1. Introduction
The presence of water in apatite (Ca
10(PO
4)
6(OH)
2 = CaApOH) has long been a topic of debate [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15]. Our detection of a
2H NMR resonance of D
2O in the lattice of both carbonated calcium hydroxylapatite (CCaApOH) and carbonated calcium fluorapatite (CCaApF) prepared in D
2O provided unambiguous evidence for the presence of water within the lattice of both of these apatites [
16]. Calcium ions reside in two types of sites in apatite, one in which calcium is bonded only to oxygens,
i.e., Ca(1), and another site in which calcium is bonded to both oxygens and a monovalent anion,
i.e., Ca(2). We have proposed that the water molecules are likely located between the monovalent anions that are positioned in the channels created by the Ca(2) ions.
Given that apatites are notoriously hygroscopic and that some workers have assumed that they contain structural water in addition to this adsorbed water, it is not surprising that previous work has been carried out on the dehydration of apatites. Early studies are accurately summarized by LeGeros
et al. [
5]. Some of these studies were accomplished on biological apatite and thus were confounded by the presence of organic matter. The study by LeGeros
et al. [
5] on both dental apatites and synthetically precipitated apatites showed that the adsorbed water was reversibly removed and readsorbed below 200 °C, whereas the water released between 200 °C and 400 °C was irreversibly lost and caused a contraction in the a-axis [
5]. Moreover, the a-axis is larger in aqueously precipitated apatites than in those “mineral or synthetic apatites from nonaqueous systems” [
5].
An aspect of apatite that has not been well documented previously is the effect of carbonate substitution in apatite on the amount of water that it structurally incorporates. The fact that there are two well-recognized mechanisms by which carbonate can enter the apatite structure [
17] makes this question particularly interesting. Carbonate can substitute for either the monovalent anion in the channels (called A-type substitution) or for tetrahedral phosphate groups (B-type susbstitution). In both cases, substitution involves a mismatch in the ionic charges, which leads to the development of vacancies in other ion sites. Intriguingly, LeGeros and colleagues [
17] said that there are no carbonate substitutes for OH in hydroxylapatite if water is involved in the synthesis. Subsequent research confirmed that only minor (at the level of 10–15%) A-type substitution of carbonate for OH occurs in low-temperature synthetic or in biological apatite [
18,
19].
The objectives of this study were to: (a) determine the relationship between the amount of structural water and the carbonate content, (b) characterize the evolution and reincorporation of structural water, and (c) determine the effect of dehydration and rehydration on the structure of carbonated hydroxyl- and fluorapatites.
2. Experimental Section
2.1. Synthesis
Samples of CCaApOH were synthesized from readily soluble sources of calcium, phosphate, and carbonate in a pH-buffered aqueous solution. All water used in the synthesis was double-deionized (Milli-Q) or deionzied and passed through a hydroxylapatite column. Nitrogen gas was bubbled through the water for ca. six hours to remove carbon dioxide. The NaOH solutions were freshly prepared after each 5–6 syntheses, and the solution bottles were sealed with Parafilm to prevent contamination by carbon dioxide. Magnetic stirrers were used for mixing.
Approximately 250 mL of water was heated to the desired temperature (typically 60 °C or 80 °C) in a large beaker. According to the desired final carbonate to phosphate ratio, a specific amount of NaHCO3 (Acros-Organics, 99.5%) was dissolved in the water. The pH was elevated to and maintained at 9.0 ± 0.3 (Hanna Instruments pH 209 meter with a Thermo Ross Sure-Flow combination reference/pH electrode) through addition of 0.1M NaOH (Acros, 97%). To the heated carbonate solution, 25 mL of a 0.15M Ca(NO3)2·4H2O (99%, Sigma-Aldrich, St. Louis, MO, USA) solution and 25 mL of a 0.09 M NaH2PO4·H2O (Aldrich, 98%) solution were added at a rate of 1 mL/min from separate burettes. After addition of the calcium and phosphate solutions, the mixture was stirred and kept at 60 °C (some syntheses were also performed at 80 °C) for 2 hours (some for 1 h). After this equilibration time, stirring was discontinued, and the mixture was allowed to cool to room temperature and then was filtered through a glass filter crucible of medium porosity. The product powder was washed three times with warm water, and then dried at 120 °C in a vacuum pistol or dried in an evacuated vacuum dessicator for a minimum of 24 h.
The synthesis of CCaApF followed the same procedure used for CCaApOH, except that a 0.6 M NaF solution was added simultaneously with and at the same rate as the addition of the calcium and phosphate solutions.
2.2. Sample Analysis and Characterization
Carbonate analysis. The weight percent carbonate in each sample was determined via C, H, N combustion elemental analysis or from the weight loss between 600 °C and 800 °C in TGA analyses. A standard curve for TGA weight loss converted to carbonate wt% was constructed using the results of combustion analyses performed by Schwarzkopf Microanalytical Laboratories (Woodside, NY, USA) or Galbraith Laboratories (Knoxville, TN, USA).
