Cyanobacteria as Biocatalysts for Carbonate Mineralization

Abstract
:1. Introduction
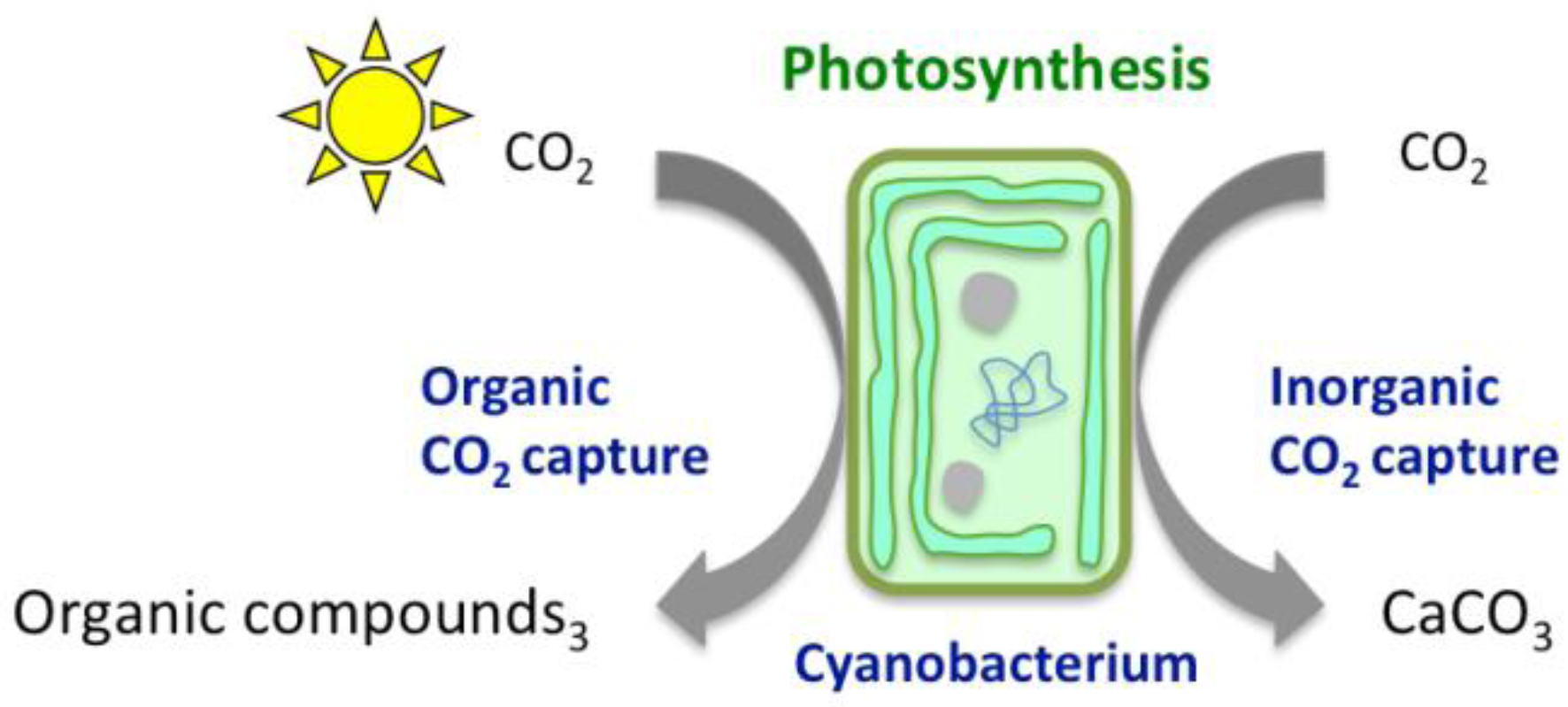
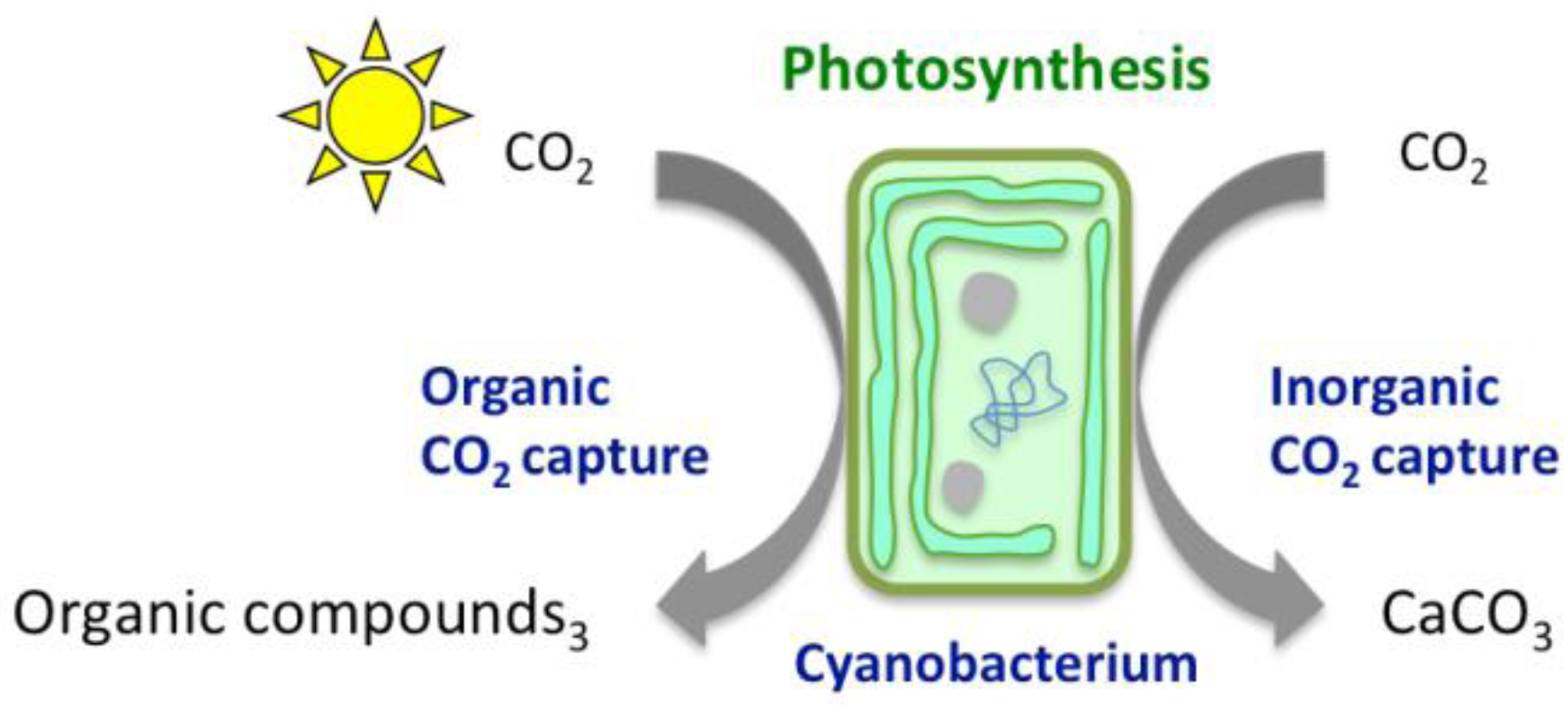
2. Chemical Reactions Governing CO2 Conversion into Carbonate Minerals
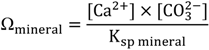
3. Cyanobacteria
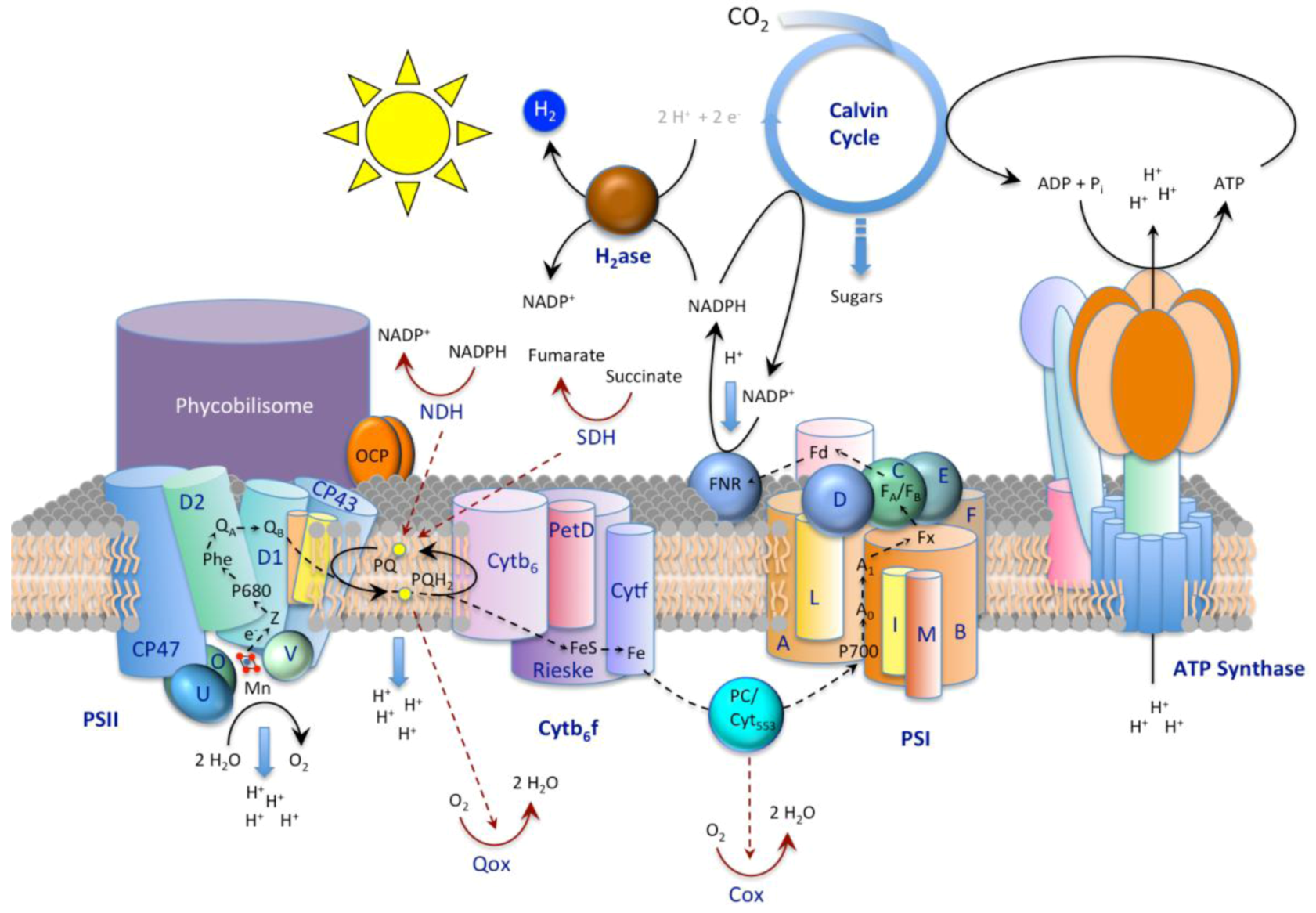
3.1. The CO2-concentrating Mechanism

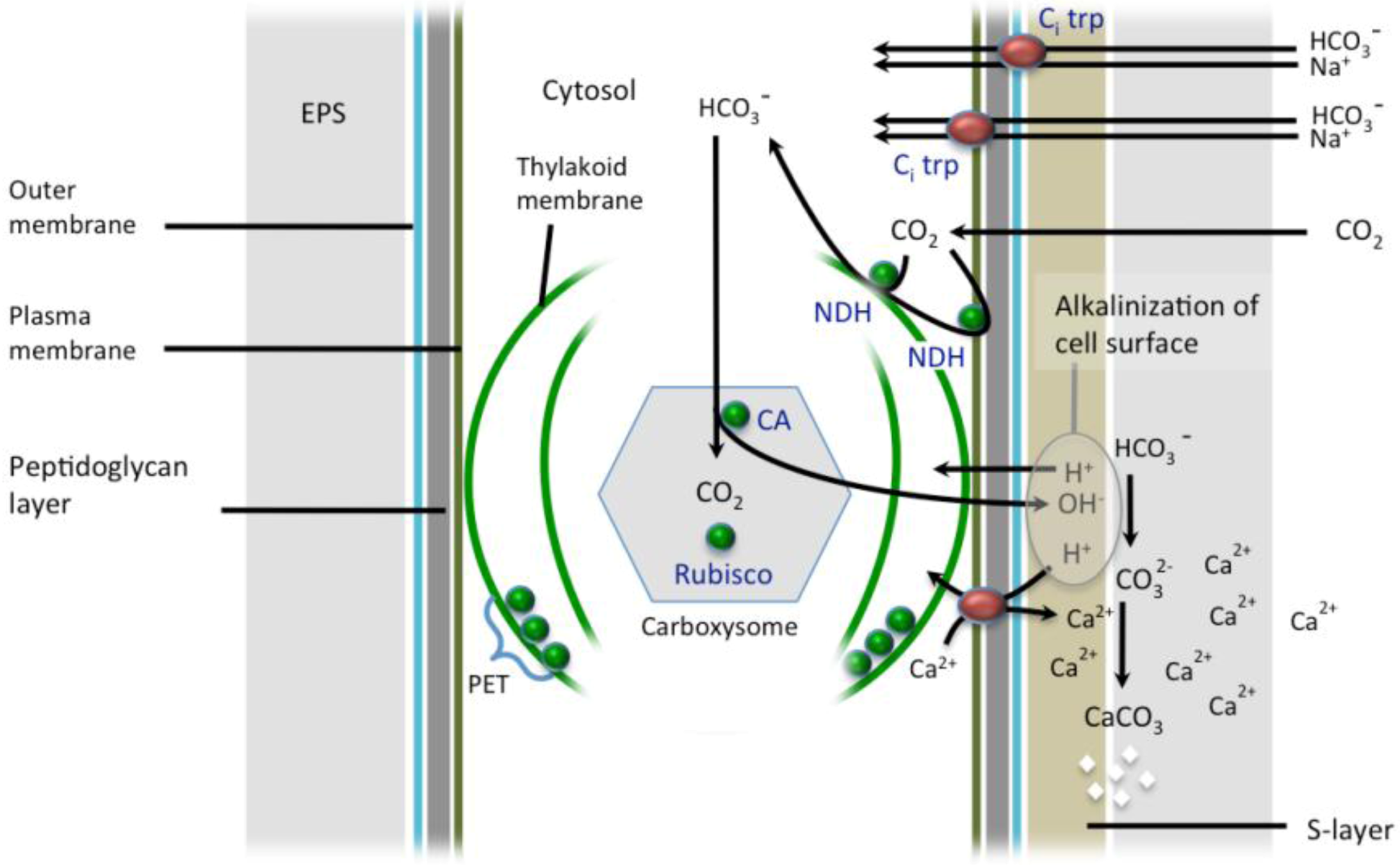
3.2. Exopolymeric Substances
3.3. S-layers
4. Cyanobacterial Carbonate Mineralization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Culture conditions | Mineral | Ref. |
| Synechococcus PCC 8806, Synechococcus PCC 8807 | Artificial seawater (ASNIII) | ND | [112] |
| Synechococcus, Planktothrix | Freshwater medium (BG11) | Cc | [113] |
| Synechococcus PCC 7942 | NaHCO3 + CaCl2 solutions | Cc | [114] |
| Synechococcus PCC 7942 | Freshwater medium (Z/10) | ACC, Ara, Cc | [115,116,117] |
| Trichodesmium erythraeum IMS101 | Artificial YBCII based seawater | Ara | [118] |
| Artificial cyanobacterial mat incl. Calothrix, Phormidium, and Pseudanabaena spp. | Seawater | Mg-Cc, Ara | [119,120] |
| Major species | Environmental conditions | Mineral | Ref. |
| Synechococcus GL24 | Meromictic lake | Ara | [121] |
| Pleurocapsa group | Soda lake | Ara | [122] |
| Phormidium spp. | Seawater lakes | Mg-Cc | [123] |
| Phormidium cf. crosbyanum, Phormidium sp. TK1,Schizothrix sp. | Seawater | ND | [124] |
| Coccoid and filamentous cyanobacteria incl. Rivularia type | Fossil and active tufa formations associated with freshwater springs, waterfall, and wet areas | Cc, Ara | [125] |
| Dichothrix spp. | Seawater | Cc, Ara | [9] |
| Rivularia haematites | Calcareous streams and freshwater lake | ND | [126] |
| Homoeothrix crustacean | Calcareous stream | Cc | [127] |
| Lyngbya sp. | Alkaline wetland | Dyp, Ara | [13] |
| Coccoid and filamentous cyanobacteria incl. Nostocales, Chroococcales, Oscillatoriales, and Pleurocapsales spp. | Alkaline brackish caldera lake | hMag, Ara | [128] |
| Diatoms and filamentoud cyanobacteria incl. Lyngbya and Gloeocapsa spp. | Alkaline lake | hMag | [129,130] |
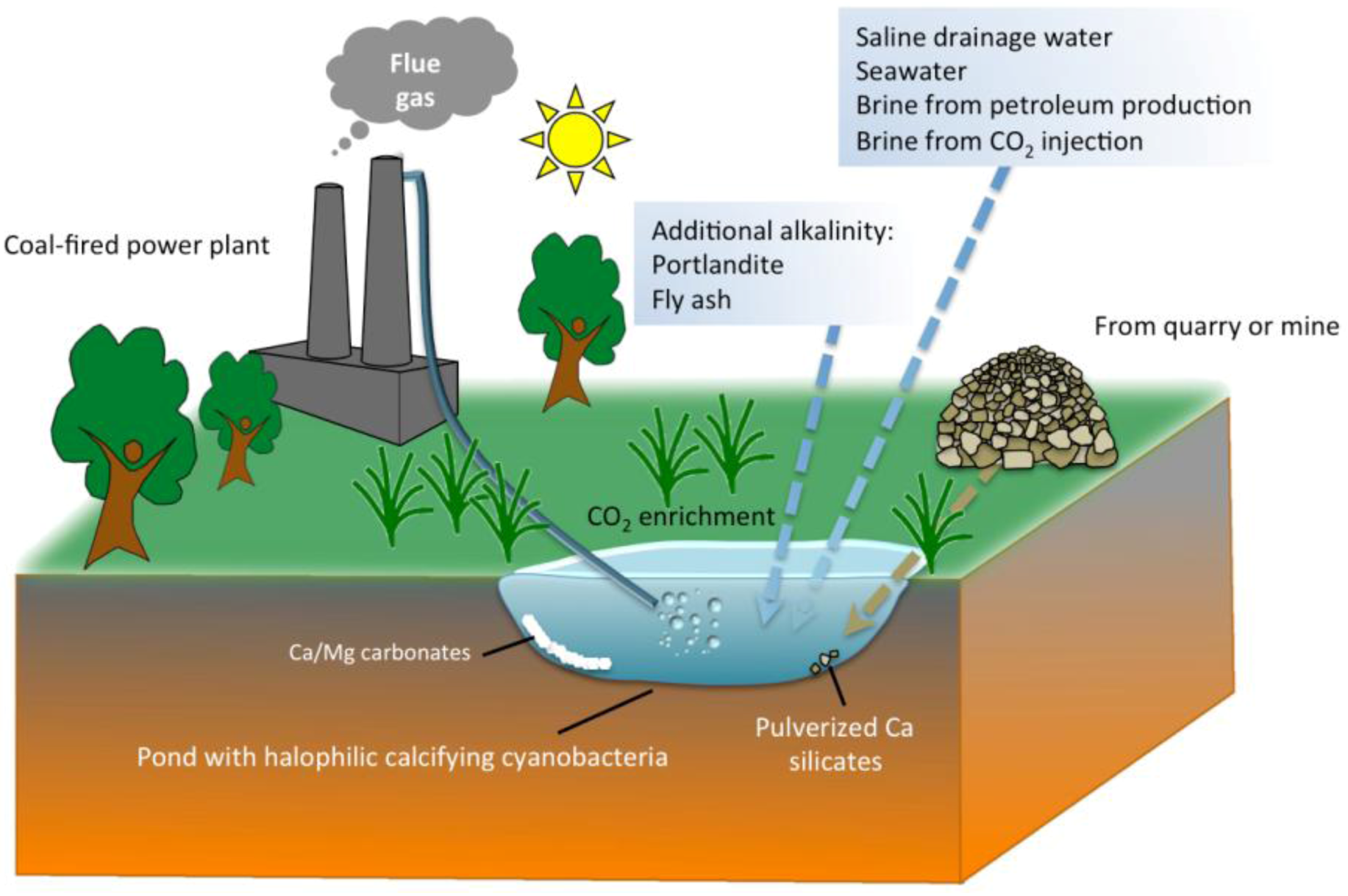
5. Practical Implications of Cyanobacterial Carbonate Mineralization
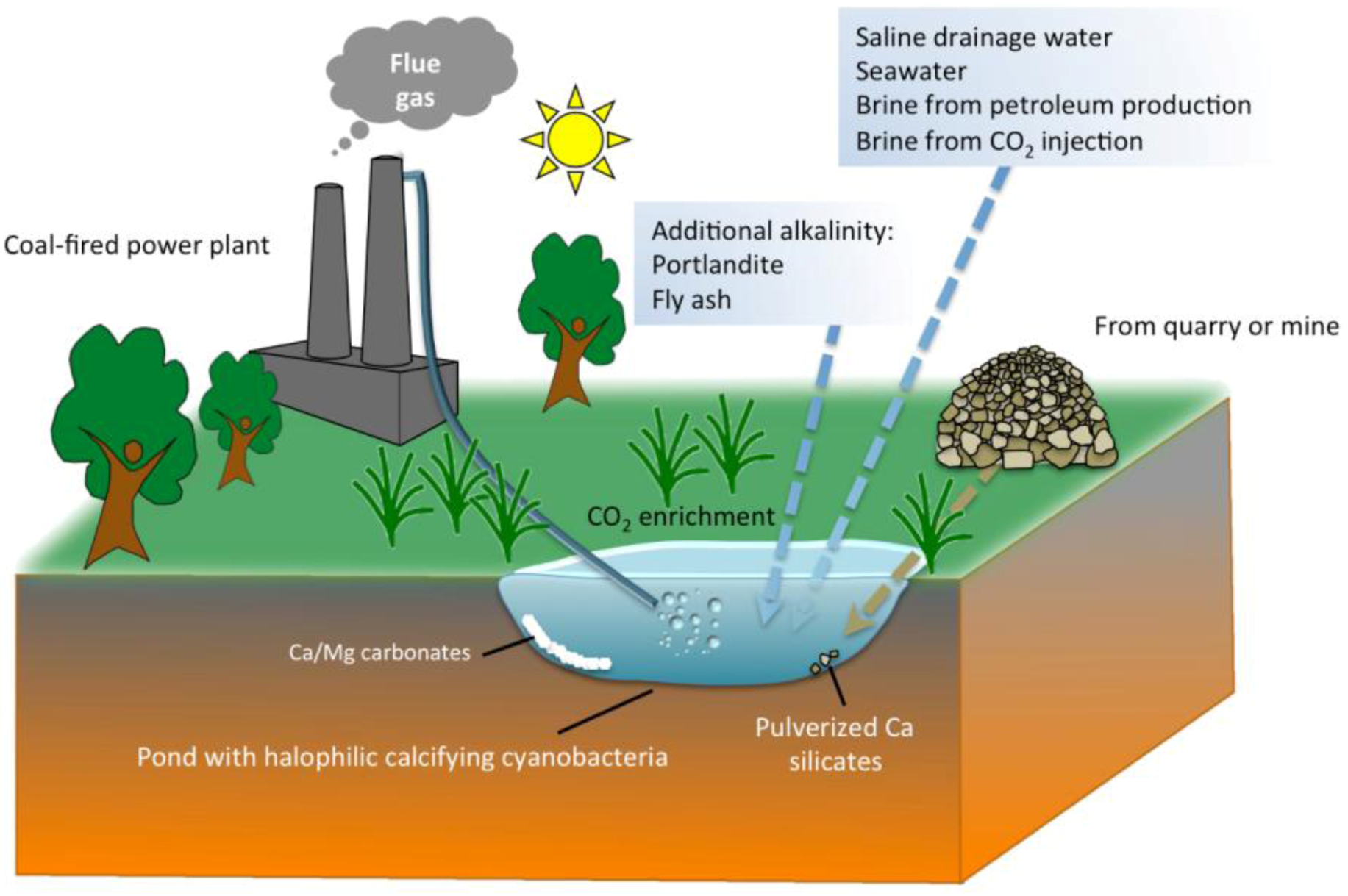
6. Conclusions
Acknowledgements
References
- Merz-Preiss, M. Calcification in cyanobacteria. In Microbial Sediments; Riding, R.E., Awramik, S.M., Eds.; Springer-Verlag: Berlin, Germany, 2000; pp. 50–56. [Google Scholar]
- Konhauser, K. Introduction to Geomicrobiology; Blackwell Publishing: Malden, MA, USA, 2007; pp. 160–166. [Google Scholar]
- Riding, R. Cyanophyte calcification and changes in ocean chemistry. Nature 1982, 299, 814–815. [Google Scholar] [CrossRef]
- Riding, R. Microbial carbonates: The geological record of calcified bacterial-algal mats and biofilms. Sedimentology 2000, 47, 179–214. [Google Scholar] [CrossRef]
- Riding, R. Cyanobacterial calcification, carbon dioxide concentrating mechanisms, and Proterozoic-Cambrian changes in atmospheric compositio. Geobiology 2006, 4, 299–316. [Google Scholar] [CrossRef]
- Riding, R. An atmospheric stimulus for cyanobacterial-bioinduced calcification ca. 350 million years ago? Palaios 2009, 24, 685–696. [Google Scholar] [CrossRef]
- Aloisi, G. The calcium carbonate saturation state in cyanobacterial mats throughout Earth’s history. Geochim. Cosmochim. Acta 2008, 72, 6037–6060. [Google Scholar] [CrossRef]
- Altermann, W.; Kazmierczak, J.; Oren, A.; Wright, D.T. Cyanobacterial calcification and its rock-building potential during 3.5 billion years of Earth history. Geobiology 2006, 4, 147–166. [Google Scholar]
- Planavsky, N.; Reid, R.P.; Lyons, T.W.; Myshrall, K.L.; Visscher, P.T. Formation and diagenesis of modern marine calcified cyanobacteria. Geobiology 2009, 7, 1–11. [Google Scholar] [CrossRef]
- Pentecost, A. Cyanobacteria associated with hot spring travertines. Can. J. Earth Sci. 2003, 40, 1447–1457. [Google Scholar] [CrossRef]
- Kremer, B.; Kazmierczak, J.; Stal, L.J. Calcium carbonate precipitation in cyanobacterial mats from sandy tidal flats of the North Sea. Geobiology 2008, 6, 46–56. [Google Scholar]
- Arp, G.; Reimer, A.; Reitner, J. Photosynthesis-induced biofilm calcification and calcium concentrations in Phanerozoic oceans. Science 2001, 292, 1701–1704. [Google Scholar]
- Power, I.M.; Wilson, S.A.; Thom, J.M.; Dipple, G.M.; Southam, G. Biologically induced mineralization of dypingite by cyanobacteria from an alkaline wetland near Atlin, British Columbia, Canada. Geochem. Trans. 2007, 8. [Google Scholar] [CrossRef]
