively minimal lattice parameter difference of 3.85% can be obtained. Therefore, the most appropriate exchange-correlation functional for the simulation of cassiterite is a generalized gradient approximation developed by Wu-Cohen (WC) [29]. Afterwards, more convergence tests were fulfilled to investigate the dependences of the cassiterite bulk cell total energy and lattice constants difference on the k-points set and cutoff energy values. The calculation results are, respectively, plotted in Figure 2 and Figure 3.
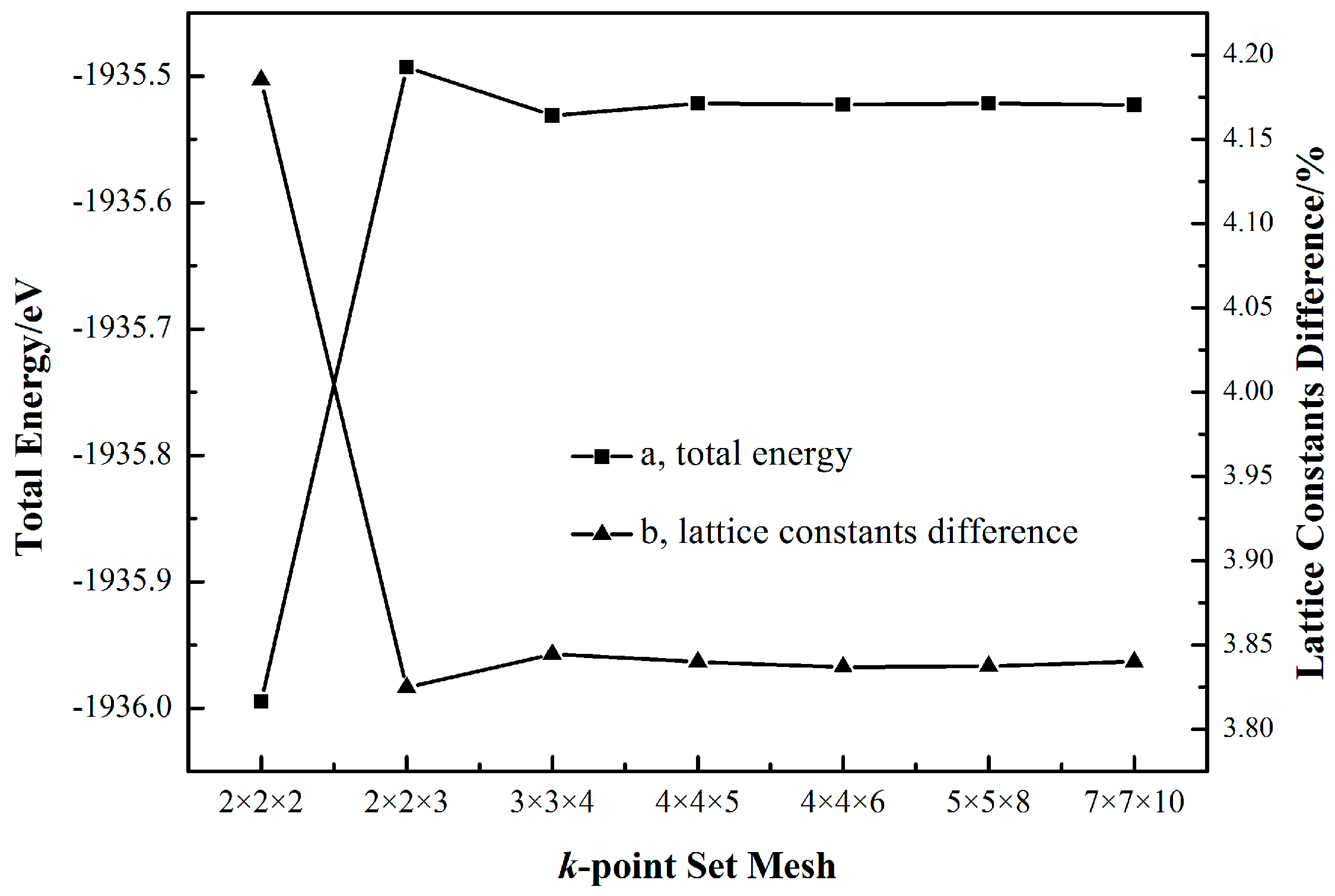
Figure 2 shows that at the cutoff energy of 340 eV, when the k-point set increases from 2 × 2 × 2 to 2 × 2 × 3, both the total energy and lattice constants difference change considerably. Once the k-point set is beyond 3 × 3 × 4, the total energy and lattice constants difference, respectively, converge to a constant value.
Figure 3 reports that when the k-point set is fixed at 3 × 3 × 4, the total energy and lattice constants difference both generally keep going down along with the increase of the cutoff energy. However, once the cutoff energy surpasses 580 eV, the total energy decreases no more than 0.05 eV and the diminution in the cell parameters difference never again exceeds 0.003%. Taking the computational cost into consideration, the results in Figure 2 and Figure 3 indicate that a k-point set of 3 × 3 × 4 and a cutoff energy of 580 eV are high enough to generate a sufficiently accurate cassiterite bulk cell for the following work.
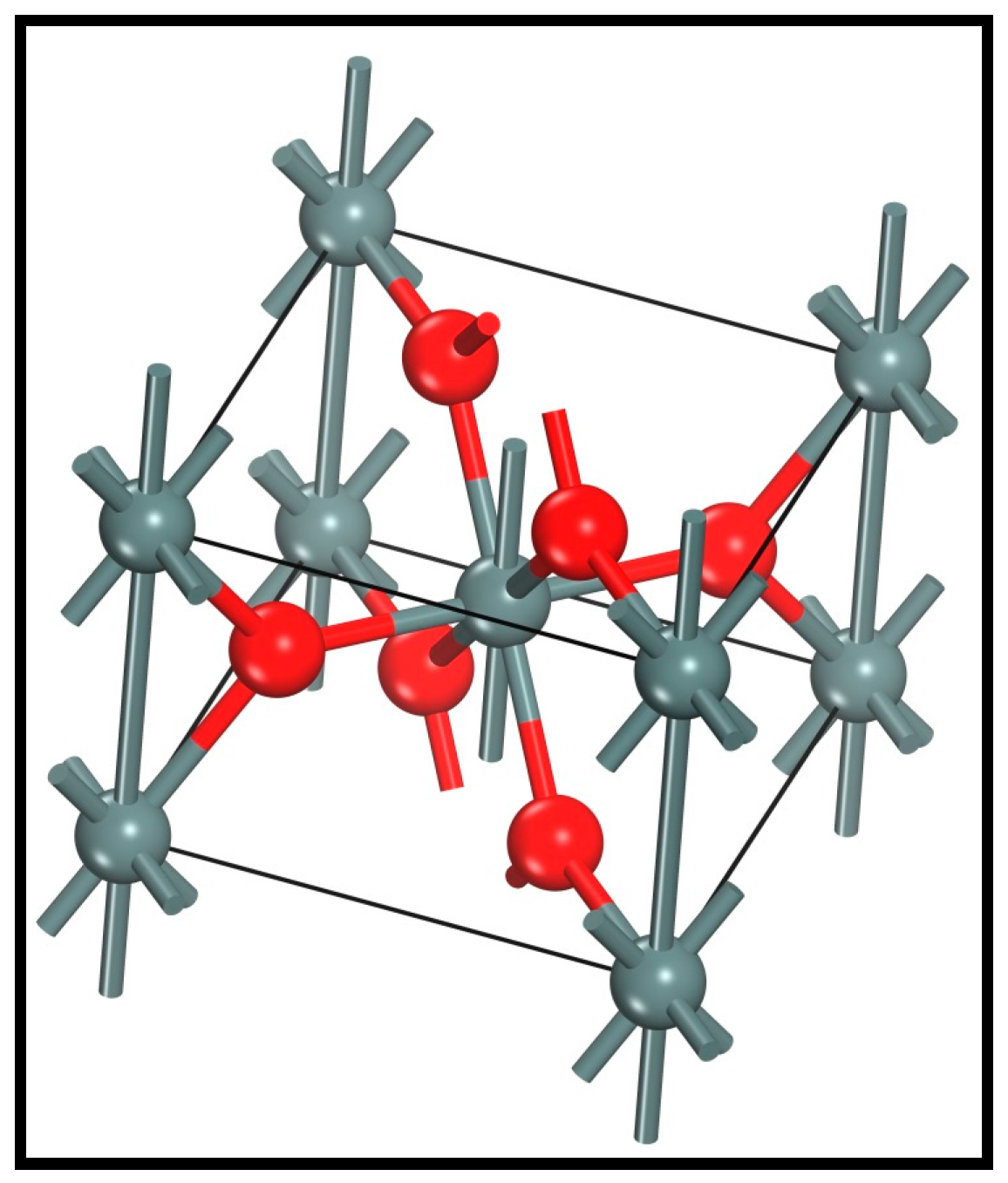
Adopting the optimized simulation parameters for a functional GGA-WC, a k-point set of 3 × 3 × 4, and cutoff energy of 580 eV, the simulated cassiterite bulk unit-cell is shown in Figure 4. The computed lattice parameters are listed in Table 3.
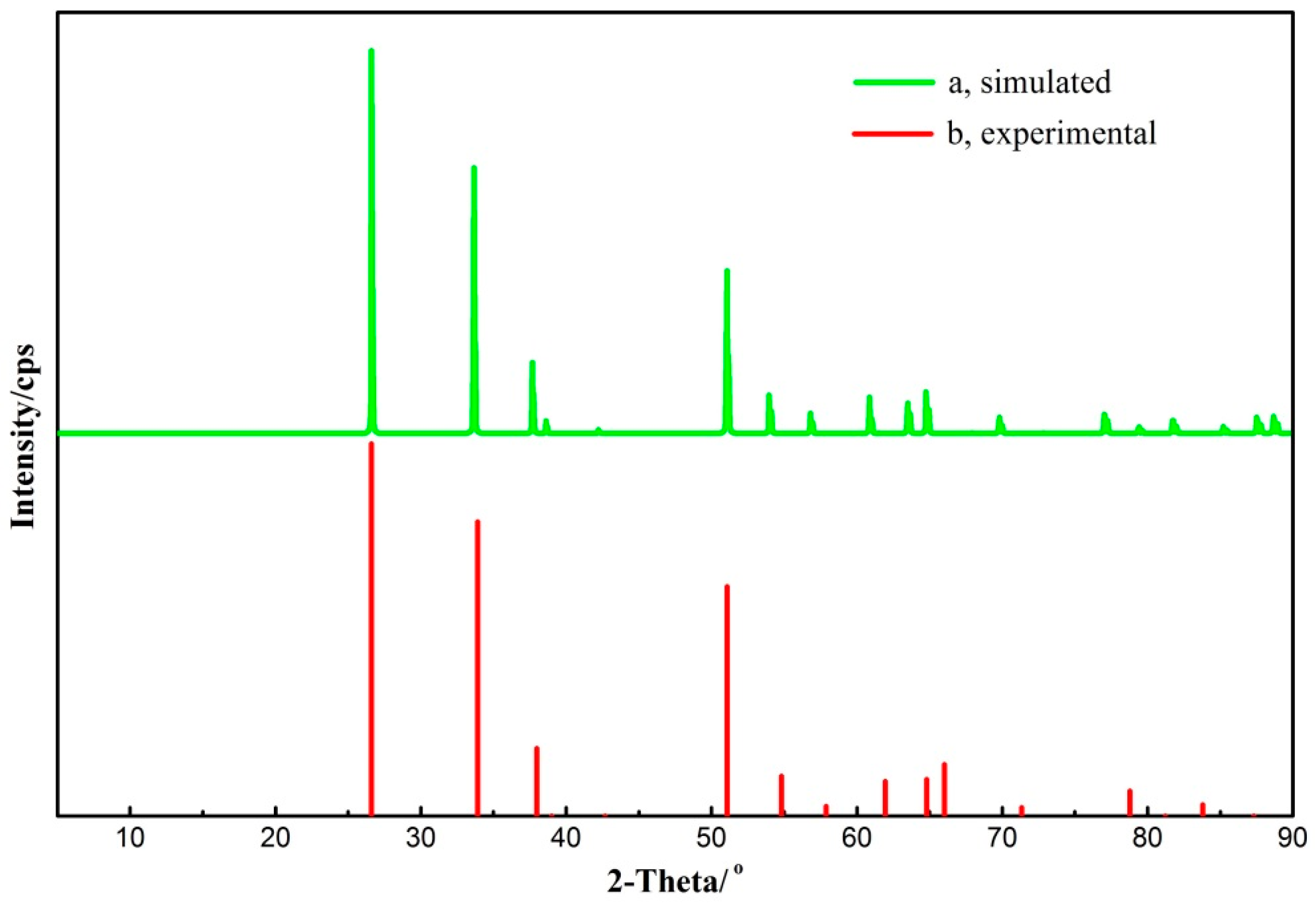
It can be seen from Table 3 that the calculated parameters tally well with the corresponding data that was obtained using neutron diffraction [28]. All the computed parameter differences are less than 3% and the modeling veracity is verified. Furthermore, the Reflex module in Accelrys Materials Studio was used to calculate the X-ray diffraction (XRD) spectra of the simulated cassiterite crystal, which was compared with the experimental one in Figure 5.