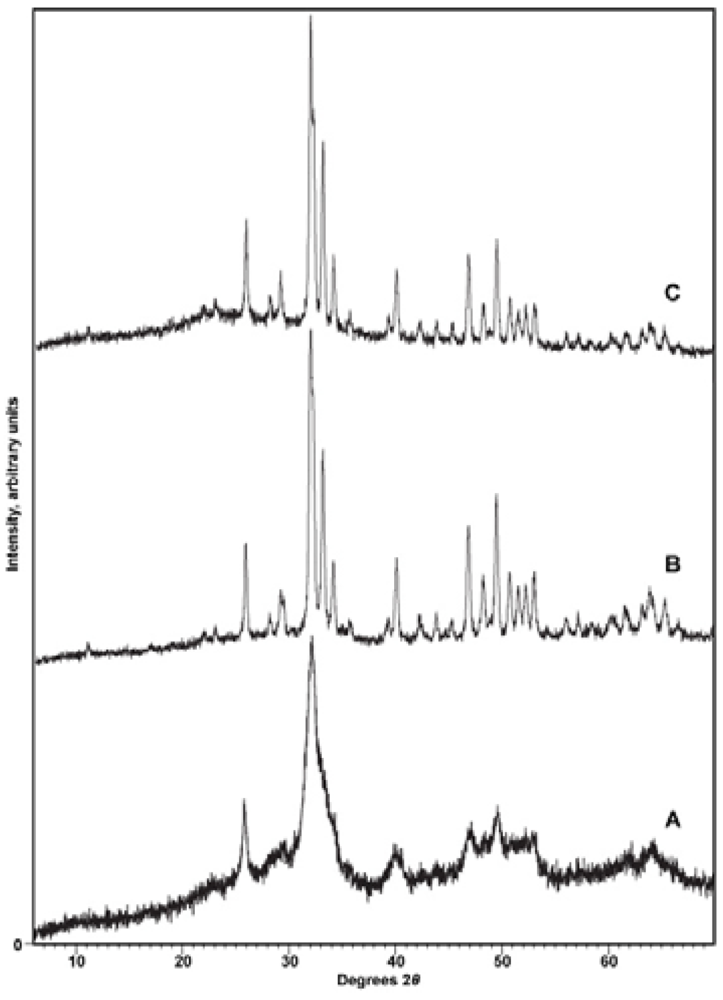
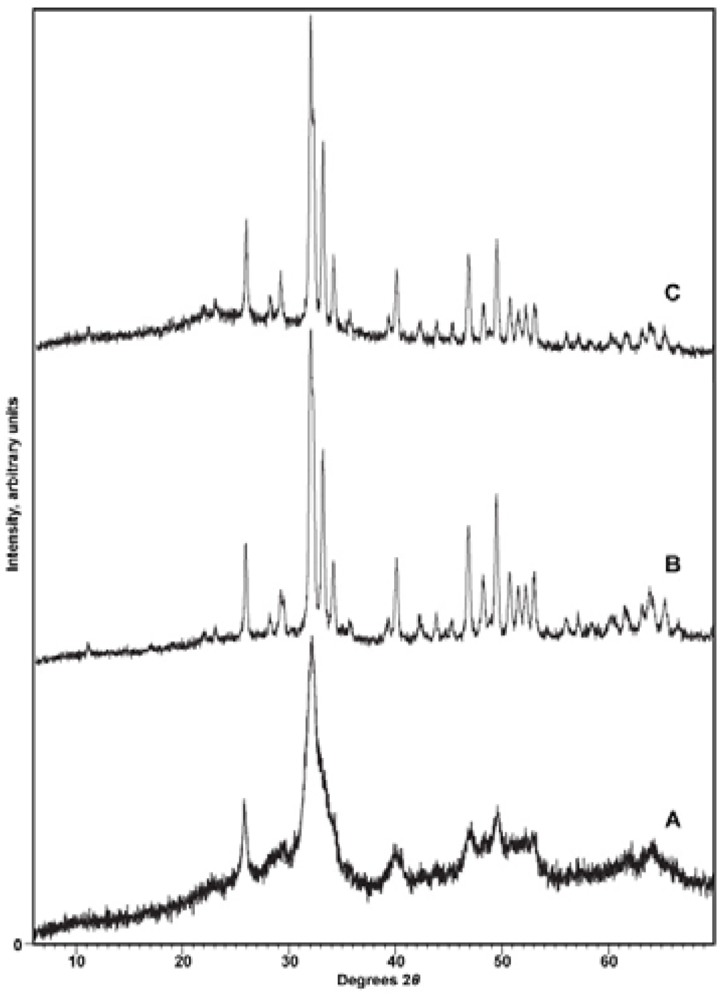
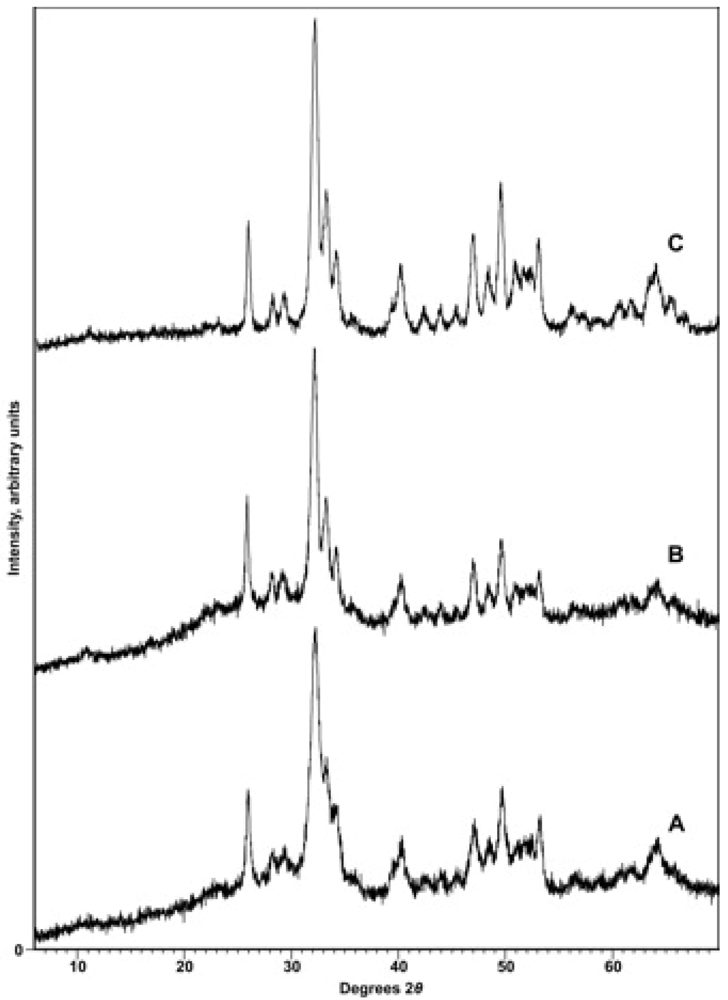
X-ray diffraction. To confirm the identity of the precipitate and to reveal any contaminating phases (particularly CaCO3), samples were subjected to X-ray diffraction analysis with either a Phillips 3520 X-ray diffractometer producing Cu-Kα radiation in conjunction with a monochromator or a PANalytical X’Pert PRO MPD (Multi-Purpose Diffractometer) Theta-Theta System with Cu-Kα radiation (λ = 1.54060 Å). Instrumental settings included a step size of 0.02° 2-theta and a dwell time of 1 s over the analysis range of 2 to 60° 2-theta. The scans were analyzed using the program PANalytical X’Pert HighScore Plus (Version 2.2e). Sample diffraction patterns were compared with the ICDD powder X-ray diffraction pattern database (PDF Release 2) using the HighScore Plus search-match algorithm. X-ray diffractograms were also used to detect other phases present in the synthetic apatites, particularly CaCO3, and to look for patterns of changing crystallinity of the samples.