- Gautret, P.; Trichet, J. Automicrites in modern cyanobacterial stromatolitic deposits of Rangiroa, Tuamotu Archipelago, French Polynesia: Biochemical parameters underlaying their formation. Sediment. Geol. 2005, 178, 55–73. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.S.; Green, S.J.; Ahrendt, S.R.; Golubic, S.; Reid, R.P.; Hetherington, K.L.; Bebout, L. Molecular and morphological characterization of cyanobacterial diversity in the stromatolites of Highborne Cay, Bahamas. ISME J. 2009, 3, 573–587. [Google Scholar] [CrossRef]
- Ludwig, R.; Al-Horani, F.A.; de Beer, D.; Jonkers, H.M. Photosynthesis-controlled calcification in a hypersaline microbial mat. Limnol. Oceanogr. 2005, 50, 1836–1843. [Google Scholar] [CrossRef]
- Strong, A.E.; Eadie, B.J. Satellite observations of calcium carbonate precipitations in the Great Lakes. Limnol. Oceanogr. 1978, 23, 877–887. [Google Scholar] [CrossRef]
- Thompson, J.B.; Schultze-Lam, S.; Beveridge, T.J.; DesMarais, D.J. Whiting events: Biogenic origin due to the photosynthetic activity of cyanobacterial picoplankton. Limnol. Oceanogr. 1997, 42, 133–141. [Google Scholar] [CrossRef]
- Yates, K.K.; Robbins, L.L. Microbial lime-mud production and its relation to climate change. In Geological Perspectives of Global Climate Change; Lee, D.G., Harrison, W.E., Hanson, B.M., Eds.; AAPG Division of Environmental Geosciences: Tulsa, OK, USA, 2001; pp. 267–283. [Google Scholar]
- Dittrich, M.; Obst, M. Are picoplankton responsible for calcite precipitation in lakes? AMBIO 2004, 33, 559–564. [Google Scholar]
- Zuddas, P.; Mucci, A. Kinetics of calcite precipitation from seawater: II. The influence of the ionic strength. Geochim. Cosmochim. Acta 1998, 62, 757–766. [Google Scholar] [CrossRef]
- Frankignoulle, M.; Gattuso, J.-P. Air-sea CO2 exchange in coastal ecosystems. In Interactions of C, N, P, and S Biogeochemical Cycles and Global Change; Wollast, R., Mackenzie, F.T., Eds.; Springer: Berlin, Germany, 1993; pp. 233–248. [Google Scholar]
- Jansson, C.; Wullschleger, S.D.; Udaya, C.K.; Tuskan, G.A. Phytosequestration: Carbon biosequestration by plants and the prospects of genetic engineering. BioScience 2010, 60, 685–696. [Google Scholar] [CrossRef]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Zeebe, R.E.; Wolf-Gladrow, D. CO2 in Seawater: Equilibrium, Kinetics, Isotopes; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Dove, P.M.; Hochella, F., Jr. Calcite precipitation mechanisms and inhibition by orthophosphate: In situ observations by Scanning Force Microscopy. Geochim. Cosmochim. Acta 1993, 57, 705–714. [Google Scholar] [CrossRef]
- Chen, T.; Neville, A.; Yuan, M. Assessing the effect of Mg2+ on CaCO3 scale formation-bulk precipitation and surface deposition. J Crystal Growth 2005, 275, e1341–e1347. [Google Scholar] [CrossRef]
- Land, L.S. Failure to precipitate dolomite at 25 °C from dilute solution sespite 1000-fold oversaturation after 32 years. Aquat. Geochem. 1998, 4, 361–368. [Google Scholar] [CrossRef]
- Morse, J.W.; Mackenzie, F.T. Geochemistry of Sedimentary Carbonates; Elsevier: New York, NY, USA, 1990. [Google Scholar]
- Kastner, M. Control of dolomite formation. Nature 1984, 410–411. [Google Scholar] [CrossRef]
- Robbins, L.L.; Blackwelder, P.L. Biochemical and ultrastructural evidence for the origin of whitings: A biologically induced calcium carbonate precipitation mechanism. Geology 1992, 20, 464–468. [Google Scholar] [CrossRef]
- Wright, D.; Oren, A. Nonphotosynthetic bacteria and the formation of carbonates and evaporites through time. Geomicrobiol. J. 2005, 22, 27–53. [Google Scholar] [CrossRef]
- Chafetz, H.S. Marine peloids-A product of bacterially induced precipitation of calcite. J. Sediment. Pet. 1986, 56, 812–817. [Google Scholar]
- Castanier, S.; Maurin, A.; Perthuisot, J.P. Experimental bacterial production of spheroidal, fibro-radial carbonate bodies-Discussions about the definition of ooids. Bull. Soc. Geol. Fr. 1989, 5, 589–595. [Google Scholar]
- Reitner, J.; Arp, G.; Thiel, V.; Gautret, P.; Galling, U.; Michaelis, W. Organic matter in Great Salt Lake ooids (Utah, USA)-First approach to a formation via organic matrices. Facies 1997, 36, 210–219. [Google Scholar]
- Verrecchia, E.P.; Freytet, P.; Verrecchia, K.E.; Dumont, J.L. Spherulites incalcrete laminar crusts: Biogenic CaCO3 precipitation as a major contributor to crust formation. J. Sediment. Res. Sec. A Sediment. Pet. Process. 1995, 65, 690–700. [Google Scholar]