It is clearly suggested in Figure 5 that all the characteristic peaks of cassiterite are locatd in the simulated X-ray diffraction spectra, which matches perfectly with the experimental spectra. Therefore, it demonstrates that the calculation parameter settings are valid and the computational cassiterite bulk cell is rational to represent the natural mineral structure.
3.2. Cassiterite Surface Model Optimization
Surface energy is defined as the work needed to be done to generate a unit new surface, and for a higher surface energy, the cleavage surface is not only arduous to create but also thermodynamically unstable. Accordingly, surface energy can be used to estimate the stability of the surface. Mulheran and Harding [30] explored various SnO2 crystallographic surfaces and made a discovery that low-index surfaces generally possess lower surface energies and a higher thermodynamic stability than high-index ones. Many researchers have focused their attention on the low-index surfaces of cassiterite. Slater et al. [31] and Oviedo and Gillan [32] have calculated the surface energies of (110), (100), (001), and (101) planes of SnO2 using the vienna ab-initio simulation package (VASP) package or general utility lattice program (GULP) code, respectively, and made the same conclusion that the (110) surface has the lowest surface energy, which is the most thermodynamically stable. Gao et al. [33] have also proved that (110) is the most stable surface of cassiterite by means of calculating the surface broken bonds density and the prediction agrees well with the surface energy calculation results of Slater et al. [31] and Oviedo et al. [32]. Nevertheless, Bandura et al. [34] claimed that the (100) surface is more stable than the (110) surface based on their DFT calculation of surface energies (see Table 4). Thus, the most stable surface of cassiterite could not be ascertained on the basis of existing literature, and thus, a battery of DFT calculations were conducted to confirm the most stable cleavage plane of cassiterite in this paper. The surface slabs used here contained three layers of Sn2O4 units and the vacuum depth was 15 Å.
As can be seen in Table 4, the calculated surface energies in this work are similar to those of Slater et al. [31] and Oviedo and Gillan [32]. The (110) surface has the lowest surface energy and it is the most stable surface of cassiterite. However, the surface energy of the (100) plane is merely a little bit higher than (110), testifying that the (100) surface is also fairly stable and should be very common in the crude SnO2 materials. As a matter of experimental fact, cassiterite grows naturally along the (110) and (100) faces and cleaves well by the two faces [35]. Therefore, the adsorption of oleate on both surfaces should be computed to confirm the strongest interaction.
Firstly, surface energy convergence tests were carried out as functions of slab thickness and vacuum depth. The tests results are presented in Table 5.
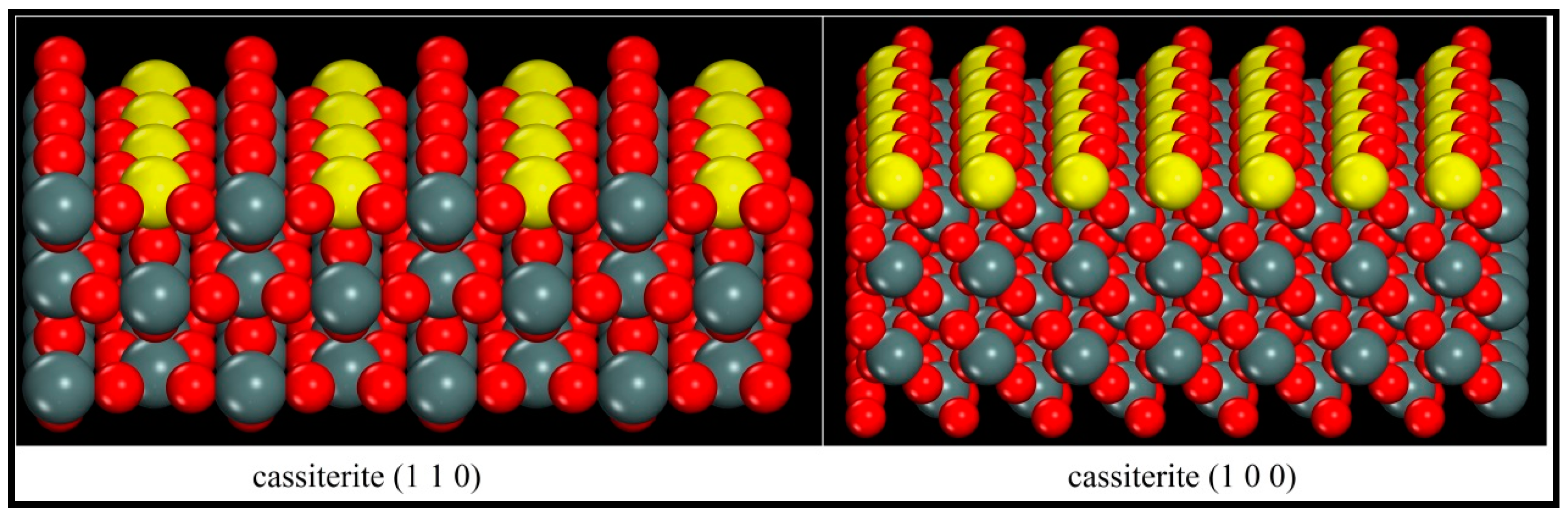
The data in Table 5 illustrate that three layers of Sn2O4 units within the slabs are sufficient to keep the variations in the surface energies of both (110) and (100) faces less than 0.01 J/m2, and a 10 Å vacuum for both faces is thick enough to prevent the interaction between the top and the bottom surfaces. Considering the length of the oleate ion and the preparation for the following adsorption modeling, the (110) and (100) surface structures containing three layers of Sn2O4 units and a 27 Å vacuum depth were optimized as shown in Figure 6, and the colour of the surface Sn atoms is changed to yellow.
It is evidently shown in Figure 6 that when the (110) and (100) surfaces were cleaved, the Sn–O bonds were broken and the surface Sn atoms lost one coordinating O atom and became five coordinated atoms. Consequently, the surface Sn atoms became active and tended to adsorb some foreign species. However, during the formation of the (110) face, the top coordination Sn–O bond was broken and the surface Sn atoms were directly exposed to the space. When the (100) plane was cleaved, the surface Sn atoms lost the upper right coordinating O atoms and the exposed Sn atoms were partially shielded by the top left surface O atoms. To sum up here, it can be predicted that when the oleate ion approaches, it will be subject to greater resistance on the (100) face than that on the (110) plane, and the adsorption on (110) may be stronger.
3.3. Adsorption of Oleate on the (110) and (100) Surfaces
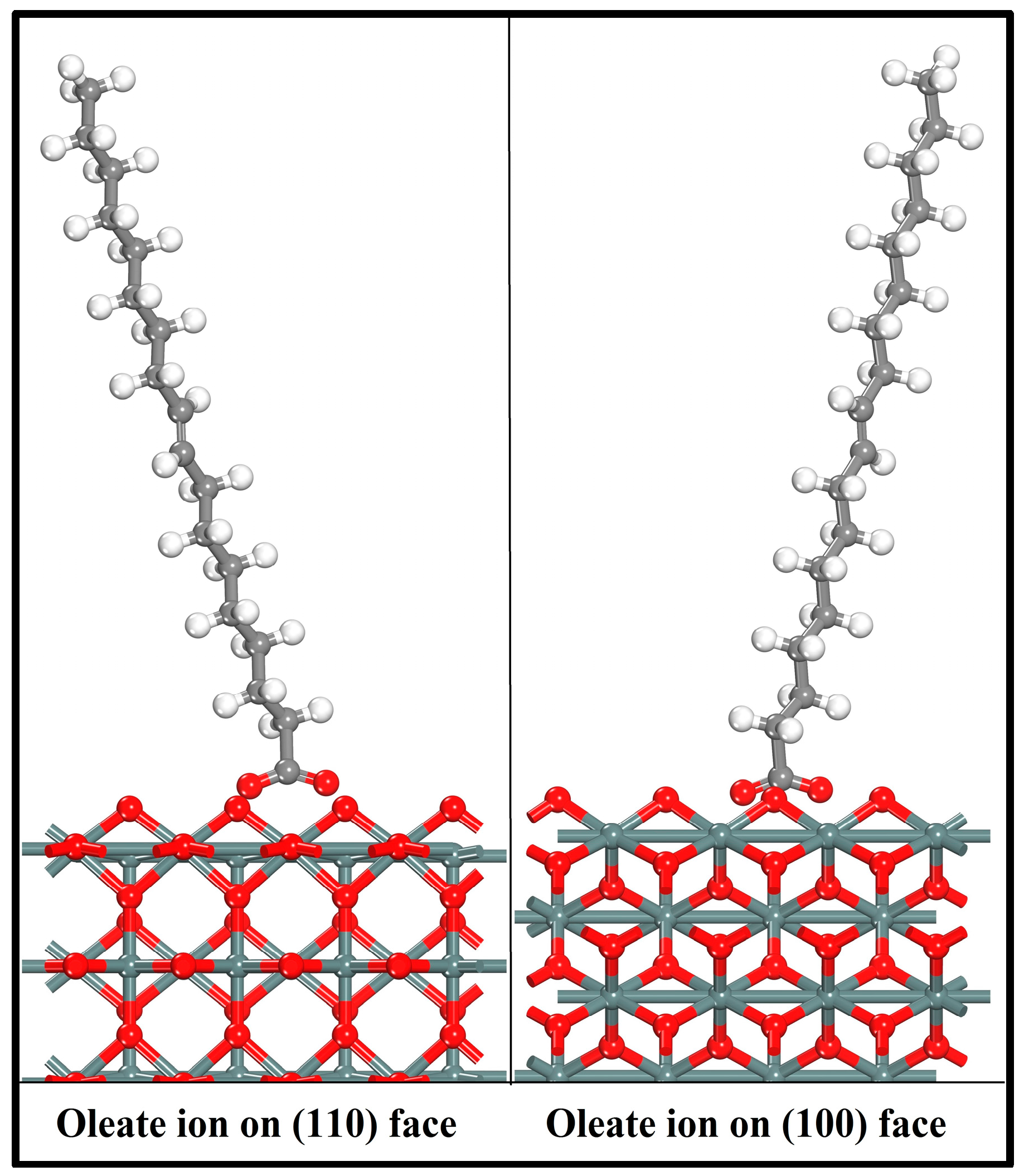
First-principles calculations were implemented to evaluate the adsorption of oleate ions on the (110) and (100) surfaces. The optimized collector ions were docked on cassiterite surfaces according to the fact that the O atoms in oleate ions interact with the metal atoms on the surfaces and form the reagent–mineral complex. Various adsorption configurations were computed, and in this paper, only the most preferable two are reported. Figure 7 displays the two geometry optimized adsorption configurations that were formed by oleate ions on cassiterite cleavage planes (110) and (100), respectively.