Cell parameters were determined using the PANalytical X’Pert PRO MPD diffractometer with samples mounted in 16 mm cavity slides with a step size of 0.02 2-theta over the range 15 to 70° 2-theta. The entire XRD pattern was profile-fitted by determining the profile for each individual peak and ultimately fitting the entire scan. The indexing method used was usually McMaille, but in some circumstances the Treor method was employed. The unit cell candidate was chosen by its high figure of merit relative to other candidates, its symmetry, and the absence of un-indexed peaks. Although the uncertainties in the cell parameters were generally within ±0.0007 Å for a single measurement, repeated measurements in some cases produced larger errors, more of the order of ±0.002 Å.
Raman microprobe analysis. Raman spectroscopy was used to further characterize the phosphate phase in the precipitate and to reveal the presence of contaminating phases more sensitively than by powder X-ray diffraction. The integrated, fiber-optically coupled microscope-spectrometer-detector confocal Raman system used in this study is from Kaiser Optical (Ann Arbor, Michigan). The HoloLab Series Research Raman Spectrometer was configured for 532-nm laser excitation, and it covered the spectral range of 0 to 4300 Δcm−1 with a resolution of 2.5 cm−1 using an Andor high-resolution, thermoelectrically cooled CCD array detector. Analyses of powders were made using an Olympus 80× objective with an N.A. of 0.75 or an Olympus 50x objective with an N.A. of 0.55. Each Raman analysis represents the average of 32 acquisitions of 4 seconds each. Every sample underwent 3 to 5 analyses. The laser power was 10 mW at the sample surface, and the diameter of the beam was one to several micrometers at the sample surface. The focus was optimized so as to obtain the maximum signal:noise ratio.