- Freytet, P.; Verrecchia, E.P. Freshwater organisms that build stromatolites: A synopsis of biocrystallization by prokaryotic and eukaryotic algae. Sedimentology 1998, 45, 535–563. [Google Scholar] [CrossRef]
- Freytet, P.; Verrecchia, E.P. Lacustrine and palustrine carbonate petrography: An overview. J. Paleolimnol. 2002, 27, 221–237. [Google Scholar] [CrossRef]
- McConnaughey, T. 13C and 18O isotopic disequilibrium in biological carbonates: I. Patterns. Geochim. Cosmochim. Acta 1989, 53, 151–162. [Google Scholar] [CrossRef]
- Craig, H. The geochemistry of the stable carbon isotopes. Geochim. Cosmochim. Acta 1953, 3, 53–92. [Google Scholar] [CrossRef]
- Christeller, J.T.; Laing, W.A.; Troughton, J.H. Isotope discrimination by Ribulose 1,5-Diphosphate Carboxylase. Plant Physiol. 1976, 57, 580–582. [Google Scholar] [CrossRef]
- Dove, P.M.; de Yoreo, J.J.; Weiner, S. Biomineralization; Mineralogical Society of America: Washington, DC, USA, 2003. [Google Scholar]
- Dupraz, C.; Reid, R.P.; Braissant, O.; Decho, A.W.; Norman, R.S.; Visscher, P.T. Processes of carbonate precipitation in modern microbial mats. Earth Sci. Rev. 2009, 96, 141–162. [Google Scholar] [CrossRef]
- Falcon, L.I.; Magallon, S.; Castillo, A. Dating the cyanobacterial ancestor of the chloroplast. ISME J. 2010, 4, 777–783. [Google Scholar] [CrossRef]
- Walker, J.C.G.; Klein, J.; Schopf, J.W.; Stevenson, D.J.; Walker, M.R. Environmental evolution of the Archean-Early Proterozoic Earth. In Earth’s Earliest Biosphere: Its Origin and Evolution; Schopf, J.W., Ed.; Princeton Univesity Press: Princeton, NJ, USA, 1983; pp. 260–290. [Google Scholar]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef]
- Whitton, B.A.; Potts, M. The Ecology of Cyanobacteria: their Diversity in Time and Space; Kluwer Academic: Norwell, MA, USA, 2000. [Google Scholar]
- Partensky, F.; Hess, W.R.; Vaulot, D. Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microb. Mol. Biol. Rev. 1999, 63, 106–127. [Google Scholar]
- Kleiner, D. Fixation of atmospheric nitrogen by microorganisms. Angew. Chem. Int. Ed. Engl. 1975, 14, 80–86. [Google Scholar] [CrossRef]
- Stal, L.J. Cyanobacterial mats and stromatolites. In The Ecology of Cyanobacteria: Their Diversity in Time and Space; Whitton, B.A., Potts, M., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; pp. 61–120. [Google Scholar]
- Awramik, S.M. Precambrian columnar stromatolite diversity: Reflection of metazoan appearance. Science 1971, 174, 825–827. [Google Scholar]
- Vincent, W.F. Effects of climate change on lakes. In Encyclopedia of Inland Waters; Likens, G.E., Ed.; Elsevier: Oxford, UK, 2009; Volume 3, pp. 55–60. [Google Scholar]
- Sutherland, I.W. Exopolysaccharides in biofilms, flocs and related structures. Water Sci. Technol. 2001, 43, 77–86. [Google Scholar]
- Nicolaus, B.; Panico, A.; Lama, L.; Romano, I.; Manca, M.C.; de Giulio, A.; Gambacorta, A. Chemical composition and production of exopolysaccharides from representative members of heterocystous and non-heterocystous cyanobacteria. Phytochemistry 1999, 52, 639–647. [Google Scholar] [CrossRef]
- Raven, J.A.; Giordano, M.; Beardall, J.; Maberly, S.C. Algal evolution in relation to atmospheric CO2: Carboxylases, carbon-concentrating mechanisms and carbon oxidation cycles. Philos. Trans. B 2012, 367, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Giordano, M.; Beardall, J.; Raven, J.A. CO2 concentrating mechanisms in algae: Mechanisms, environmental modulation, and evolution. Annu. Rev. Plant Biol. 2005, 56, 99–131. [Google Scholar] [CrossRef]
- Price, G.D.; Howitt, S.M. The cyanobacterial bicarbonate transporter BicA: Its physiological role and the implications of structural similarities with human SLC26 transporters. Biochem. Cell Biol. 2011, 89, 178–188. [Google Scholar] [CrossRef]
- Kaplan, A.; Reinhold, L. CO2 concentrating mechanisms in photosynthetic microorganisms. Annu. Rev. Plant Phys. 1999, 50, 539–570. [Google Scholar] [CrossRef]
- Badger, M.R.; Price, G.D. CO2 concentrating mechanisms in cyanobacteria: Molecular components, their diversity and evolution. J. Exp. Bot. 2003, 54, 609–622. [Google Scholar] [CrossRef]
- Price, G.D.; Badger, M.R.; Woodger, F.J.; Long, B.M. Advances in understanding the cyanobacterial CO2-concentrating-mechanism (CCM): Functional components, Ci transporters, diversity, genetic regulation and prospects for engineering into plans. J. Exp. Bot. 2008, 59, 1441–1461. [Google Scholar]