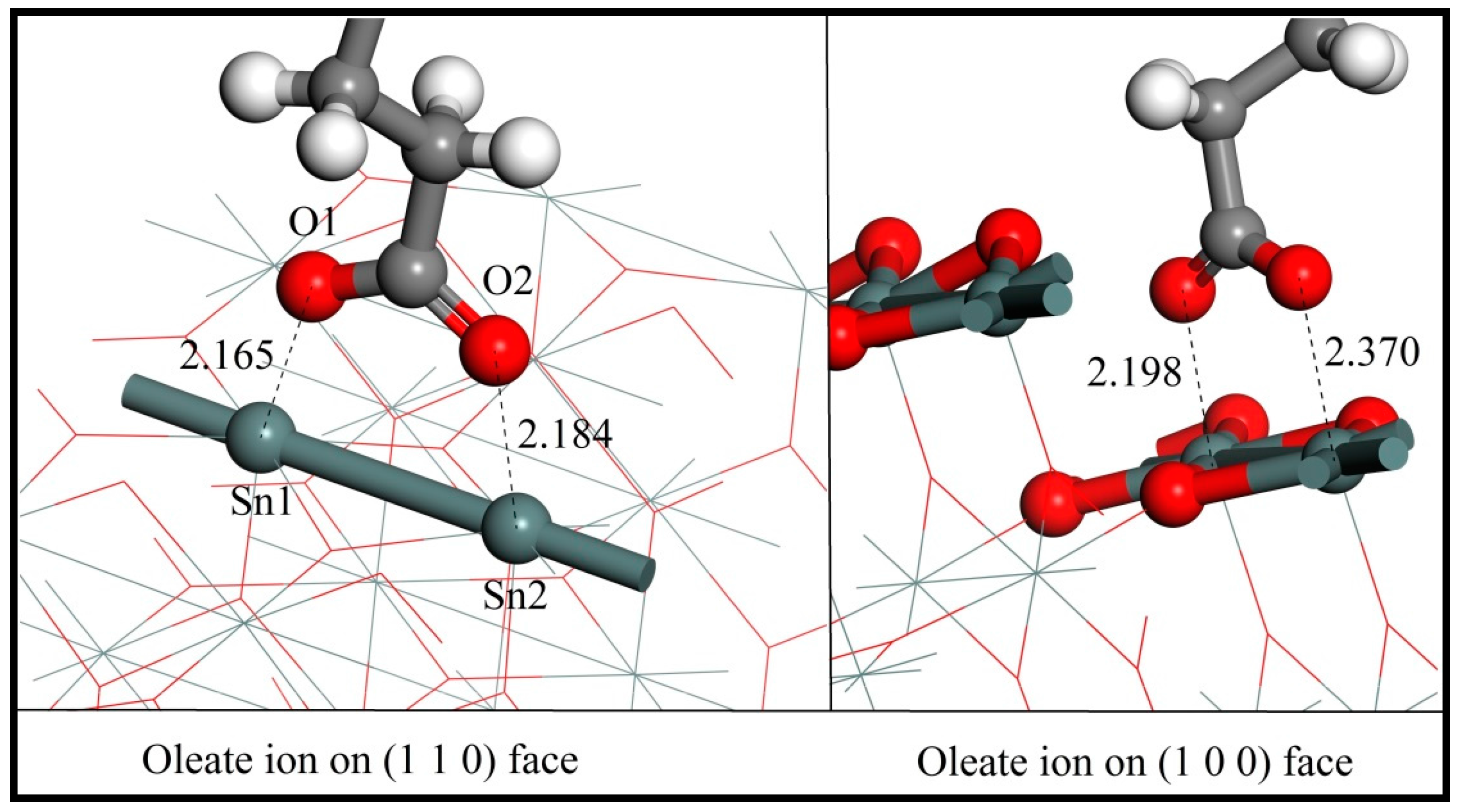
As shown in Figure 7, the two oxygen atoms of the oleate ions bond with two adjacent surface Sn atoms over either the (110) or (100) plane and result in the formation of a chelating ring, respectively. The bonding details of the oleate anion on cassiterite (110) and (100) surfaces are shown in Figure 8. The lengths of the two Sn–O bond formed on (110) are 2.165 Å and 2.184 Å, respectively, and both are shorter than the equivalent bond lengths of 2.370 Å and 2.198 Å formed on (100), as shown in Figure 8. The computed interaction energies of oleate ions on planes (110) and (100) are −244.70 kJ/mol and −195.29 kJ/mol, respectively (Table 6), which convincingly demonstrates that the oleate ion is more inclined to be adsorbed on surface (110), i.e., the most stable cleavage plane of cassiterite. Hence, the conclusion that can be drawn here is that the adsorption of an oleate ion is stronger on plane (110). Consequently, the following calculations were conducted on this surface.
3.4. Mechanism of Oleate–Cassiterite Interaction
3.4.1. Adsorption Energy Comparison
Flotation is a separation technology taking place in aqueous media, whose success relies on regulating the wanted mineral surfaces hydrophobic using collectors and the unwanted mineral surfaces hydrophilic using inhibitors [36,37,38]. In flotation, cassiterite particles are firstly dispersed in water. Thus, the adsorption energies of H2O and OH− on the SnO2 (110) plane were calculated, and the results are listed in Table 6. It can be seen that the adsorption energy of the H2O molecule on the (110) plane is −76.74 kJ/mol, which tallies well with the results of the contact angle measurements [22] showing that the SnO2 surfaces have a strong affinity for water, indicating that hydration films will be easily formed on cassiterite surfaces. Besides, the adsorption energy of −115.22 kJ/mol of OH− in Table 6 reveals that the OH− ions can adsorb on cassiterite (110) surfaces spontaneously and strongly. These two adsorption energies coincide well with the results of the flotation tests and zeta potential measurements which show that the initial floatability of cassiterite is extremely poor and the surfaces are negatively charged [22]. By comparison, the interaction energy of the oleate ion on the (110) surface is over two times more than that of OH− and over three times more than that of H2O, with the more negative adsorption energy suggesting that the oleate ion can adsorb on the (110) surface much more strongly. At this point, the interaction process can be imagined as follows: Firstly, when the cassiterite particles are placed into water, the H2O molecules and OH− ions promptly get adsorbed onto the surfaces and make the cassiterite particles hydrated. Then, when the oleate ions approach, its stronger interaction energy makes it easy for oleate ions to replace the adsorbed H2O and OH−, which results in the hydrophobicity of the mineral surfaces.
3.4.2. Mulliken Population Analysis
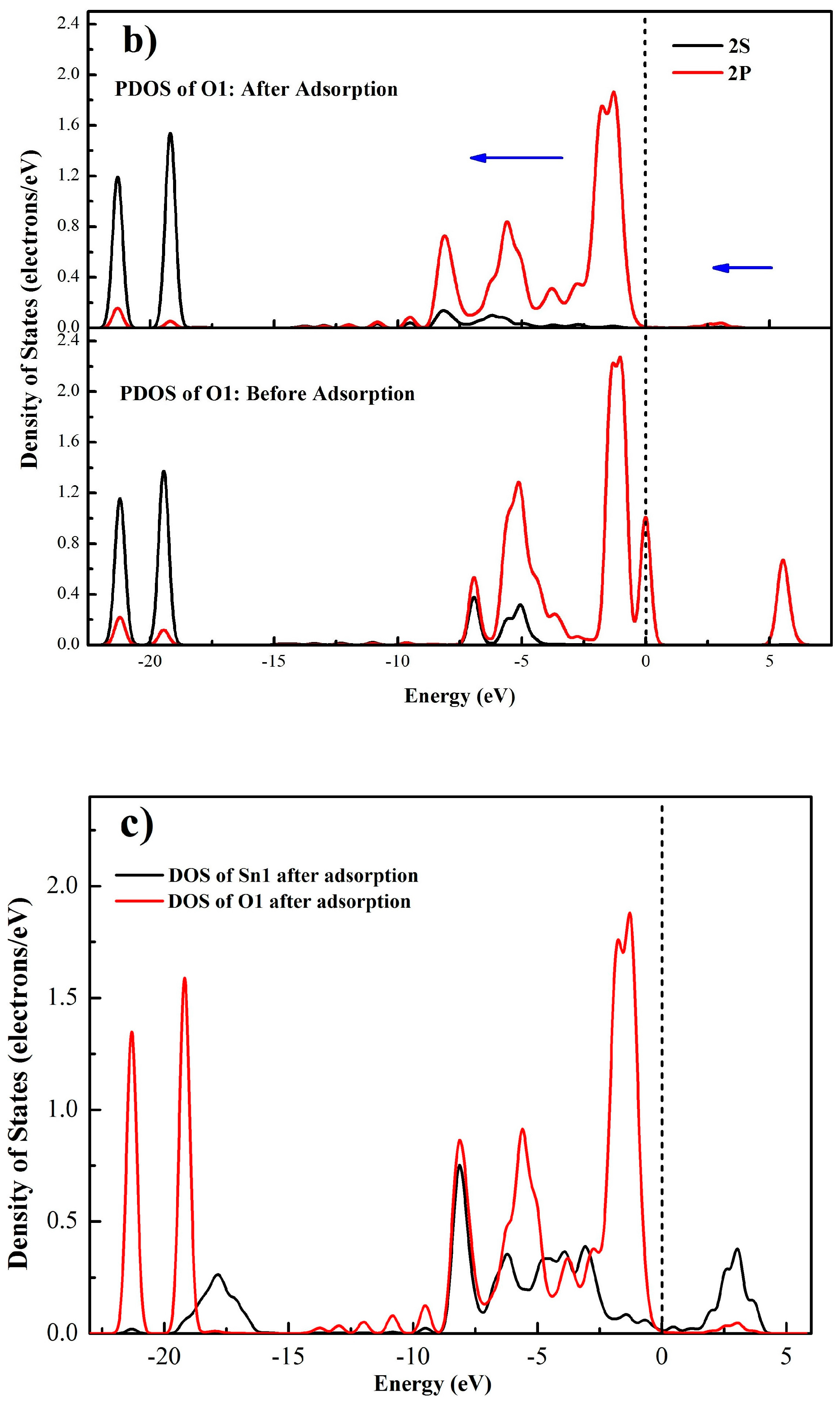
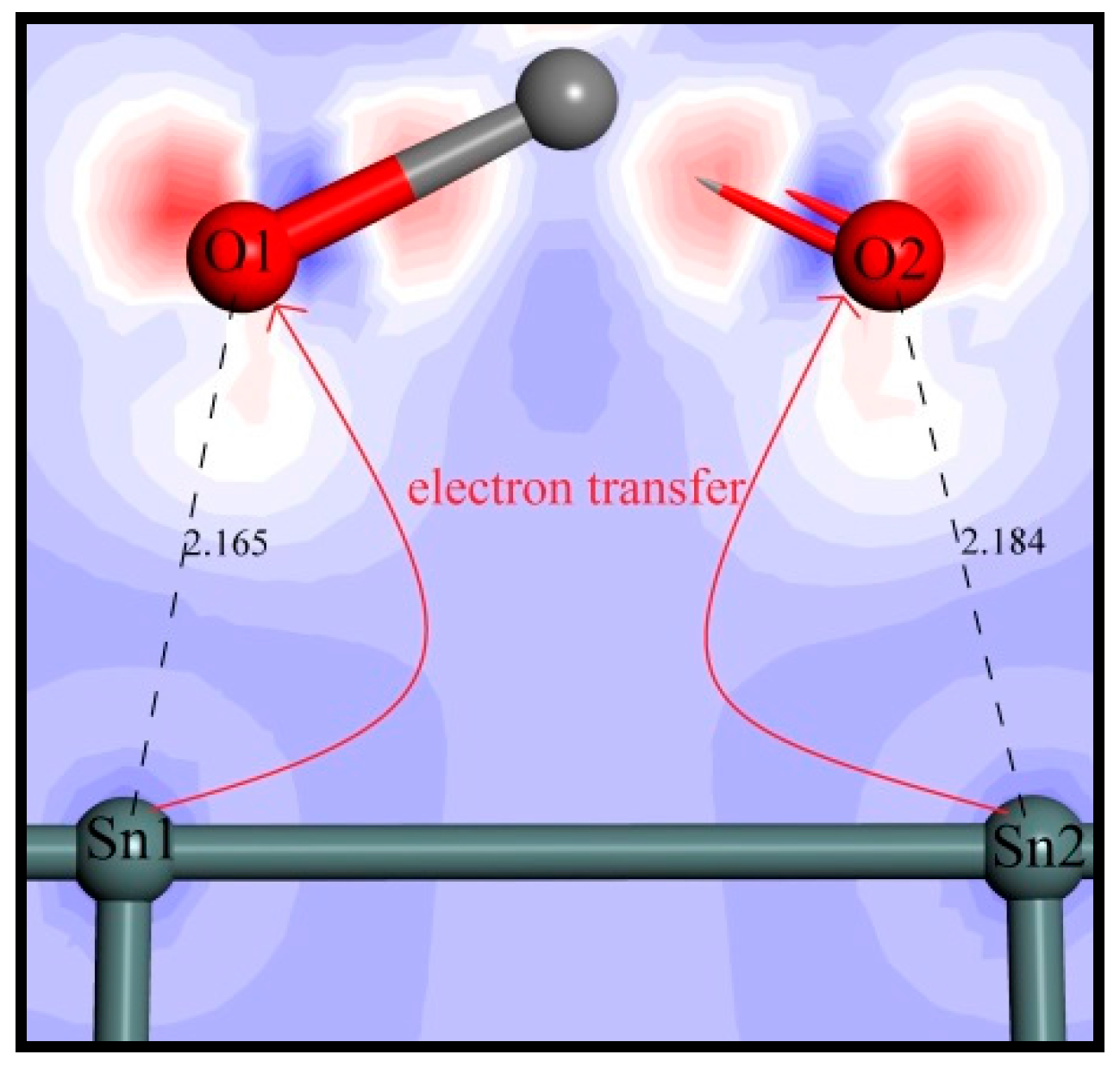
Mulliken population calculations can show the electron transfer and provide intuitive evidence for understanding a chemical reaction [39]. Moreover, the magnitude of bond populations is a good measure of the covalency of a bond, where the greater the value, the stronger the covalency of the bond [40]. The C–C bond in diamond crystal is evidently a strong covalent bond, whose bond population is calculated as 0.75. The mulliken populations of the pertinent atoms and bonds were computed to further clarify the adsorption process of oleate ions on the SnO2 (110) surface. The involved atoms are numbered as shown in Figure 8 and the calculated populations are listed in Table 7, where negative values represent getting electrons and positive values indicate losing them. Obviously, it can be seen that after adsorption, the mulliken charges of O1 and O2 decrease from −0.49 to −0.58 and −0.46 to −0.58, respectively. Meanwhile, the charges of Sn1 and Sn2 increase from 1.79 to 1.84 and 1.81, respectively. These charge changes indicate electron transfers from Sn1 and Sn2 atoms to O1 and O2 atoms, and two new chemical bonds come into being. On the other hand, the computed overlap populations of O1–Sn1 and O2–Sn2 bonds are 0.30 and 0.29, respectively, which are much smaller than that of the C–C bond in diamond, indicating that the O1–Sn1 and O2–Sn2 bonds are of a low covalency.