The program GRAMS 32 (Galactic, Salem, NH, USA) was used to deconvolve appropriate Raman peaks into their component spectral bands. The bands were modeled as mixed Gaussian and Lorentzian in form, in order to derive the wavenumber position, width at half-maximum intensity, and area of the bands.
2.3. Thermogravimetric Analysis
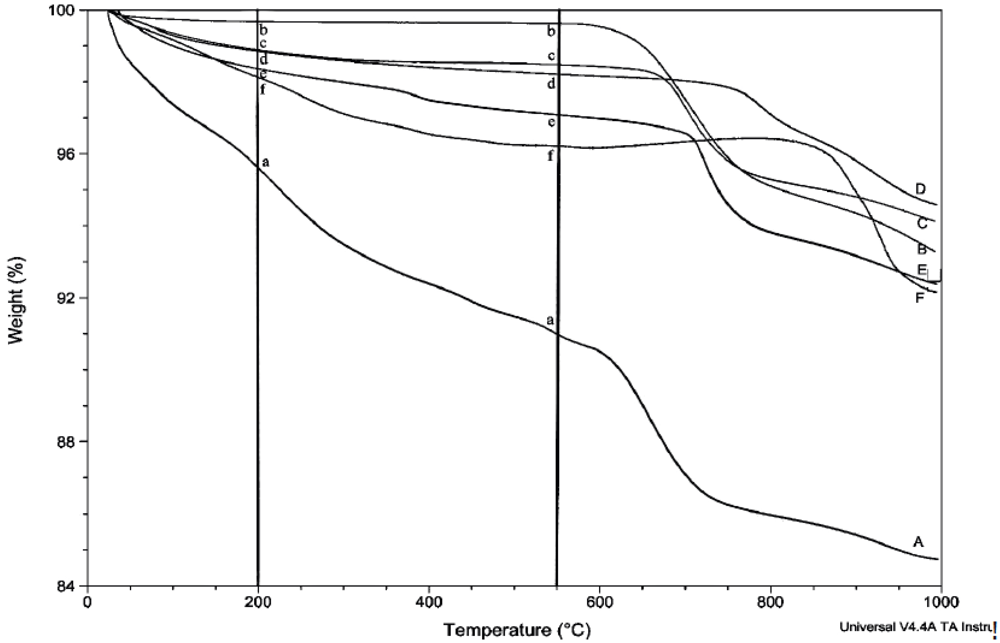
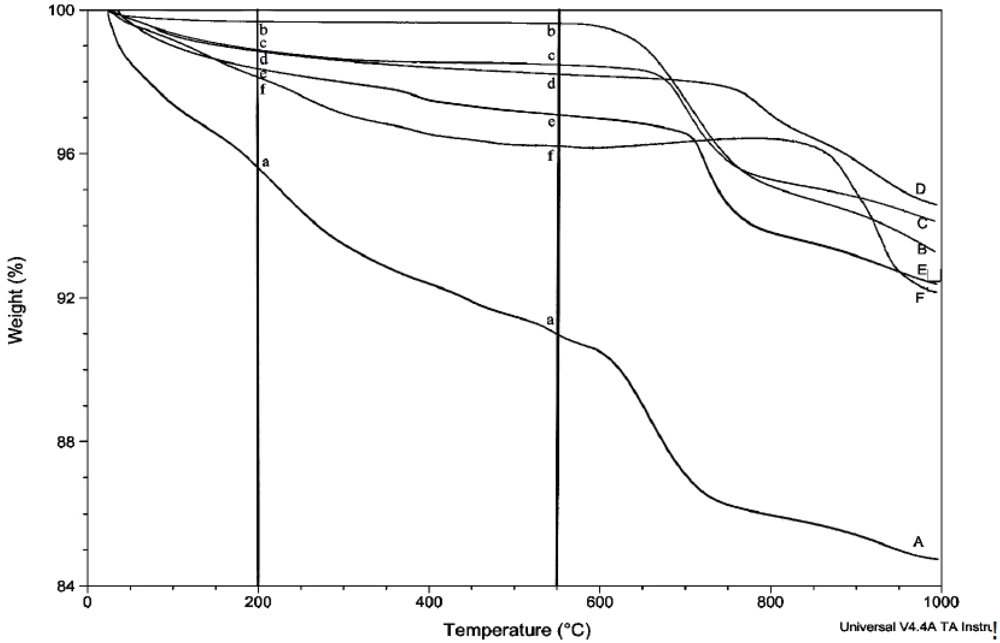
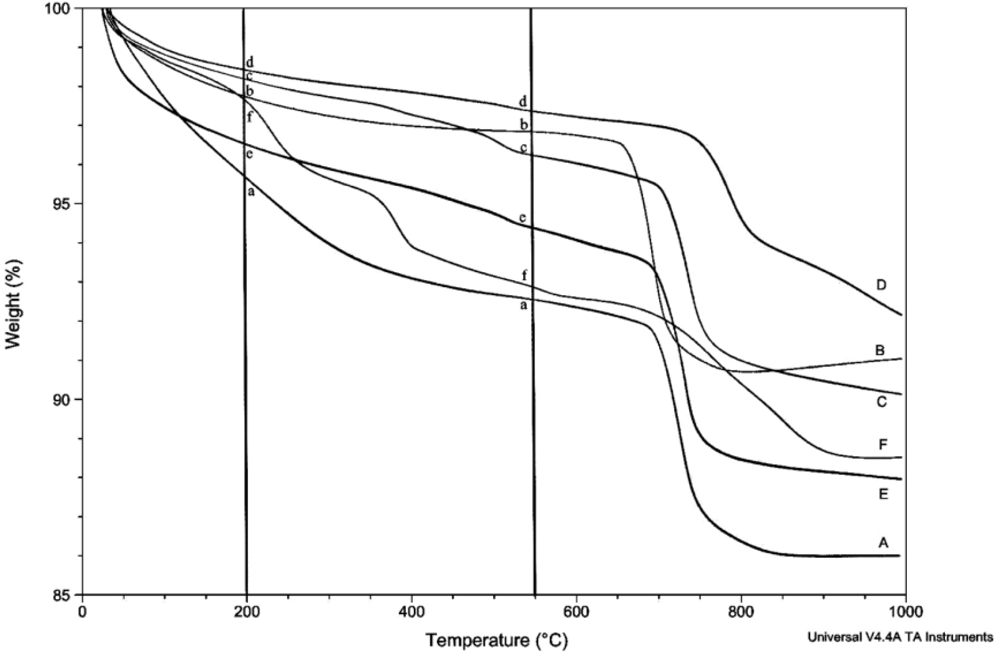
The amount of structural and adsorbed water in the samples was determined by thermogravimetric analysis (TGA) using the TA Instruments TGA Q500. Unless otherwise stated, thermograms were obtained using a balance flow of 40 mL/min and a sample flow of 60 mL/min of dry nitrogen gas. Samples of 7–20 mg were heated to 1,000 °C at a rate of 15 °C/min. The relative uncertainty for the measurement of weight loss in multiple scans is estimated to be about 8%. Linear regression lines are shown for most plots involving weight loss, but correlation coefficients are not given because of their generally low values and limited utility.
2.4. Rehydration Experiments
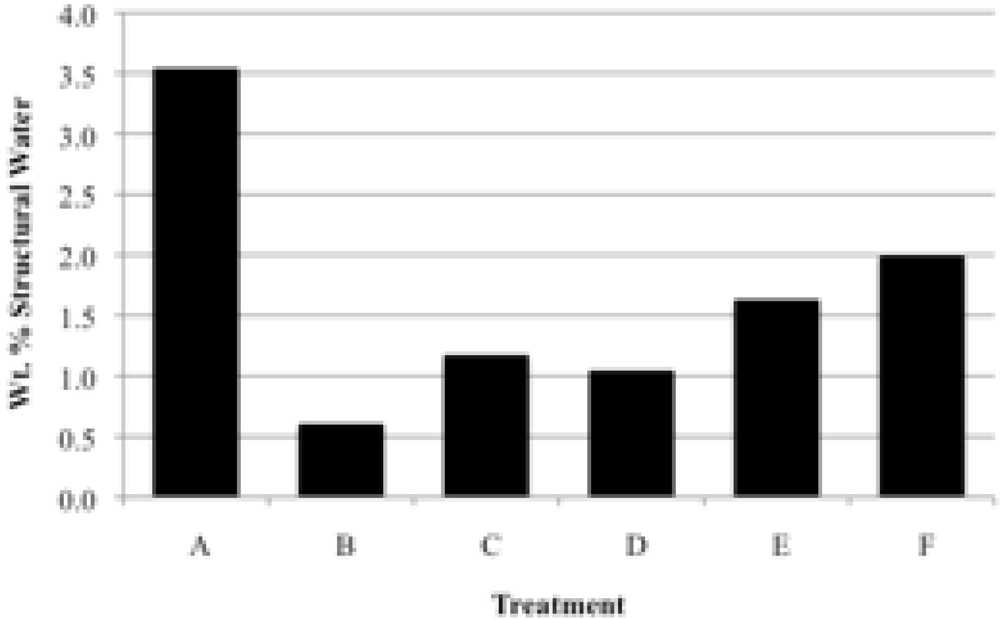
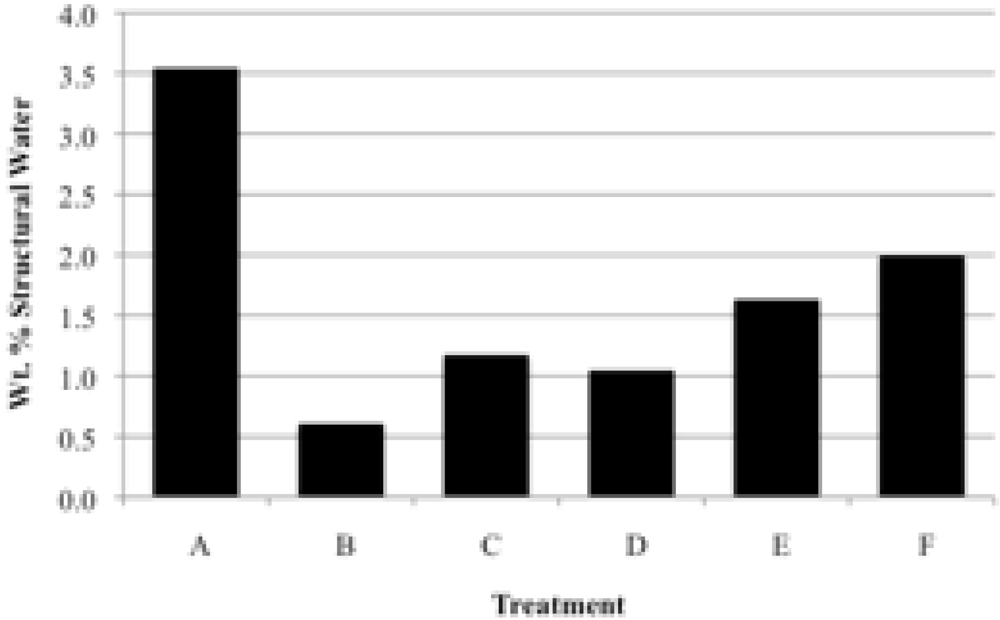
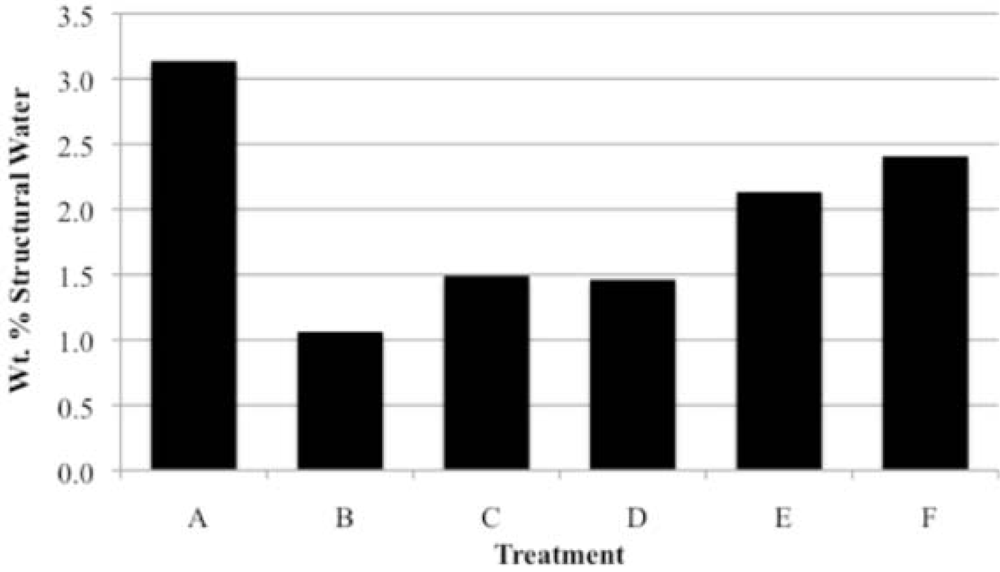
Prior to rehydration experiments, the purity of the phase was checked by XRD, and after vacuum drying for 24 h, TGA was used to measure the amount of water in the apatite. The weight percent lost between room temperature and 200 °C was taken to be adsorbed water, while the weight percent lost between 200 °C and 550 °C indicated the loss of structural water.