- Jansson, C.; Northen, T. Calcifying cyanobacteria-The potential of biomineralization for carbon capture and storage. Curr. Opin. Biotechnol. 2010, 21, 365–371. [Google Scholar] [CrossRef]
- Merz, M. The biology of carbonate precipitation by cyanobacteria. Facies 1992, 26, 81–101. [Google Scholar] [CrossRef]
- Kim, Y.; Oh, S.; Kim, S.H. Released exopolysaccharide (r-EPS) produced from probiotic bacteria reduce biofilm formation of enterohemorrhagic Escherichia coli O157:H7. Biochem. Biophys. Res. Commun. 2009, 379, 324–329. [Google Scholar]
- Richert, L.; Golubic, S.; Le Guedes, R.; Ratiskol, J.; Payri, C.; Guezennec, J. Characterization of exopolysaccharides produced by cyanobacteria isolated from polynesian microbial mats. Curr. Microbiol. 2005, 51, 379–384. [Google Scholar]
- Monsan, P.; Bozonnet, S.; Albenne, C.; Joucla, G.; Willemot, R.M.; Remaud-Simeon, M. Homopolysaccharides from lactic acid bacteria. Int. Dairy J. 2001, 11, 675–685. [Google Scholar] [CrossRef]
- Pereira, S.; Zille, A.; Micheletti, E.; Moradas-Ferreira, P.; de Philippis, R.; Tamagnini, P. Complexity of cyanobacterial exopolysaccharides: Composition, structures, inducing factors and putative genes involved in their biosynthesis and assembly. FEMS Microbiol. Rev. 2009, 33, 917–941. [Google Scholar] [CrossRef]
- De Philippis, R.; Sili, C.; Paperi, R.; Vincenzini, M. Exopolysaccharide-producing cyanobacteria and their possible exploitation: A review. J. Appl. Phycol. 2001, 13, 293–299. [Google Scholar] [CrossRef]
- Tamaru, Y.; Takani, Y.; Yoshida, T.; Sakamoto, T. Crucial role of extracellular polysaccharides in desiccation and freezing tolerance in the terrestrial cyanobacterium Nostoc commune. Appl. Environ. Microb. 2005, 71, 7327–7333. [Google Scholar]
- De Philippis, R.; Faraloni, C.; Sili, C.; Vincenzini, M. Algal biocenosis in the benthic mucilaginous aggregates of the Tyrrhenian sea, with emphasis on the exopolysaccharide-producing microalgal community. Arch. Hydrobiol. Suppl. 2003, 148, 487–498. [Google Scholar]
- De Philippis, R.; Vincenzini, M. Exocellular polysaccharides from cyanobacteria and their possible applications. Fems. Microbiol. Rev. 1998, 22, 151–175. [Google Scholar] [CrossRef]
- Sharma, N.; Tiwari, S.P.; Tripathi, K.; Rai, A. Sustainability and cyanobacteria (blue-green algae): Facts and challenges. J. Appl. Phycol. 2011, 23, 1059–1087. [Google Scholar] [CrossRef]
- Li, J.Y.; Luan, Z.K.; Zhu, B.X.; Gong, X.Y.; Peng, D.C. Effects of colloidal organic matter on nitrification and composition of extracellular polymeric substances in biofilms. J. Chem. Technol. Biotechnol. 2002, 77, 1333–1339. [Google Scholar] [CrossRef]
- Baptista, M.S.; Vasconcelos, M.T. Cyanobacteria metal interactions: Requirements, toxicity, and ecological implication. Crit. Rev. Microbiol. 2006, 32, 127–137. [Google Scholar] [CrossRef]
- Colica, G.; Mecarozzi, P.C.; de Philippis, R. Treatment of Cr(VI)-containing wastewaters with exopolysaccharide-producing cyanobacteria in pilot flow through and batch systems. Appl. Microbiol. Biotechnol. 2010, 87, 1953–1961. [Google Scholar] [CrossRef]
- Sharma, M.; Kaushik, A.; Somvir; Bala, K.; Kamra, A. Sequestration of chromium by exopolysaccharides of Nostoc and Gloeocapsa from dilute aqueous solutions. J. Hazard. Mater. 2008, 157, 315–318. [Google Scholar] [CrossRef]
- Kiran, B.; Kaushik, A. Chromium binding capacity of Lyngbya putealis exopolysaccharides. Biochem. Eng. J. 2008, 38, 47–54. [Google Scholar] [CrossRef]
- Micheletti, E.; Colica, G.; Viti, C.; Tamagnini, P.; de Philippis, R. Selectivity in the heavy metal removal by exopolysaccharide-producing cyanobacteria. J. Appl. Microbiol. 2008, 105, 88–94. [Google Scholar] [CrossRef]
- Ozturk, S.; Aslim, B. Relationship between chromium (VI) resistance and extracellular polymeric substances (EPS) concentration by some cyanobacterial isolates. Environ. Sci. Pollut. Res. 2008, 15, 478–480. [Google Scholar] [CrossRef]
- Ozturk, S.; Aslim, B.; Suludere, Z. Evaluation of chromium (VI) removal behaviour by two isolates of Synechocystis sp. in terms of exopolysaccharide (EPS) production and monomer composition. Bioresour. Technol. 2009, 100, 5588–5593. [Google Scholar] [CrossRef]
- Ozturk, S.; Aslim, B.; Suludere, Z. Cadmium (II) sequestration characteristics by two isolates of Synechocystis sp. in terms of exopolysaccharide (EPS) production and monomer composition. Bioresour. Technol. 2010, 101, 9742–9748. [Google Scholar] [CrossRef]