The electrons transmission between Sn atoms and O atoms is delineated in Figure 9 by analyzing the electron density difference of oleate–cassiterite adsorption configuration. It can be intuitively seen that the Sn1 and Sn2 atoms lose electrons to O1 and O2 atoms. Combining the bond population calculation and the electron density difference analysis, it is clearly certified that the oleate ions chemically adsorb on the (110) surface by ionic bonds.
3.4.3. Electrons Density Analysis
The electron density distributions of O1, O2, Sn1, and Sn2 atoms before and after the adsorption of OL− on the cassiterite (110) plane are listed in Table 8. It is noticeable that after the adsorption, the electrons in s orbitals of all the four atoms are reduced and the electrons in p orbitals of all the four atoms are increased. Additionally, the total electrons of two Sn atoms are reduced and those of the two O atoms are increased. These changes may be attributed to the point when the O atoms of OL− get close to the Sn atoms, part of the electrons in s orbitals of the O and Sn atoms are attracted and excited by the other nucleuses, and these excited electrons in the s orbital move up to a higher energy level. Moreover, the electronegativity of O atoms is stronger than Sn atoms, making the electron clouds between O–Sn bonds lean towards O atoms. Therefore, the Sn atoms exhibit a loss of electrons.
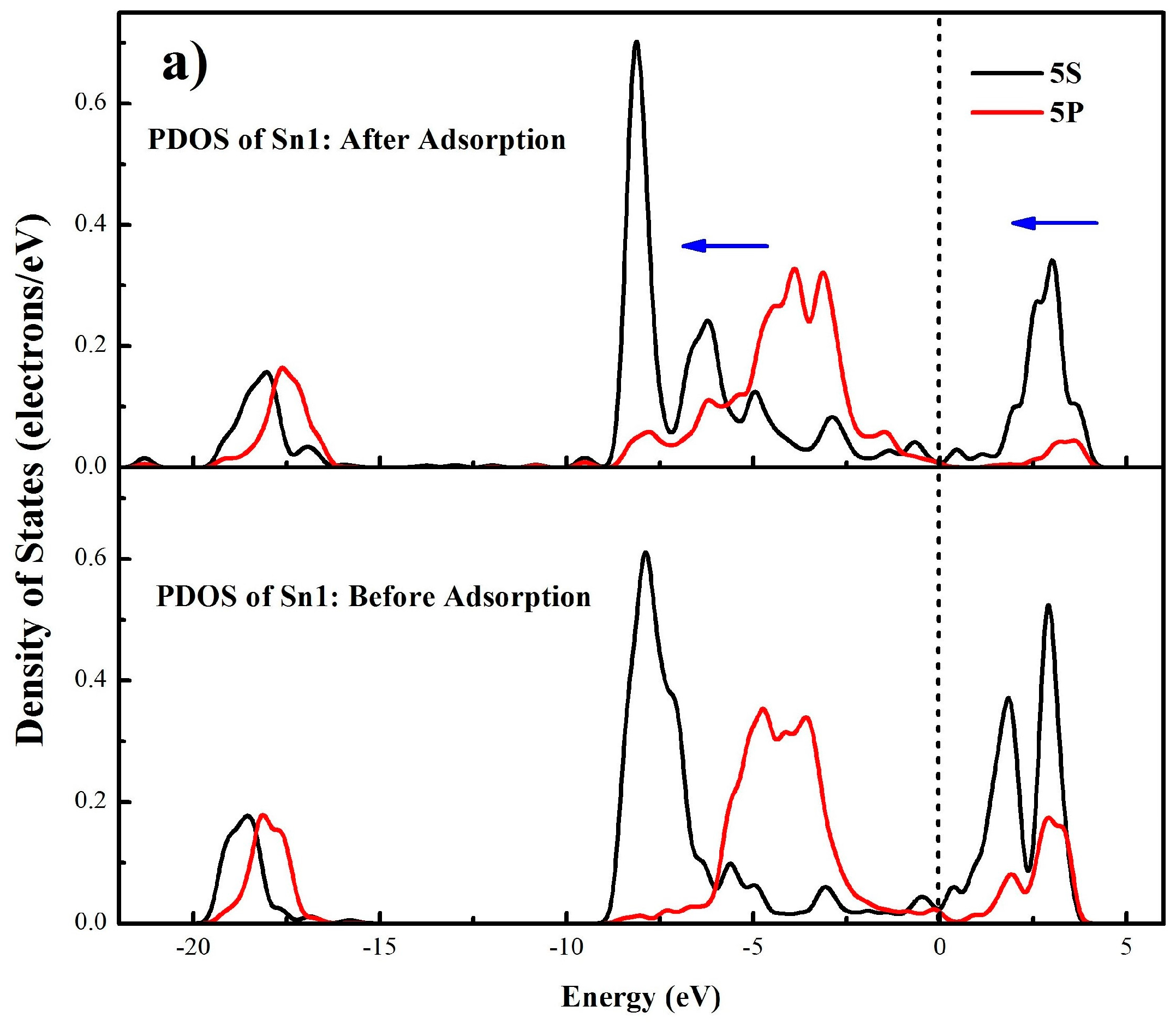
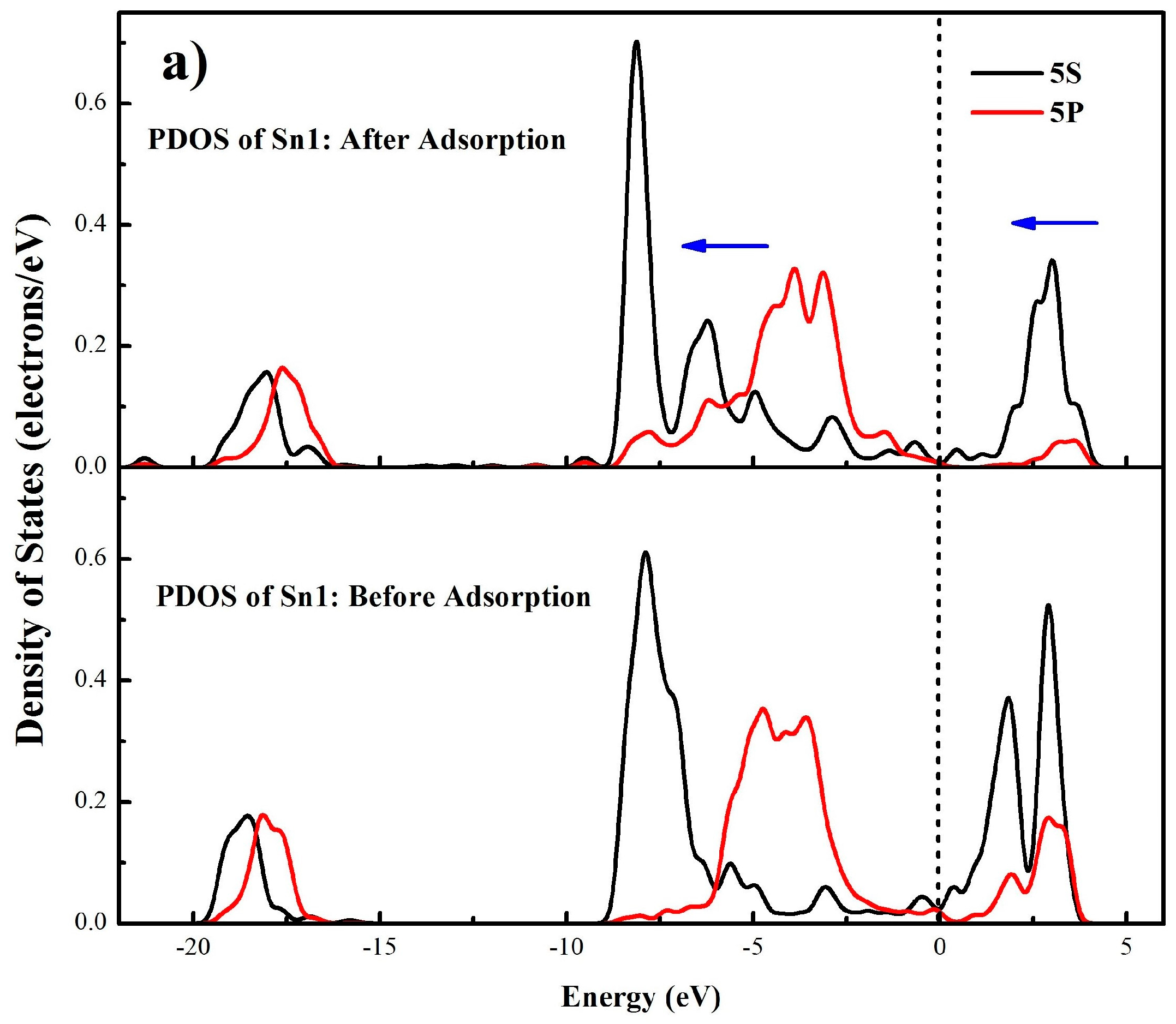
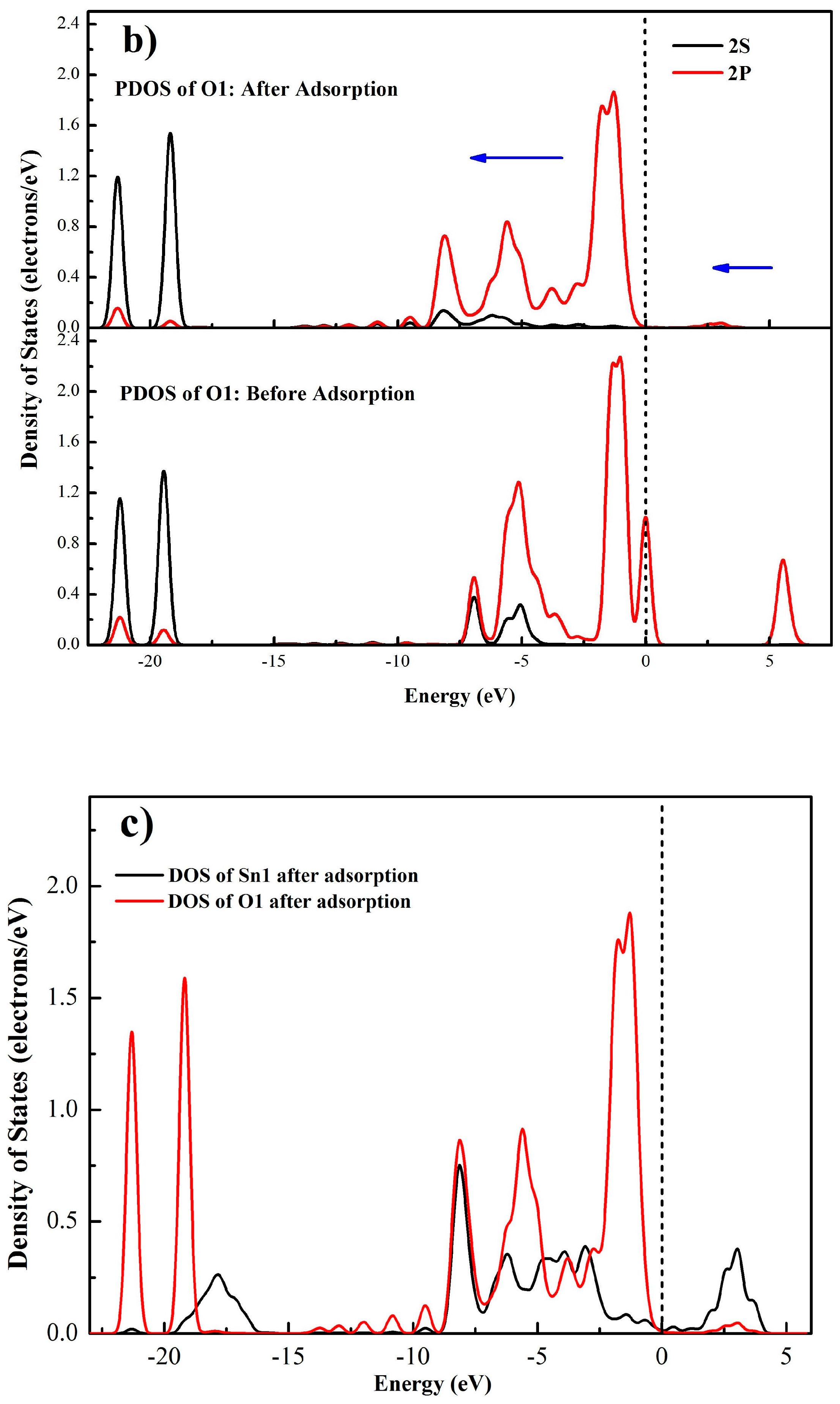
A more detailed bonding mechanism during the interaction between OL− and cassiterite (110) surface is obtained in Figure 10 by comparing the density of states (DOS) of Sn1 and O1 atoms before and after the adsorption of OL− on the (110) surface. In Figure 10a,b, it can be seen before adsorption that the bands of Sn1 around the Fermi level are dominated by the 5s and 5p orbitals, and the DOS of O1 near the Fermi level mainly results from the 2p orbital, implying that the electrons in 5s and 5p orbitals of Sn1 and the 2p orbital of O1 are active, meaning that the Sn1 and O1 atoms tend to bond with each other. Besides, the locality of the states of the two atoms is high before the adsorption. After adsorption, for the O1 atom, the 2p orbital at the Fermi level disappears and the electronic states generally shift to lower energy states; for the Sn1 atom, the peaks of the DOS around the Fermi level move to a lower energy direction and the intensities of the peaks all decrease. These changes demonstrate that the 5s and 5p orbitals of Sn1 and the 2p orbital of O1 play great roles in the bonding process and the O1 and Sn1 atoms are stabilized after the interaction. Meanwhile, for both the O1 and Sn1 atoms, the non-locality of the s and p orbitals between −10 eV and −2.5 eV increases and some new peaks come into being after the adsorption. In Figure 10c, strong resonance appears between −10 eV and −2.5 eV in the DOS of both the Sn1 and O1 atoms, which can be attributed to the hybridization of the 5s and 5p orbitals of Sn1 and the 2p orbital of O1. Thus, the formation of the O1–Sn1 bond mainly results from the 5s and 5p orbitals of Sn1 and the 2p orbital of O1. Similar changes are observed in the DOS of O2 and Sn2 atoms.