For each rehydration experiment four 50 mg samples of an apatite were dehydrated by heating in four ceramic crucibles in a muffle furnace for 2 h at 550 °C. These samples were then either immersed in water for 2 h at room temperature (22 °C), immersed in boiling water for 2 h, immersed in water for 4 days at room temperature, or immersed in water for 4 days at 100 °C. The rehydrations were performed in 100 mL round-bottom flasks with magnetic stirring using double-deionized (Milli-Q) water previously saturated with nitrogen gas. The samples were filtered by suction with Buchner funnels and dried in vacuo (4 torr) for at least 24 hours. The dried samples were analyzed by TGA and XRD.
4. Discussion
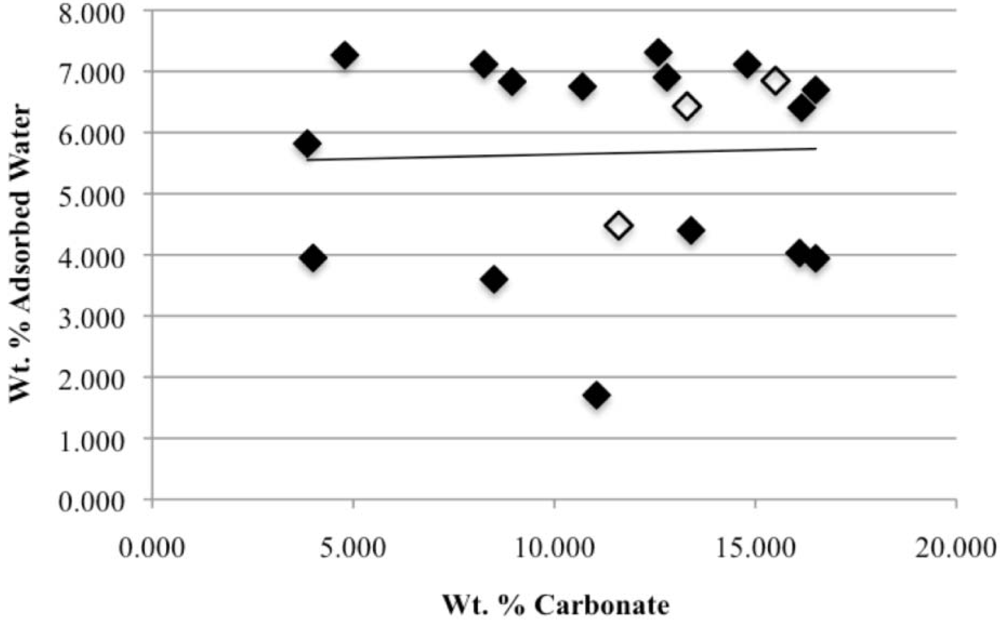
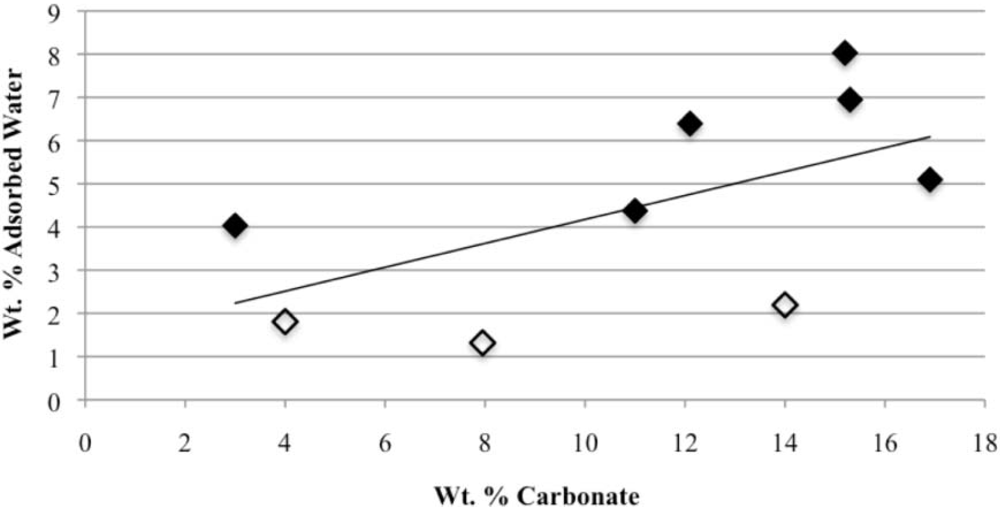
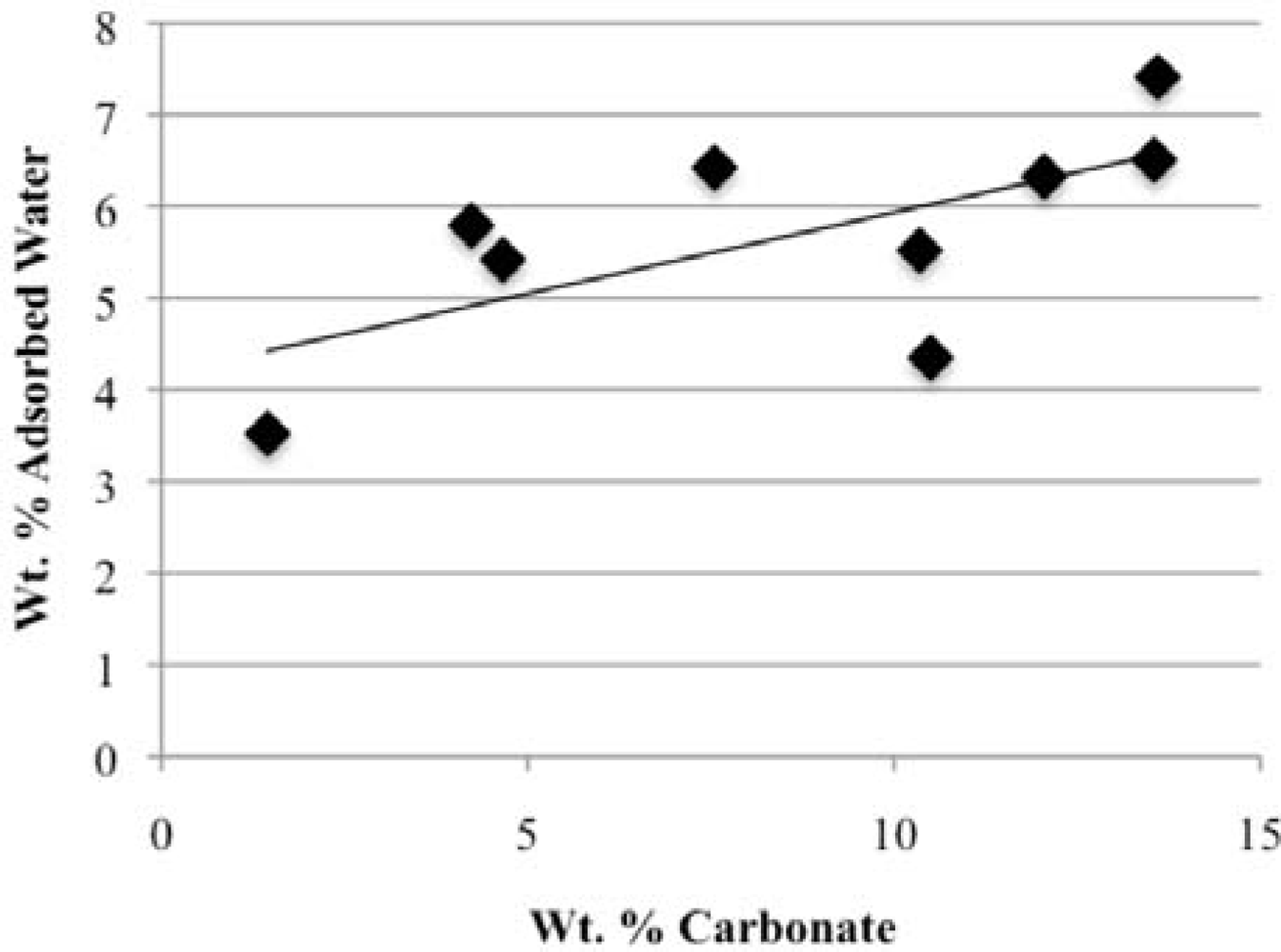
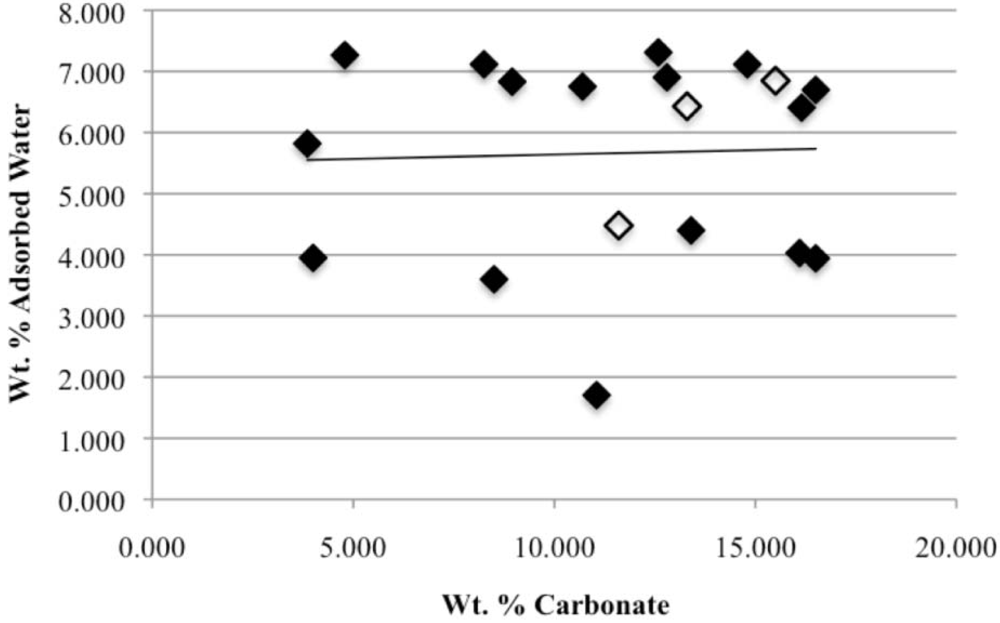
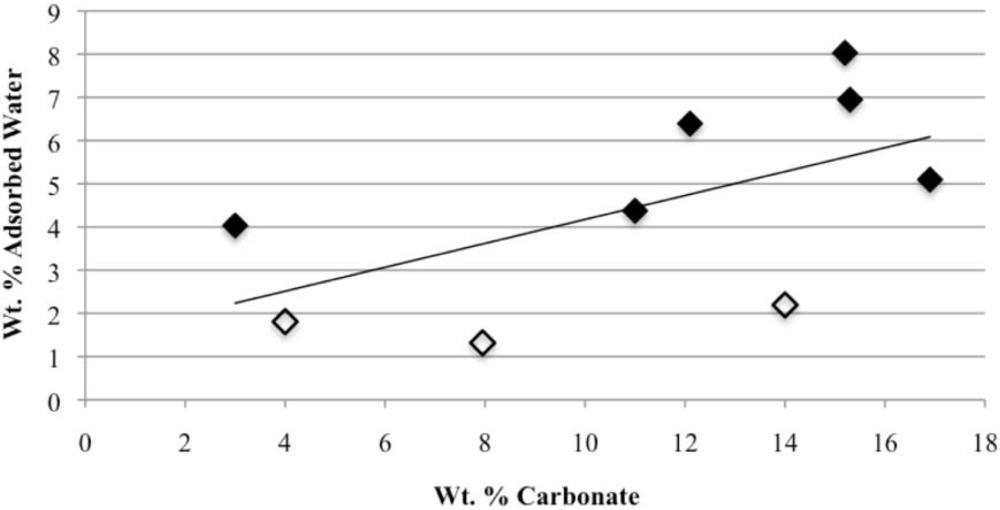
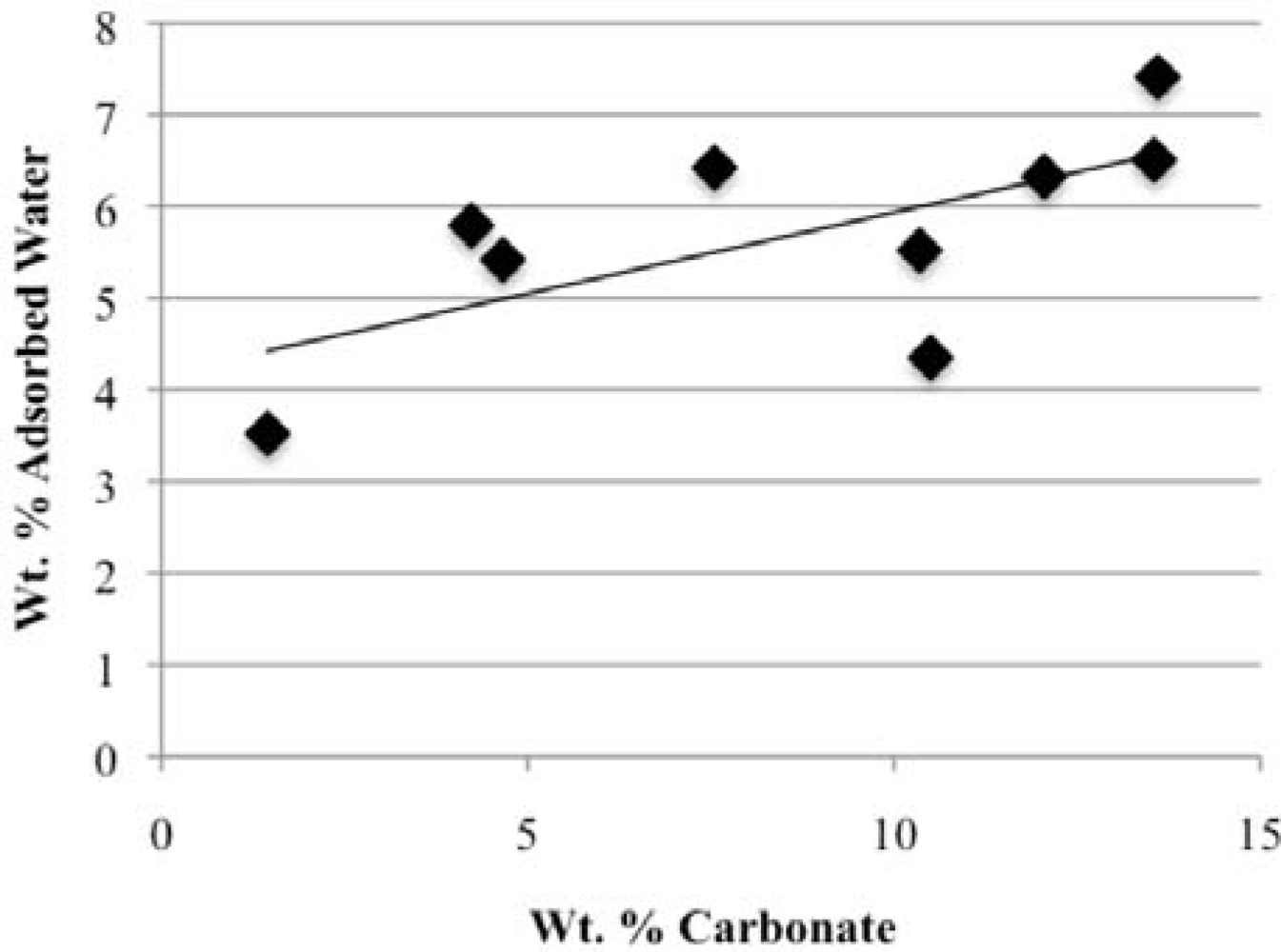
Our study has revealed a number of important characteristics of calcium hydroxyl- and fluorapatites. First, there is a positive correlation between the concentrations of adsorbed water and carbonate. This correlation most likely is the result of a decrease in crystallite size (and consequent increase in surface area) as carbonate content increases [
27,
28]. This correlation with adsorbed water is seen in our CCaApF samples prepared with a two-hour digestion time and in CCaApOH samples prepared with a one-hour digestion time. Presumably, a shorter time for crystal maturation amplifies this relationship between carbonate concentration and abundance of adsorbed water.