- Pereira, S.; Micheletti, E.; Zille, A.; Santos, A.; Moradas-Ferreira, P.; Tamagnini, P.; de Philippis, R. Using extracellular polymeric substances (EPS)-producing cyanobacteria for the bioremediation of heavy metals: Do cations compete for the EPS functional groups and also accumulate inside the cell? Microbiology 2011, 157, 451–458. [Google Scholar] [CrossRef]
- Ruangsomboon, S.; Chidthaisong, A.; Bunnag, B.; Inthorn, D.; Harvey, N.W. Lead (Pb2+) adsorption characteristics and sugar composition of capsular polysaccharides of cyanobacterium Calothrix marchica. Songklanakarin J. Sci. Technol. 2007, 29, 529–541. [Google Scholar]
- Yee, N.; Benning, L.G.; Phoenix, V.R.; Ferris, F.G. Characterization of metal-cyanobacteria sorption reactions: A combined macroscopic and infrared spectroscopic investigation. Environ. Sci. Technol. 2004, 38, 775–782. [Google Scholar]
- Arp, G.; Reimer, A.; Reitner, J. Calcification in cyanobacterial biofilms of alkaline salt lakes. Eur. J. Phycol. 1999, 34, 393–403. [Google Scholar] [CrossRef]
- Défarge, C. Organomineralization. In Encyclopedia of Geobiology, 1st; Reitner, J., Thiel, V., Eds.; Springer: Dordrecht, The Netherlands, 2011; pp. 697–701. [Google Scholar] [Green Version]
- Trichet, J.; Défarge, C. Non-biologically supported organomineralization. Inst. Oceanogr. Bull. 1995, 14, 203–236. [Google Scholar]
- Dupraz, C.; Visscher, P.T.; Baumgartner, L.K.; Reid, R.P. Microbe-mineral interactions: Early carbonate precipitation in a hypersaline lake (Eleuthera Island, Bahamas). Sedimentology 2004, 51, 745–765. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Decho, A.W. A laboratory investigation of cyanobacterial extracellular polymeric secretions (EPS) in influencing CaCO3 polymorphism. J. Cryst. Growth 2002, 240, 230–235. [Google Scholar] [CrossRef]
- Houwink, A.L. A macromolecular mono-layer in the cell wall of Spirillum spec. Biochim. Biophys. Acta 1953, 10, 360–366. [Google Scholar] [CrossRef]
- Sleytr, U.B.; Messner, P.; Pum, D.; Sára, M. Crystalline bacterial cell surface layers. Mol. Microbiol. 1993, 10, 911–916. [Google Scholar] [CrossRef]
- Messner, P.; Sleytr, U.B. Crystalline bacterial cell-surface layers. In Advances in Microbial Physiology; Academic Press: San Diego, CA, USA, 1992. [Google Scholar]
- Pavkov-Keller, T.; Howorka, S.; Keller, W. The structure of bacterial S-layer proteins. Prog. Mol. Biol. Transl. Sci. 2011, 103, 73–130. [Google Scholar] [CrossRef]
- Sleytr, U.B. Heterologous reattachment of regular arrays of glycoproteins on bacterial surfaces. Nature 1975, 257, 400–402. [Google Scholar] [CrossRef]
- Sleytr, U.B.; Plohberger, R. The dynamic process of assembly of two-dimentional arrays of macromolecules on bacterial cell walls. In Electron Microscopy at Molecular Dimensions; Baumeister, W., Vogell, W., Eds.; Springer-Verlag: Berlin, Germany, 1980; pp. 36–47. [Google Scholar]
- Sleytr, U.B.; Messner, P.; Pum, D.; Sára, M. Crystalline bacterial cell surface layers (S layers): From supramolecular cell structure to biomimetics and nanotechnology. Angew. Chem. Int. Ed. 1999, 38, 1034–1054. [Google Scholar] [CrossRef]
- Sára, M.; Sleytr, U.B. S-layer proteins. J. Bacteriol. 2000, 182, 859–868. [Google Scholar] [CrossRef]
- Messner, P. Chemical composition and biosynthesis of S-layers. In Crystalline Bacterial Cell Surface Layer Proteins (S-layers); Sleytr, U.B., Messner, P., Pum, D., Sára, M., Eds.; Academic Press: Austin, TX, USA, 1996; pp. 35–76. [Google Scholar]
- Brahamsha, B. An abundant cell-surface polypeptide is required for swimming by the nonflagellated marine cyanobacterium Synechococcus. Proc. Natl. Acad. Sci. 1996, 93, 6504–6509. [Google Scholar] [CrossRef]
- Gilchrist, A.; Fisher, J.A.; Smit, J. Nucleotide sequence analysis of the gene encoding the Caulobacter crescentus paracrystalline surface layer protein. Can. J. Microbiol. 1992, 38, 193–202. [Google Scholar] [CrossRef]
- Tsukagoshi, N.; Tabata, R.; Takemura, T.; Yamagata, H.; Udaka, S. Molecular cloning of a major cell wall protein gene from protein-producing Bacillus brevis 47 and its expression in Escherichia coli and Bacillus subtilis. J. Bacteriol. 1984, 158, 1054–1060. [Google Scholar]
- Beveridge, T.J.; Pouwels, P.H.; Sara, M.; Kotiranta, A.; Lounatmaa, K.; Kari, K.; Kerosuo, E.; Haapasalo, M.; Egelseer, E.M.; Schocher, I.; et al. Functions of S-layers. FEMS Microbiol. Rev. 1997, 20, 99–149. [Google Scholar] [CrossRef]