4. Conclusions
Quantum chemical calculations were carried out to study the adsorption of oleate on cassiterite surfaces. The most suitable parameter settings for cassiterite bulk cell optimization are the exchange-correlation functional GGA-WC, a k-point set of 3 × 3 × 4, and a cutoff energy of 580 eV, and the optimized cassiterite bulk cell parameter differences are all no more than 3%. The surface energy of four low index planes follows the increasing sequence (110) < (100) < (101) < (001), indicating that (110) is the most stable cleavage plane of cassiterite, but (100) is only slightly more unstable than (110). The (110) and (100) surface slabs containing three layers of Sn2O4 and a 10 Å vacuum are adequate for the construction of rational surface structures.
The adsorption energies of oleate ions on (110) and (100) planes are −244.70 kJ/mol and −195.29 kJ/mol, respectively, suggesting that the adsorption on (110) is stronger and the (110) plane was selected as the adsorption surface. Comparing the interaction energies of OL−, OH−, and H2O on cassiterite (110) plane, it is found that OL− is able to replace the adsorbed H2O and OH− on the surface. Mulliken population calculations together with the electron density difference analysis reveal that the oleate ions chemically adsorb on the (110) surface by ionic bonds. DOS analysis demonstrates that the formation of the O1–Sn1 bond mainly results from the 5s and 5p orbitals of the Sn1 atom and the 2p orbital of the O1 atom. The results of the DFT simulation in this paper are in good agreement with those of the experiments.
Acknowledgments
The authors are greatly thankful and appreciative for the financial support from the National Natural Science Foundation of China (Grant number 51674066, 51204035).
Author Contributions
Jie Liu and Guichen Gong conceived and designed the experiments; Guichen Gong performed the experiments; Guichen Gong and Jie Liu analyzed the data; Yuexin Han and Yinmin Zhu contributed reagents/materials/analysis tools; Guichen Gong wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Feng, Q.C.; Zhao, W.J.; Wen, S.M.; Cao, Q.B. Activation mechanism of lead ions in cassiterite flotation with salicylhydroxamic acid as collector. Sep. Purif. Technol. 2017, 178, 193–199. [Google Scholar] [CrossRef]
- Falcon, L.M. The gravity recovery of cassiterite. J. S. Afr. Inst. Min. Metall. 1982, 4, 112–117. [Google Scholar]
- Baldauf, H.; Schoenherr, J.; Schubert, H. Alkane dicarboxylic acids and aminonaphthol sulfnoic acids—A new reagent regime for cassiterite flotation. Int. J. Miner. Process. 1985, 15, 117–133. [Google Scholar] [CrossRef]
- Sreevinas, T.; Manohar, C. Adsorption of octyl hydroxamic acid/salt on cassiterite. Miner. Process. Extr. Metall. Rev. 2000, 20, 503–519. [Google Scholar]
- Sreenivas, T.; Padmanabhan, N.P.H. Surface chemistry and flotation of cassiterite with alkyl hydroxamates. Colloids Surf. A Physicochem. Eng. Asp. 2002, 205, 47–59. [Google Scholar] [CrossRef]
- Zhou, Y.C.; Tong, X.; Song, S.X.; Wang, X.; Deng, Z.G. Beneficiation of cassiterite fines from a tin tailing slime by froth flotation. Sep. Sci. Technol. 2014, 49, 458–463. [Google Scholar] [CrossRef]
- Leistner, T.; Embrechts, M.; Leißner, T.; Chelgani, S.C.; Osbahr, I. A study of the reprocessing of fine and ultrafine cassiterite from gravity tailing residues by using various flotation techniques. Miner. Eng. 2016, 96–97, 94–98. [Google Scholar] [CrossRef]
- Oliveira, J.F.; Adamian, R. Physico-chemical factors affecting the separation of cassiterite and fluorite by froth flotation. Miner. Process. Extr. Metall. Rev. 1992, 9, 125–134. [Google Scholar] [CrossRef]
- Gruner, H.; Bilsing, U. Cassiterite flotation using styrene phosphonic acid to produce high-grade concentrates at high recoveries from finely disseminated ores- comparison with other collectors and discussion of effective circuit configurations. Miner. Eng. 1992, 5, 429–434. [Google Scholar] [CrossRef]
- Angadi, S.I.; Sreenivas, T.; Jeon, H.S.; Baek, S.H.; Mishra, B.K. A review of cassiterite beneficiation fundamentals and plant practices. Miner. Eng. 2015, 70, 178–200. [Google Scholar] [CrossRef]
- Xu, Y.B.; Qin, W.Q. Surface analysis of cassiterite with sodium oleate in aqueous solution. Sep. Sci. Technol. 2012, 47, 502–506. [Google Scholar] [CrossRef]
- Quast, K. Surface chemistry and oleate flotation of three South Australian micaceous hematites. Miner. Eng. 2016, 85, 123–129. [Google Scholar] [CrossRef]
- Peck, A.S.; Raby, L.H.; Wadsworth, M.E. An infrared study of the flotation of hematite with oleic acid and sodium oleate. Soc Min Eng. 1966, 235, 301–307. [Google Scholar]
- Rath, S.S.; Sinha, N.; Sahoo, H.; Das, B.; Mishra, B.K. Molecular modeling studies of oleate adsorption on iron oxides. Appl. Surf. Sci. 2014, 295, 115–122. [Google Scholar] [CrossRef]
- Rai, B.; Sathish, P.; Tanwar, J.; Pradip; Moon, K.S.; Fuerstenau, D.W. A molecular dynamics study of the interaction of oleate and dodecylammonium chloride surfactants with complex aluminosilicate minerals. J. Colloid Interface Sci. 2011, 362, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.M.; Luo, B.B.; Sun, C.Y.; Liu, J.; Sun, H.T. Density functional theory study of α-Bromolauric acid adsorption on the α-quartz (101) surface. Miner. Eng. 2016, 92, 72–77. [Google Scholar] [CrossRef]
- Xu, L.H.; Hu, Y.H.; Wu, H.Q.; Tian, J.; Liu, J. Surface crystal chemistry of spodumene with different size fractions and implications for flotation. Sep. Purif. Technol. 2016, 169, 33–42. [Google Scholar] [CrossRef]
- Pradip; Rai, B. Design of tailor-made surfactants for industrial applications using a molecular modelling approach. Colloids Surf. A 2002, 205, 139–148. [Google Scholar] [CrossRef]
- Pradip; Rai, B. Molecular modeling and rational design of flotation reagents. Int. J. Miner. Process. 2003, 72, 95–110. [Google Scholar] [CrossRef]
- Rai, B.; Pradip. Design of highly selective industrial performance chemicals: A molecular modelling approach. Mol. Simul. 2008, 34, 1209–1214. [Google Scholar] [CrossRef]
- Tan, X.; He, F.Y.; Shang, Y.B.; Yin, W.Z. Flotation behavior and adsorption mechanism of (1-hydroxy-2-methyl-2-octenyl) phosphonic acid to cassiterite. Trans. Nonferr. Met. Soc. China 2016, 26, 2469–2478. [Google Scholar] [CrossRef]
- Liu, J.; Gong, G.C.; Han, Y.X. Influences of organic depressants on the floatability of fine cassiterite. J. China Univ. Min. Technol. 2016, 45, 610–614. (In Chinese) [Google Scholar]
- Mineral Commodity Summaries. Tin. 2017. Available online: http://minerals.usgs.gov/ (accessed on 5 March 2017).
- Sahoo, H.; Sinha, N.; Rath, S.S.; Das, B. Ionic liquids as novel quartz collectors: Insights from experiments and theory. Chem. Eng. J. 2015, 273, 46–54. [Google Scholar] [CrossRef]
- Madsen, G.K.H.; Novák, P. Charge order in Magnetite. An LDA + U study. Physics 2004, 69, 777–783. [Google Scholar] [CrossRef]
- Anisimov, V.I.; Elfimov, I.S.; Hamada, N.; Terakura, K. Charge-ordered insulating state of Fe3O4 from first-principles electronic structure calculations. Phys. Rev. B 1996, 54, 4387–4390. [Google Scholar] [CrossRef]
- Kovács, S.Á.; Cynthia, S.L. Electronic structure and charge ordering in magnetite: Implications for the Fe3O4 (001)–water interface. Mol. Simul. 2010, 36, 1289–1296. [Google Scholar] [CrossRef]
- Bolzan, A.A.; Fong, C.; Kennedy, B.J.; Howard, C.J. Structural studies of rutile-type metal dioxides. Acta Crystallogr. 1997, 53, 373–380. [Google Scholar] [CrossRef]
- Wu, Z.G.; Cohen, R.E. A more accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, 235116. [Google Scholar] [CrossRef]
- Mulheran, P.A.; Harding, J.H. The stability of SnO2 surfaces. Model. Simul. Mater. Sci. Eng. 1992, 1, 39–43. [Google Scholar] [CrossRef]
- Slater, B.; Catlow, C.R.A.; Gay, D.H.; Williams, D.E.; Dusastre, V. Study of surface segregation of antimony on SnO2 surfaces by computer simulation techniques. J. Phys. Chem. B 1999, 103, 10644–10650. [Google Scholar] [CrossRef]
- Oviedo, J.; Gillan, M.J. Energetics and structure of stoichiometric SnO2 surfaces studied by first-principles calculations. Surf. Sci. 2000, 463, 93–101. [Google Scholar] [CrossRef]
- Gao, Z.Y.; Sun, W.; Hu, Y.H. Mineral cleavage nature and surface energy: Anisotropic surface broken bonds consideration. Trans. Nonferr. Met. Soc. China 2014, 24, 2930–2937. [Google Scholar] [CrossRef]
- Bandura, A.V.; Sofo, J.O.; Kubicki, J.D. Derivation of force field parameters for SnO2-H2O surface systems from plane-wave density functional theory calculations. J. Phys. Chem. B 2006, 110, 8386–8397. [Google Scholar] [CrossRef] [PubMed]
- Henrich, V.E.; Cox, P.A.; Diebold, U. The surface science of metal oxides. Phys. Today 1994, 191, 8386–8397. [Google Scholar] [CrossRef]
- Beneventi, D.; Allix, J.; Zeno, E.; Nortier, P.; Carré, B. Simulation of surfactant contribution to ink removal selectivity in flotation deinking lines. Sep. Purif. Technol. 2009, 64, 357–367. [Google Scholar] [CrossRef]
- Weng, X.Q.; Mei, G.J.; Zhao, T.T.; Zhu, Y. Utilization of novel ester-containing quaternary ammonium surfactant as cationic collector for iron ore flotation. Sep. Purif. Technol. 2013, 103, 187–194. [Google Scholar] [CrossRef]
- Sahoo, H.; Rath, S.S.; Das, B. Use of the ionic liquid-tricaprylmethyl ammonium salicylate (TOMAS) as a flotation collector of quartz. Sep. Purif. Technol. 2014, 136, 66–73. [Google Scholar] [CrossRef]
- Oertzen, G.U.V.; Skinner, W.M. Ab initio and X-ray photoemission spectroscopy study of the bulk and surface electronic structure of pyrite (100) with implications for reactivity. Phys. Rev. B 2005, 72, 235427. [Google Scholar] [CrossRef]
- Guo, X.J.; Li, L.; Liu, Z.Y.; Li, D.L.; He, J.L. Hardness of covalent compounds: Roles of metallic component and d valence electrons. J. Appl. Phys. 2008, 104, 23503. [Google Scholar] [CrossRef]
Figure 1.
Optimized oleate ion structure with partial Mulliken charges (colour codes: Gray—C, White—H, Red—O).
Figure 1.
Optimized oleate ion structure with partial Mulliken charges (colour codes: Gray—C, White—H, Red—O).
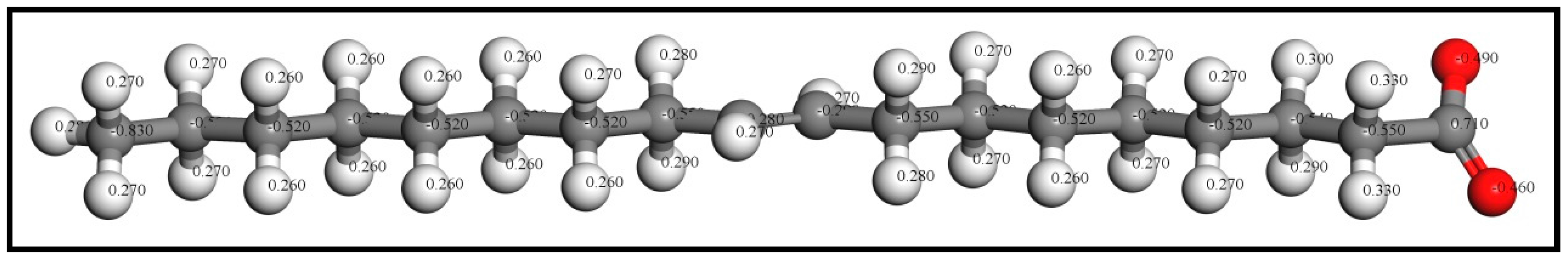
Figure 2.
The dependence of the total energy (a) and lattice constants difference (b) on the k-point set with a fixed cutoff energy of 340 eV.
Figure 2.
The dependence of the total energy (a) and lattice constants difference (b) on the k-point set with a fixed cutoff energy of 340 eV.
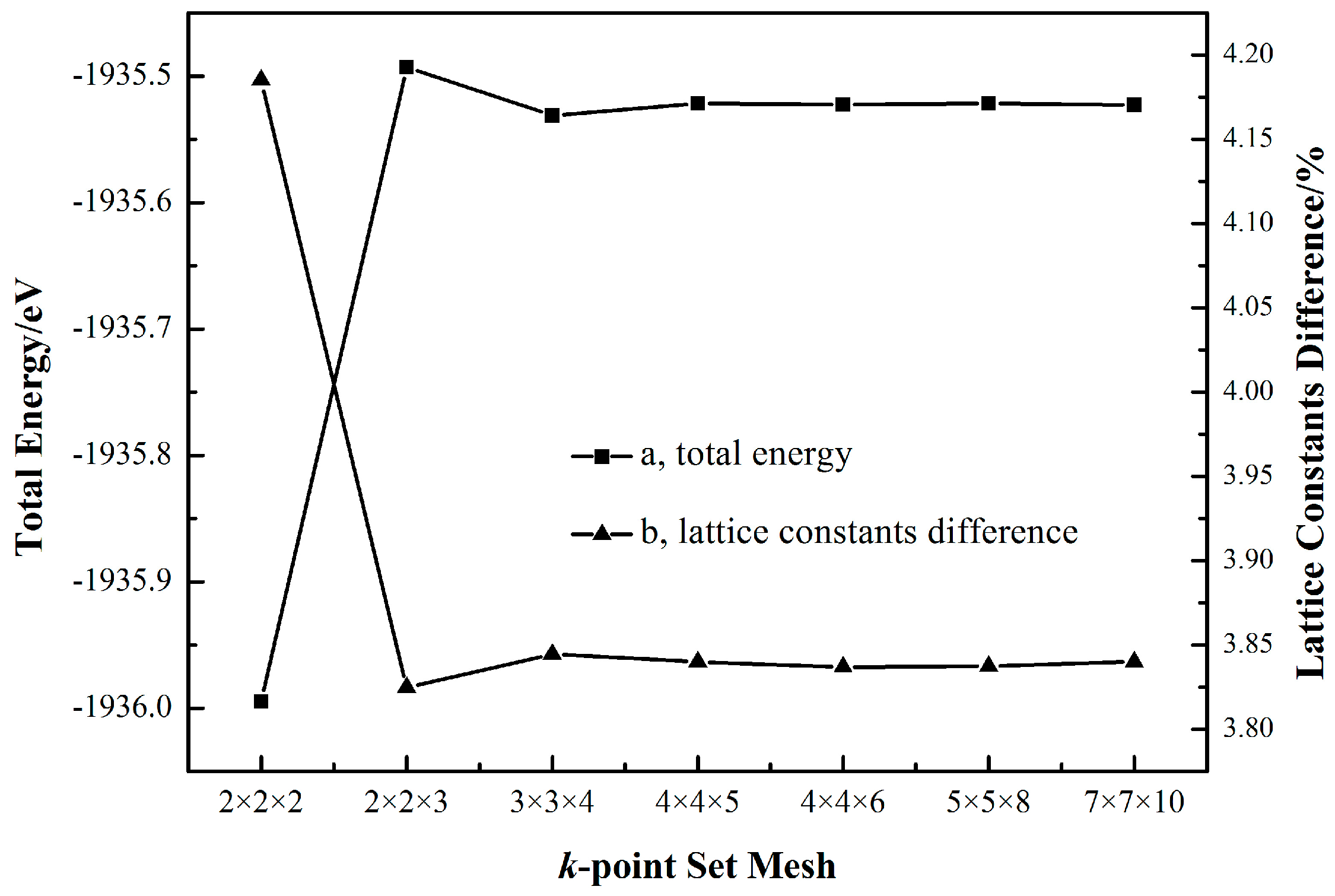
Figure 3.
The dependence of the total energy (a) and lattice constants difference (b) on the cutoff energy with a fixed k-point set of 3 × 3 × 4.
Figure 3.
The dependence of the total energy (a) and lattice constants difference (b) on the cutoff energy with a fixed k-point set of 3 × 3 × 4.
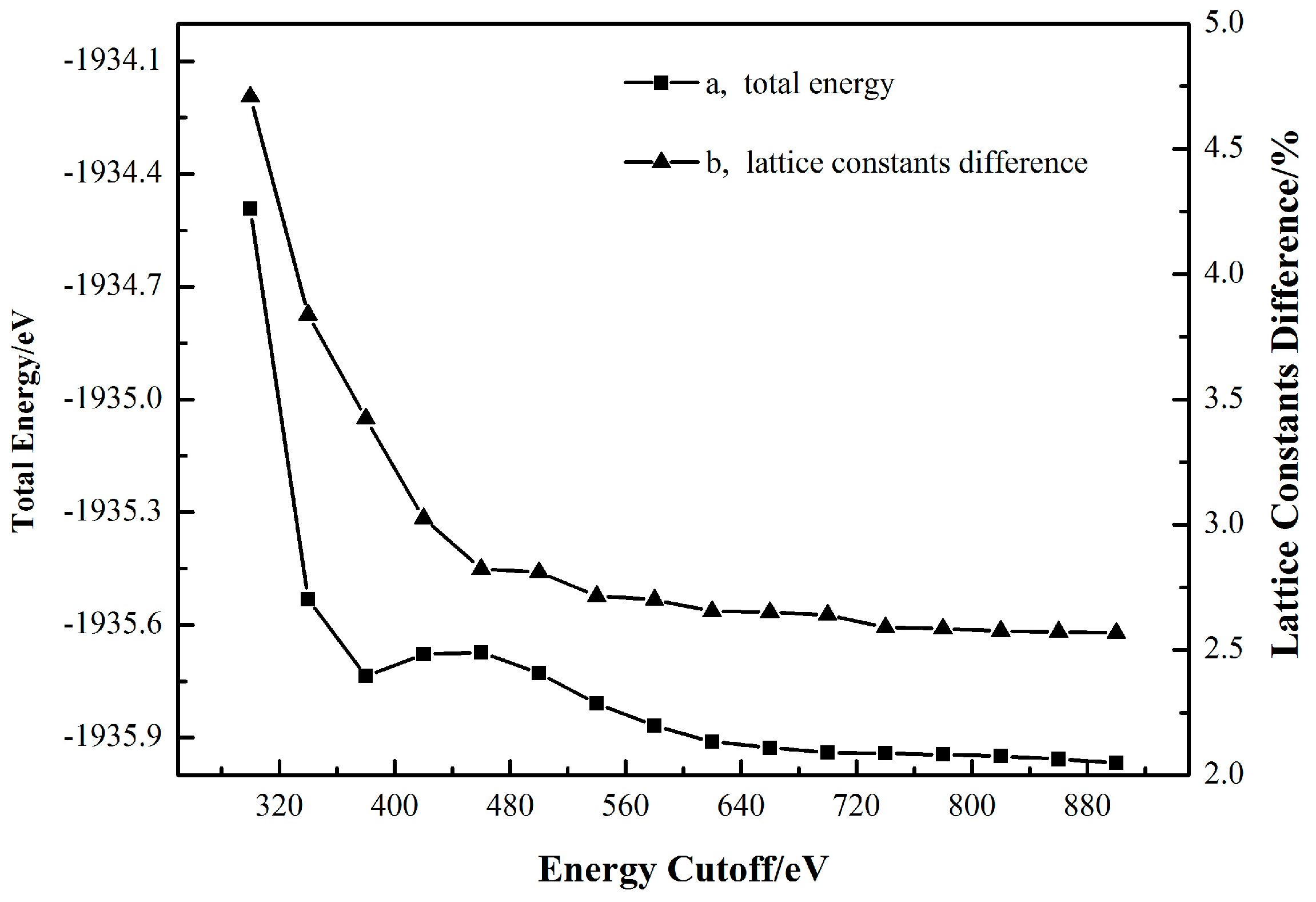
Figure 4.
The optimized cassiterite bulk unit-cell (colour codes: Gray—Sn, Red—O).

Figure 5.
Contrast of the simulated (a) X-ray diffraction spectra with the experimental (b) one.