- Claus, H.; Akca, E.; Debaerdemaeker, T.; Evrard, C.; Declercq, J.P.; Harris, J.R.; Schlott, B.; Konig, H. Molecular organization of selected prokaryotic S-layer proteins. Can. J. Microbiol. 2005, 51, 731–743. [Google Scholar] [CrossRef]
- Breitwieser, A.; Gruber, K.; Sleytr, U.B. Evidence for an S-layer protein pool in the peptidoglycan of Bacillus stearothermophilus. J. Bacteriol. 1992, 174, 8008–8015. [Google Scholar]
- Sturm, E.; Egelseer, E.; Sára, M.; Sleytr, U.B. Can S-layers of Bacillaceae control the release of their own exoproteins? In Advances in Bacterial Paracrystalline Surface Layers; Beveridge, T.J., Koval, S.F., Eds.; Plenum: New York, NY, USA, 1993; pp. 297–302. [Google Scholar]
- Beveridge, T.J. Bacterial S-layers. Curr. Opin. Struc. Biol. 1994, 4, 204–212. [Google Scholar] [CrossRef]
- Bahl, H.; Scholz, H.; Bayan, N.; Chami, M.; Leblon, G.; Gulik-Krzywicki, T.; Shechter, E.; Fouet, A.; Mesnage, S.; Tosi-Couture, E.; et al. Molecular biology of S-layers. FEMS Microbiol. Rev. 1997, 20, 47–98. [Google Scholar]
- Smarda, J.; Smajs, D.; Komrska, J.; Krzyzanek, V. S-layers on cell walls of cyanobacteria. Micron 2002, 33, 257–277. [Google Scholar] [CrossRef]
- McCarren, J.; Heuser, J.; Roth, R.; Yamada, N.; Martone, M.; Brahamsha, B. Inactivation of swmA results in the loss of an outer cell layer in a swimming Synechococcus strain. J. Bacteriol. 2005, 187, 224–230. [Google Scholar]
- Strom, S.L.; Brahamsha, B.; Fredrickson, K.A.; Apple, J.K.; Rodríguez, A.G. A giant cell surface protein in Synechococcus WH8102 inhibits feeding by a dinoflagellate predator. Environ. Microbiol. 2011, 18, 807–816. [Google Scholar]
- Schultze-Lam, S.; Beveridge, T.J. Nucleation of celestite and strontianite on a cyanobacterial S-layer. Appl. Environ. Microb. 1994, 60, 447–453. [Google Scholar]
- Schultze-Lam, S.; Harauz, G.; Beveridge, T.J. Participation of a cyanobacterial S layer in fine-grain mineral formation. J. Bacteriol. 1992, 174, 7971–7981. [Google Scholar]
- Lee, B.D.; Apel, W.A.; Walton, M.R. Screening of cyanobacterial species for calcification. Biotechnol. Progr. 2004, 20, 1345–1351. [Google Scholar] [CrossRef]
- Martinez, R.E.; Gardés, E.; Pokrovsky, O.S.; Schott, J.; Oelkers, E.H. Do photosynthetic bacteria have a protective mechanism against carbonate precipitation at their surfaces? Geochim. Cosmochim. Acta 2010, 74, 1329–1337. [Google Scholar] [CrossRef]
- Dittrich, M.; Müller, B.; Mavrocordatos, D.; Wehrli, B. Induced calcite precipitation by cyanobacterium Synechococcus. Acta Hydrochim. Hydrobiol. 2003, 31, 162–169. [Google Scholar] [CrossRef]
- Obst, M.; Wehrli, B.; Dittrich, M. CaCO3 nucleation by cyanobacteria: laboratory evidence for a passive, surface-induced mechanism. Geobiology 2009, 7, 324–347. [Google Scholar] [CrossRef]
- Obst, M.; Dynes, J.J.; Lawrence, J.R.; Swerhone, G.D.W.; Benzerara, K.; Karunakaran, C.; Kaznatcheev, K.; Tyliszczak, T.; Hitchcock, A.P. Precipitation of amorphous CaCO3 (aragonite-like) by cyanobacteria: A STXM study of the influence of EPS on the nucleation process. Geochim. Cosmochim. Acta 2009, 73, 4180–4198. [Google Scholar]
- Obst, M.; Dittrich, M.; Kuehn, H. Calcium adsorption and changes of the surface microtopography of cyanobacteria studied by AFM, CFM, and TEM with respect to biogenic calcite nucleation. Geochem. Geophy. Geosyst. 2006, 7. [Google Scholar] [CrossRef]
- Kranz, S.A.; Wolf-Gladrow, D.; Nehrke, G.; Langer, G.; Rost, B. Calcium carbonate precipitation induced by the growth of the marine cyanobacteria Trichodesmium. Limnol. Oceanogr. 2010, 55, 2563–2569. [Google Scholar] [CrossRef] [Green Version]
- Fenchel, T.; Kühl, M. Artificial cyanobacterial mats: Growth, structure, and vertical zonation patterns. Microbial. Ecol. 2000, 40, 85–93. [Google Scholar]
- Kühl, M.; Fenchel, T.; Kazmierczak, J. Growth, structure and calcification potential of an artificial cyanobacterial mat. In Fossil and Recent Biofilms: A Natural History of Life on Earth; Krumbein, W., Paterson, D.M., Zavarzin, G.A., Eds.; Kluwer Academic Publisher: Dordrecht, The Netherlands, 2003; pp. 77–102. [Google Scholar]
- Douglas, S.; Beveridge, T.J. Mineral formation by bacteria in natural microbial communities. Fems. Microbiol. Ecol. 1998, 26, 79–88. [Google Scholar] [CrossRef]
- Kempe, S.; Kazmierczak, J.; Landmann, G.; Konuk, T.; Reimer, A.; Lipp, A. Largest known microbialites discovered in Lake Van, Turkey. Nature 1991, 349, 605–608. [Google Scholar]