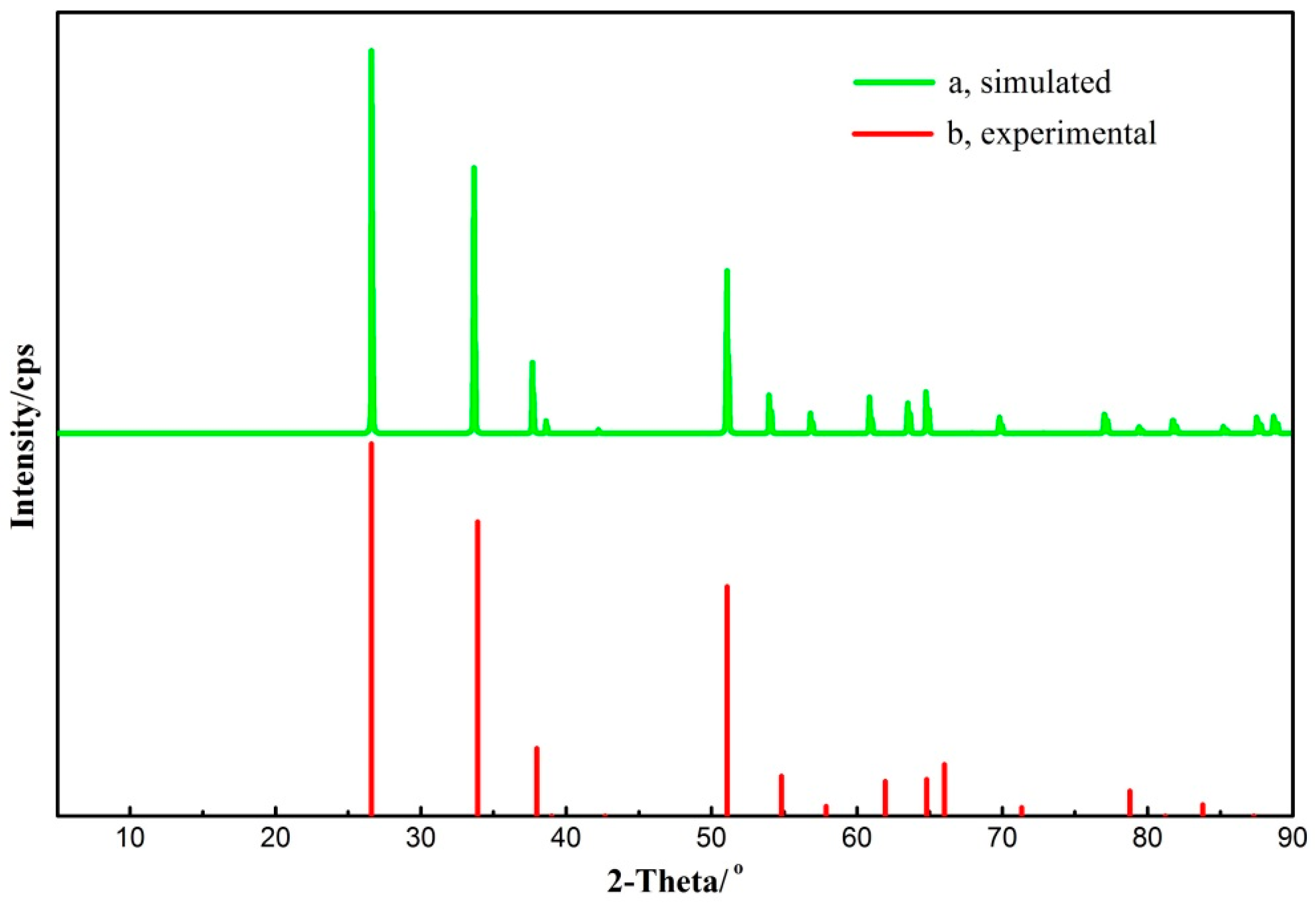
Figure 6.
Optimized cassiterite (110) and (100) surfaces (colour codes: Gray—Sn, Red—O, Yellow—surface Sn).
Figure 6.
Optimized cassiterite (110) and (100) surfaces (colour codes: Gray—Sn, Red—O, Yellow—surface Sn).
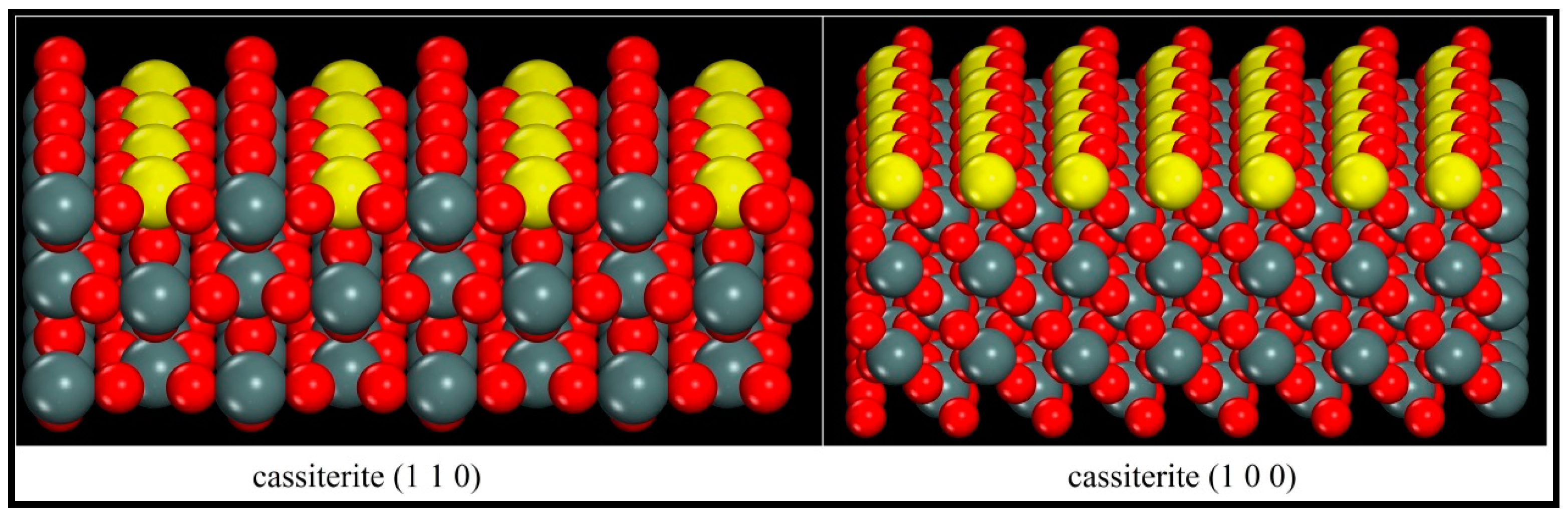
Figure 7.
Optimized adsorption configurations of oleate over cassiterite (110) and (100) surfaces (colour codes: Gray—C, White—H, Red—O, Dark Gray—Sn).
Figure 7.
Optimized adsorption configurations of oleate over cassiterite (110) and (100) surfaces (colour codes: Gray—C, White—H, Red—O, Dark Gray—Sn).
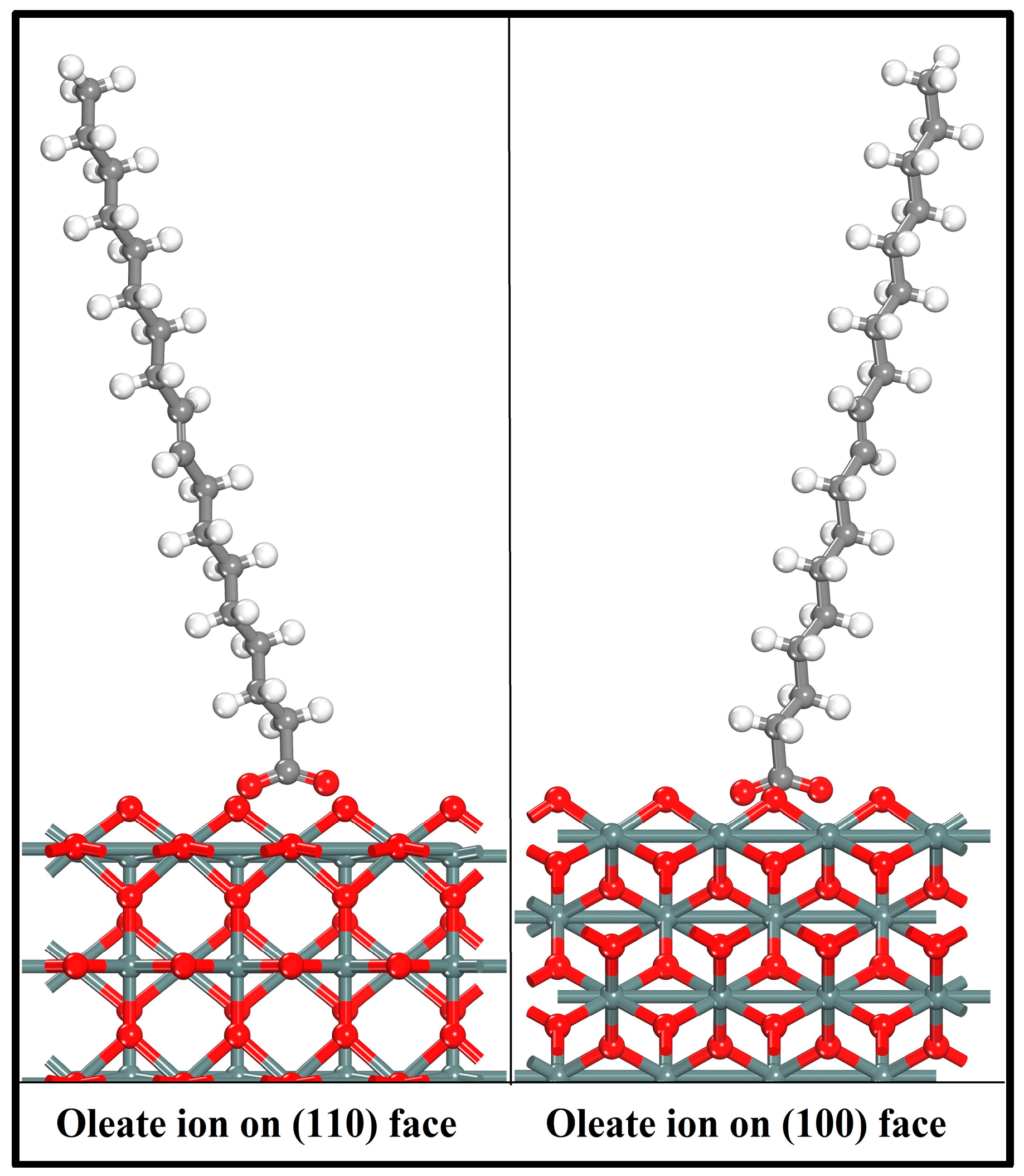
Figure 8.
The bonding details of an oleate anion on cassiterite (110) and (100) surfaces (colour codes: Gray—C, White—H, Red—O, Dark Gray—Sn).
Figure 8.
The bonding details of an oleate anion on cassiterite (110) and (100) surfaces (colour codes: Gray—C, White—H, Red—O, Dark Gray—Sn).
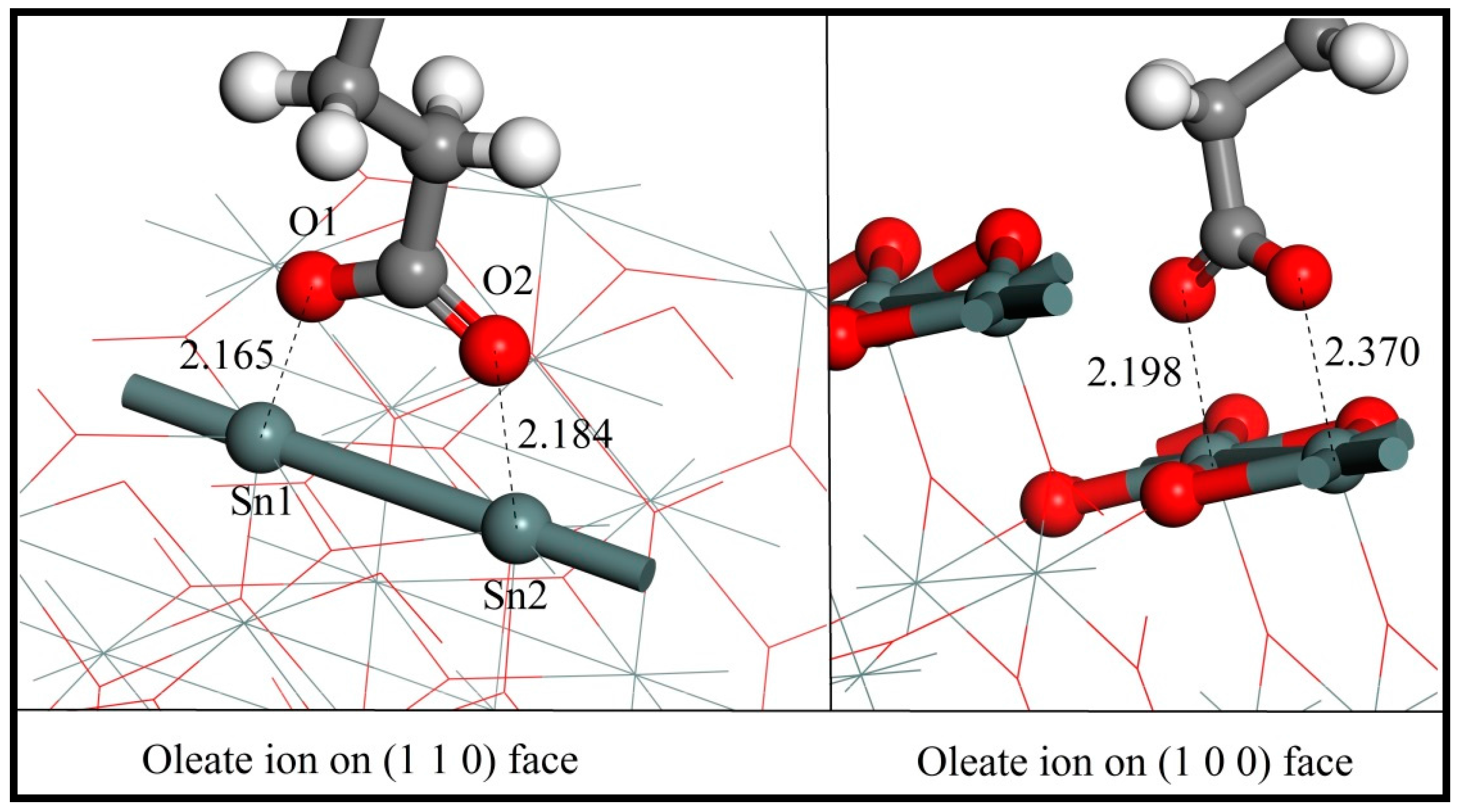
Figure 9.
Electron density difference of oleate–cassiterite adsorption configuration. Blue area represents losing electrons, and red contours indicate an electron density increase (colour codes: Gray—C, Red—O, Dark Gray—Sn).
Figure 9.
Electron density difference of oleate–cassiterite adsorption configuration. Blue area represents losing electrons, and red contours indicate an electron density increase (colour codes: Gray—C, Red—O, Dark Gray—Sn).
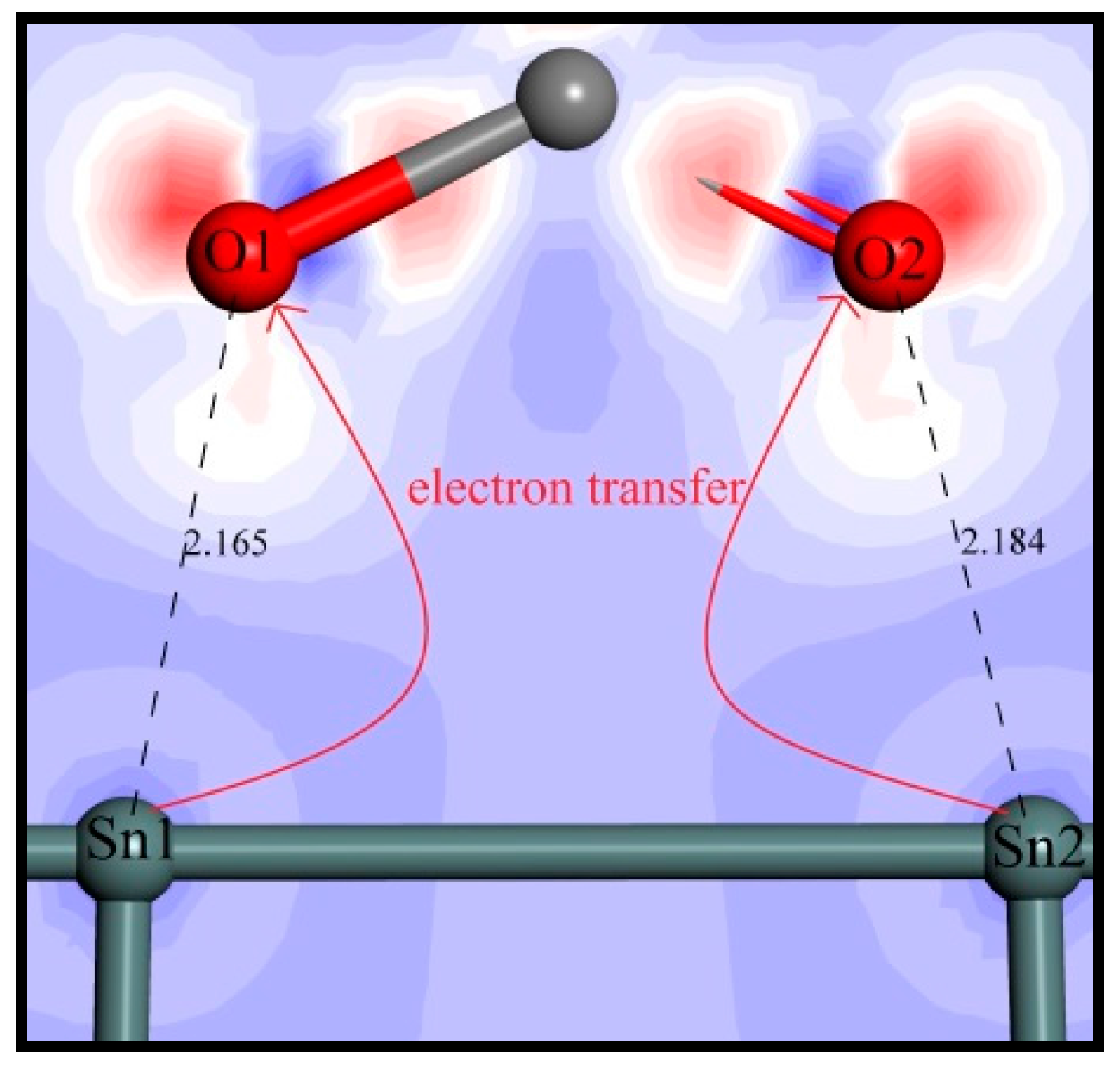
Figure 10.
Partial density of states (PDOS) of Sn1 (a) and O1 (b) atoms before and after adsorption, density of states of Sn1 and O1 atoms after adsorption (c), (the blue arrows illustrates the peak shift directions).
Figure 10.
Partial density of states (PDOS) of Sn1 (a) and O1 (b) atoms before and after adsorption, density of states of Sn1 and O1 atoms after adsorption (c), (the blue arrows illustrates the peak shift directions).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Convergence criteria for the simulation work.
| Items | Units | Values |
|---|---|---|
| Energy tolerance | eV/atom | 2 × 10−5 |
| Max. force | eV/Å | 0.05 |
| Max. stress | GPa | 0.1 |
| Max. displacement | Å | 0.002 |
| SCF tolerance | eV/atom | 1 × 10−6 |
Table 2.
Computed cassiterite bulk cell parameters from different GGA functionals compared with experimental values [28].
Table 2.
Computed cassiterite bulk cell parameters from different GGA functionals compared with experimental values [28].
| Data Sources | Functionals | a/Å | b/Å | c/Å | Total Difference Value/Å | Difference/% |
|---|---|---|---|---|---|---|
| Experimental | - | 4.737 | 4.737 | 3.186 | - | - |
| Computed | GGA-WC | 4.929 | 4.929 | 3.29 | 0.488 | 3.85 |
| GGA-PBE | 4.957 | 4.957 | 3.291 | 0.545 | 4.30 | |
| GGA-RPBE | 5.006 | 5.006 | 3.281 | 0.633 | 5.00 | |
| GGA-PW91 | 4.952 | 4.952 | 3.285 | 0.529 | 4.18 | |
| GGA-PBESOL | 4.927 | 4.927 | 3.294 | 0.489 | 3.86 |
Table 3.
Computed and experimental cassiterite lattice parameters.
| Parameters | a/Å | b/Å | c/Å | Sn–O Bond Length/Å | Sn–O–Sn Bond Angle/° |
|---|---|---|---|---|---|
| Experimental | 4.737 | 4.737 | 3.186 | 2.048–2.058 | 129.263 |
| Computed | 4.860 | 4.860 | 3.277 | 2.107–2.110 | 129.070 |
| Difference/% | 2.59 | 2.59 | 2.83 | 2.53–2.88 | 0.15 |
Table 4.
Surface energies of four low-index surfaces of SnO2 obtained by different researchers.
| Surface Index | Surface Energy, J/m2 | |||
|---|---|---|---|---|
| Ref [31] | Ref [32] | Ref [34] | This Work | |
| (110) | 1.40 | 1.04 | 1.01 | 1.03 |
| (100) | 1.65 | 1.14 | 0.92 | 1.04 |
| (001) | 2.36 | 1.72 | 1.74 | 1.83 |
| (101) | 1.54 | 1.33 | 1.28 | 1.38 |
Table 5.
(110) and (100) surface energy tests results as functions of slab thickness and vacuum depth.
Table 5.
(110) and (100) surface energy tests results as functions of slab thickness and vacuum depth.
| Layers of Sn2O4 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| (110) surface energy, J/m2 | 0.977 | 1.030 | 1.038 | 1.044 | 1.049 |
| (100) surface energy, J/m2 | 1.031 | 1.036 | 1.043 | 1.047 | 1.053 |
| Vacuum depth, Å | 10 | 12 | 14 | 16 | 18 |
| (110) surface energy, J/m2 | 1.045 | 1.037 | 1.036 | 1.038 | 1.036 |
| (100) surface energy, J/m2 | 1.042 | 1.042 | 1.041 | 1.038 | 1.040 |
Table 6.
Comparison of adsorption energies of different adsorbates on cassiterite (110) or (100) surfaces.
Table 6.
Comparison of adsorption energies of different adsorbates on cassiterite (110) or (100) surfaces.
| Adsorbate Complex | OL− on (110) | OL− on (100) | H2O on (110) | OH− on (110) |
|---|---|---|---|---|
| Interaction energy, kJ/mol | −244.70 | −195.29 | −76.74 | −115.22 |
Table 7.
Mulliken populations of involved atoms and bonds during the adsorption process of oleate ions on the SnO2 (110) surface.
Table 7.
Mulliken populations of involved atoms and bonds during the adsorption process of oleate ions on the SnO2 (110) surface.
| States | Before Adsorption | After Adsorption | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Atoms/bonds | O1 | O2 | Sn1 | Sn2 | O1 | O2 | Sn1 | Sn2 | O1–Sn1 bond | O2–Sn2 bond |
| Mulliken charge | −0.49 | −0.46 | 1.79 | 1.79 | −0.58 | −0.58 | 1.84 | 1.81 | 0.30 | 0.29 |
Table 8.
Electron density distributions of the pertinent atoms before and after the adsorption of oleate ions on the SnO2 (110) surface.
Table 8.
Electron density distributions of the pertinent atoms before and after the adsorption of oleate ions on the SnO2 (110) surface.
| States | Before Adsorption | After Adsorption | ||||||
|---|---|---|---|---|---|---|---|---|
| Atoms | O1 | O2 | Sn1 | Sn2 | O1 | O2 | Sn1 | Sn2 |
| s | 1.84 | 1.84 | 1.02 | 1.02 | 1.81 | 1.80 | 0.95 | 0.98 |
| p | 4.66 | 4.61 | 1.19 | 1.19 | 4.77 | 4.77 | 1.21 | 1.21 |
| total | 6.50 | 6.45 | 2.21 | 2.21 | 6.58 | 6.57 | 2.16 | 2.19 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, J.; Gong, G.; Han, Y.; Zhu, Y. New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study. Minerals 2017, 7, 236. https://doi.org/10.3390/min7120236
AMA Style
Liu J, Gong G, Han Y, Zhu Y. New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study. Minerals. 2017; 7(12):236. https://doi.org/10.3390/min7120236
Chicago/Turabian StyleLiu, Jie, Guichen Gong, Yuexin Han, and Yimin Zhu. 2017. "New Insights into the Adsorption of Oleate on Cassiterite: A DFT Study" Minerals 7, no. 12: 236. https://doi.org/10.3390/min7120236
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.