- Défarge, C.; Trichet, J. From biominerals to ‘organominerals’: The example of the modern lacustrine calcareous stromatolites from polynesian atolls. In Procceedings of 7th International Symposium on Biomineralization, MC, Monaco, 17-20 November 1993.
- Gautret, P.; Camoin, G.; Golubic, S.; Sprachta, S. Biochemical control of calcium carbonate precipitation in modern lagoonal microbialites, Tikehau Atoll, French Polynesia. J. Sediment. Res. 2004, 74, 462–478. [Google Scholar] [CrossRef]
- Sanders, D.; Unterwurzacher, M.; Rüf, B. Microbially induced calcium carbonate in tufas of the western Eastern Alps: A first overview. Geogr. Alps 2006, 3, 167–189. [Google Scholar]
- Pentecost, A. Growth and calcification of the freshwater cyanobacterium Rivularia haematites. Proc. R. Soc. London Ser. B Biol. Sci. 1987, 232, 125–136. [Google Scholar] [CrossRef]
- Pentecost, A. Growth and calcification of the cyanobacterium Homoeothrix crustacea. J. Gen. Microbiol. 1988, 134, 2665–2671. [Google Scholar]
- Kazmierczak, J.; Kempe, S.; Kremer, B.; Lopez-Garcia, P.; Moreira, D.; Tavera, R. Hydrochemistry and microbialites of the alkaline crater lake Alchichica, Mexico. Facies 2011, 57, 543–570. [Google Scholar] [CrossRef]
- Braithwaite, C.J.R.; Zedef, V. Hydromagnesite stromatolites and sediments in an alkaline lake, Salda Golu, Turkey. J. Sediment. Res. 1996, 66, 991–1002. [Google Scholar]
- Braithwaite, C.J.R.; Zedef, V. Living hydromagnesite stromatolites from Turkey. Sediment. Geol. 1994, 92, 1–5. [Google Scholar] [CrossRef]
- Sutherland, I.W. The biofilm matrix-An immobilized but dynamic microbial environment. Trends Microbiol. 2001, 9, 222–227. [Google Scholar] [CrossRef]
- Shiraishi, F.; Bissett, A.; de Beer, D.; Reimer, A.; Arp, G. Photosynthesis, respiration and exopolymer calcium-binding in biofilm calcification (Westerhfer and deinschwanger creek, germany). Geomicrobiol. J. 2008, 25, 83–94. [Google Scholar] [CrossRef]
- Roberts, J.A.; Bennett, P.C.; Gonzalez, L.A.; Macpherson, G.L.; Milliken, K.L. Microbial precipitation of dolomite in methanogenic groundwater. Geology 2004, 32, 277–280. [Google Scholar] [CrossRef]
- Rogers, J.R.; Bennett, P.C.; Macpherson, G.L. Microbial precipitation of dolomite in groundwater. Geochim. Cosmochim. Acta 2003, 67, A400. [Google Scholar]
- Van Lith, Y.; Vasconcelos, C.; Warthmann, R.; Martins, J.C.F.; McKenzie, J.A. Bacterial sulfate reduction and salinity: Two controls on dolomite precipitation in Lagoa Vermelha and Brejo do Espinho (Brazil). Hydrobiologia 2002, 485, 35–49. [Google Scholar] [CrossRef]
- Warthmann, R.; van Lith, Y.; Vasconcelos, C.; McKenzie, J.A.; Karpoff, A.M. Bacterially induced dolomite precipitation in anoxic culture experiments. Geology 2000, 28, 1091–1094. [Google Scholar] [CrossRef]
- Wright, D.T. The role of sulphate-reducing bacteria and cyanobacteria in dolomite formation in distal ephemeral lakes of the Coorong region, South Australia. Sediment. Geol. 1999, 126, 147–157. [Google Scholar] [CrossRef]
- Wright, D.T.; Wacey, D. Precipitation of dolomite using sulphate-reducing bacteria from the Coorong Region, South Australia: Significance and implications. Sedimentology 2005, 52, 987–1008. [Google Scholar] [CrossRef]
- Yates, K.K.; Robbins, L.L. Production of carbonate sediments by a unicellular green alga. Am. Mineral 1998, 83, 1503–1509. [Google Scholar]
- McConnaughey, T.A.; Falk, R.H. Calcium-proton exchange during algal calcification. Biol. Bull. 1991, 180, 185–195. [Google Scholar] [CrossRef]
- McConnaughey, T.A.; Whelan, J.F. Calcification generates protons for nutrient and bicarbonate uptake. EarthSci. Rev. 1997, 42, 95–117. [Google Scholar] [CrossRef]
- Lucas, W.J.; Keifer, D.W.; Sanders, D. Bicarbonate transport in Chara corallina: Evidence for co-transport of HCO3− with H+. J. Membr. Biol. 1983, 73, 263–274. [Google Scholar] [CrossRef]
- Rowland, S.M.; Gangloff, R.A. Structureand paleoecology of lower cambrian reefs. Palaios 1988, 3, 111–135. [Google Scholar] [CrossRef]
- Banks, E.D.; Barton, H.A.; Taylor, N.M.; Gulley, J.; Lubbers, B.R.; Giarrizo, J.G.; Bullen, H.A.; Hoehler, T.M. Bacterial calcium carbonate precipitation in cave environments: A function of calcium homeostasis. Geomicrobiol. J. 2010, 27, 444–454. [Google Scholar] [CrossRef]
- Kamennaya, N.A.; Holman, E.A.; Mahoney, L.; Zemla, M.; Cappuccio, J.; Chavarría, L.A.; Swarbreck, S.M.; Ajo-Franklin, C.; Auer, M.; Northen, T.; Holman, H.-Y.; Jansson, C. Unpublished Work, Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2012.
- Dittrich, M.; Sibler, S. Cell surface groups of two picocyanobacteria strains studied by zeta potential investigations, potentiometric titration, and infrared spectroscopy. J. Colloid Interface Sci. 2005, 286, 487–495. [Google Scholar] [CrossRef]
- Couradeau, E.; Benzerara, K.; Gerard, E.; Moreira, D.; Bernard, S.; Brown, G.E.; Lopez-Garcia, P. An early-branching microbialite cyanobacterium forms intracellular carbonates. Science 2012, 336, 459–462. [Google Scholar]
- Kazmierczak, J.; Kempe, S. Genuine modern analogues of Precambrian stromatolites from caldera lakes of Niuafo’ou Island, Tonga. Naturwissenschaften 2006, 93, 119–126. [Google Scholar] [CrossRef]
- Lyons, W.B.; Long, D.T.; Hines, M.E.; Gaudette, H.E.; Armstrong, P.B. Calcification of cyanobacterial mats in Solar Lake, Sinai. Geology 1984, 12, 623–626. [Google Scholar] [CrossRef]
- Robbins, L.L.; Hansen, M.E.; Kleypas, J.A.; Meylan, S.C. CO2calc: A User-friendly Seawater Carbon Calculator for Windows, Max OS X, and iOS (iPhone); U.S. Geological Survey Open-File Report 2010–1280; U.S. Department of the Interior, U.S. Geological Survey: Reston, VA, USA, 2010. [Google Scholar]
- Lewis, E.; Wallace, D.W.R. Program Developed for CO2 System Calculations; Carbona Dioxide Information Analysis Center, Oak Ridge National Laboratory, US Departments of Energy: Oak Ridge, TN, USA, 1998. [Google Scholar]
- Parkhurst, D.L.; Thorstenson, D.C.; Plummer, L.N.; Geological, S. PHREEQE: A Computer Program for Geochemical Calculations; U.S. Geological Survey: Reston, VA, USA, 1990. [Google Scholar]
- Suzuki, A. Combined effects of photosynthesis and calcification on the partial pressure of carbon dioxide in seawater. J. Oceanogr. 1998, 53, 1–7. [Google Scholar] [CrossRef]
- Frankignoulle, M.; Canon, C. Marine calcification as a source of carbon-dioxide: Positive feedback of increasing atmospheric CO2. Limnol. Oceanogr. 1994, 39, 458–462. [Google Scholar] [CrossRef]
- Buitenhuis, E.; vanBleijswijk, J.; Bakker, D.; Veldhuis, M. Trends in inorganic and organic carbon in a bloom of Emiliania huxleyi in the North Sea. Mar. Ecol. Prog. Ser. 1996, 143, 271–282. [Google Scholar] [CrossRef]
- Bennett, P.C.; Rogers, J.R.; Choi, W.J. Silicates, silicate weathering, and microbial ecology. Geomicrobiol. J. 2001, 18, 3–19. [Google Scholar] [CrossRef]
- Buss, H.L.; Luttge, A.; Brantley, S.L. Etch pit formation on iron silicate surfaces during siderophore-promoted dissolution. Chem. Geol. 2007, 240, 326–342. [Google Scholar] [CrossRef]
- Barker, W.W.; Banfield, J.F. Zones of chemical and physical interaction at interfaces between microbial communities and minerals: A model. Geomicrobiol. J. 1998, 15, 223–244. [Google Scholar] [CrossRef]
- Barker, W.W.; Welch, S.A.; Banfield, J.F. Biogeochemical weathering of silicate minerals. Rev. Mineral. 1997, 35, 391–428. [Google Scholar]
- Chan, C.S.; Fakra, S.C.; Edwards, D.C.; Emerson, D.; Banfield, J.F. Iron oxyhydroxide mineralization on microbial extracellular polysaccharides. Geochim. Cosmochim. Acta 2009, 73, 3807–3818. [Google Scholar]
- Santelli, C.M.; Welch, S.A.; Westrich, H.R.; Banfield, J.F. The effect of Fe-oxidizing bacteria on Fe-silicate mineral dissolution. Chem. Geol. 2001, 180, 99–115. [Google Scholar] [CrossRef]
- Ferris, F.G.; Wiese, R.G.; Fyfe, W.S. Precipitation of carbonate minerals by microorganisms: Implications for silicate weathering and the global carbon-dioxide budget. Geomicrobiol. J. 1994, 12, 1–13. [Google Scholar] [CrossRef]
- Kasting, J.F. Earths early atmosphere. Science 1993, 259, 920–926. [Google Scholar]
- Sarmiento, J.L.; Bender, M. Carbon biogeochemistry and climate change. Photosynth. Res. 1994, 39, 209–234. [Google Scholar] [CrossRef]
- Sandberg, P.A. Nonskeletal aragonite and pCO2 in the Phanerozoic and Proterozoic. In The Carbon Cycle and Atmospheric CO2: Natural Variations Archean to Present, Geophysical Monograph Series; Sundquist, E.T., Brocker, W.S., Eds.; AGU: Washington, DC, USA, 1985; Volume 32, pp. 585–594. [Google Scholar]
- Schewiakoff, W. Über einen neuen bakterienähnlichen Organismus des Süßwassers. Habilitationsschrift; University of Heidelberg: Heidelberg, Baden, Germany, 1893. [Google Scholar]
- Riding, R. A hard life for cyanobacteria. Science 2012, 336, 427–428. [Google Scholar] [CrossRef]
- Jansson, C. Metabolic engineering of cyanobacteria for direct conversion of CO2 to hydrocarbon biofuels. Prog. Bot. 2012, 73, 81–93. [Google Scholar] [CrossRef]
- Lee, B.D.; Apel, W.A.; Walton, M.R. Whitings as a Potential Mechanism for Controlling Atmospheric Carbon Dioxide Concentrations; Final Project Report INL/EXT-06-01351; Idaho Falls, ID, USA, 2006. [Google Scholar]
- Robbins, L.L.; Yates, K.K. Direct Measurements of CO2 Fluxes in Marine Whitings; US Geological Survey: St Petersburg, FL, USA, 2001. [Google Scholar]
- National Research Council of the National Academies, Novel Approaches to Carbon Management:Separation, Capture, Sequestration, and Conversion to Useful Products-Workshop Report; The National Academies Press: Washington, DC, USA, 2003.
- Fabry, V.J. Calcium Carbonate Production by Coccolithophorid Algae in Long Term, Carbon Dioxide Sequestration; Quarterly Progress Report; California State University: San Marcos, CA, USA, 2004. [Google Scholar]
- Power, I.M.; Wilson, S.A.; Small, D.P.; Dipple, G.M.; Wan, W.K.; Southam, G. Microbially mediated mineral carbonation: Roles of phototrophy and heterotrophy. Environ. Sci. Technol. 2011, 45, 9061–9068. [Google Scholar]
- Power, I.M.; Dipple, G.M.; Southam, G. Bioleaching of ultramafic tailings by acidithiobacillus spp. for CO2 sequestration. Environ. Sci. Technol. 2010, 44, 456–462. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kamennaya, N.A.; Ajo-Franklin, C.M.; Northen, T.; Jansson, C. Cyanobacteria as Biocatalysts for Carbonate Mineralization. Minerals 2012, 2, 338-364. https://doi.org/10.3390/min2040338
Kamennaya NA, Ajo-Franklin CM, Northen T, Jansson C. Cyanobacteria as Biocatalysts for Carbonate Mineralization. Minerals. 2012; 2(4):338-364. https://doi.org/10.3390/min2040338
Chicago/Turabian StyleKamennaya, Nina A., Caroline M. Ajo-Franklin, Trent Northen, and Christer Jansson. 2012. "Cyanobacteria as Biocatalysts for Carbonate Mineralization" Minerals 2, no. 4: 338-364. https://doi.org/10.3390/min2040338