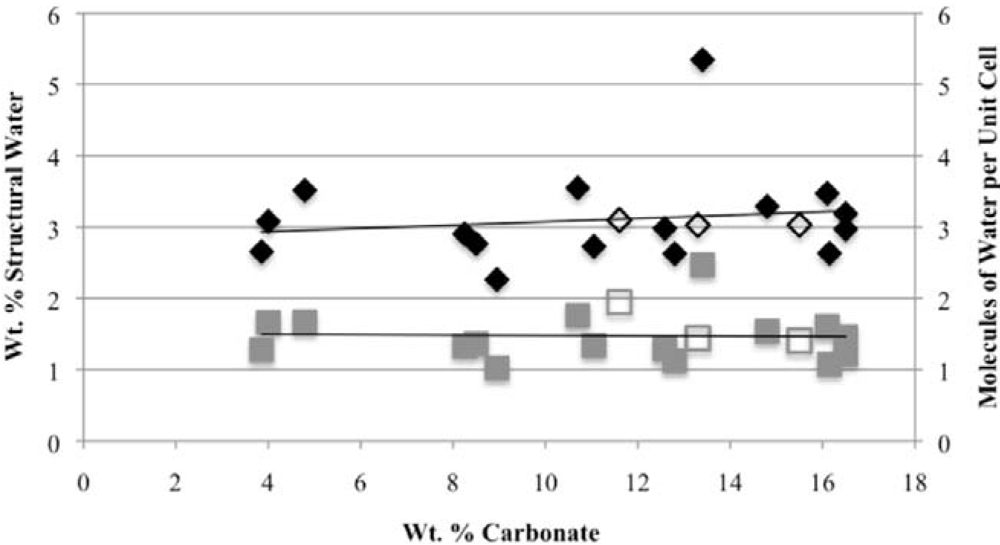
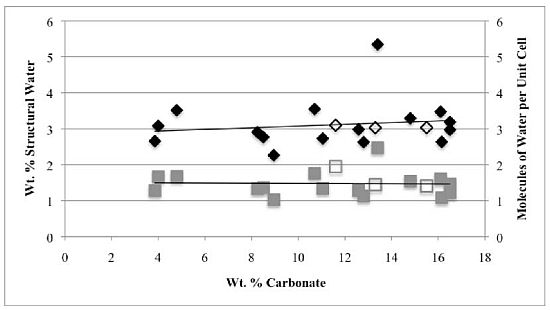
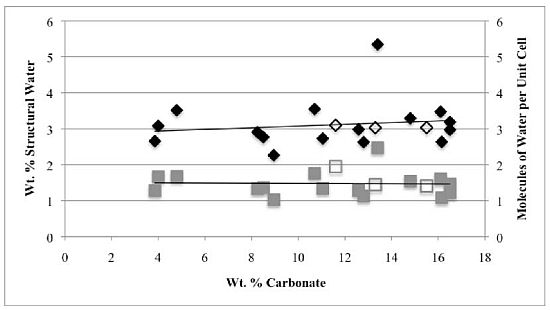
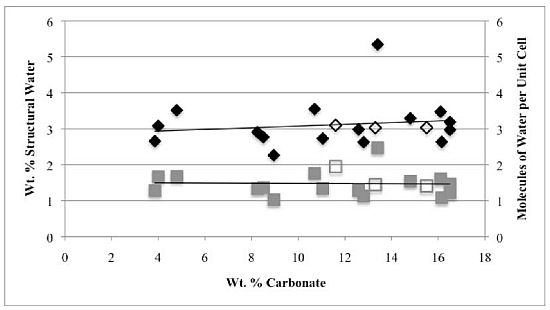
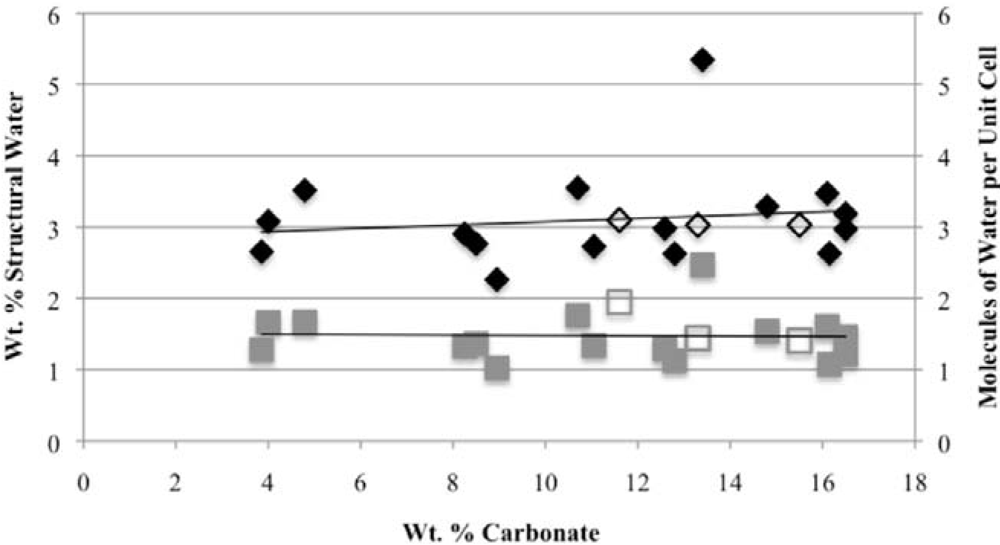
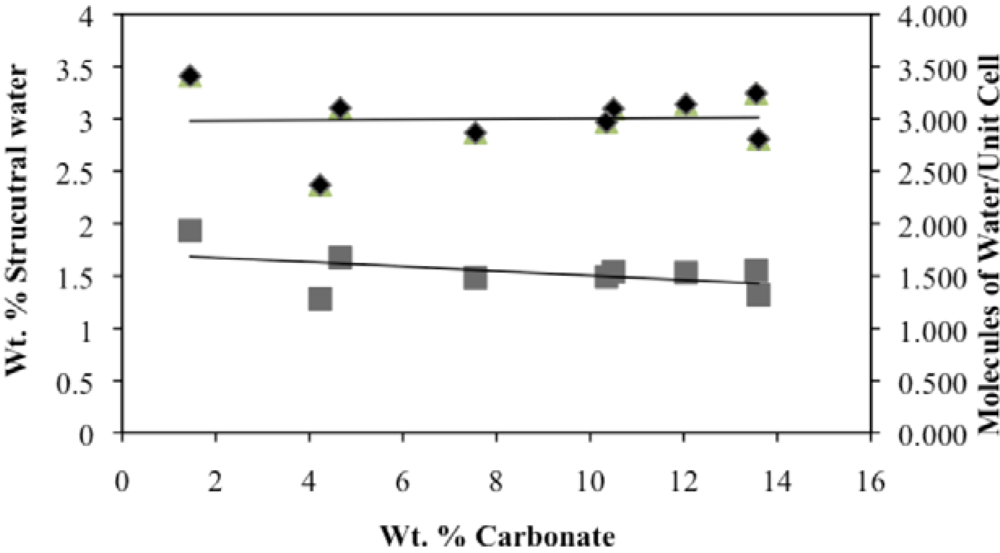
Second, the number of molecules of structural water per unit cell is independent of the carbonate concentration, with an average value of 1.5–1.6 for both hydroxyl- and fluorapatites. These values, based on direct TGA measurements, compare favorably with the modeled values of Wilson
et al. [
8,
29] of 1.05 for a Na-bearing CCaApOH with 12.5 wt% carbonate and 1.97 for a Ca-deficient hydroxylapatite based on Rietveld refinement of XRD data. Because the Ca
10(PO
4)
6(X)
2 unit cell contains a total of two X ions (X = F
− or OH
−), our number of proposed water molecules per unit cell is close to the total number of X groups in the uncarbonated apatite. If, as we suggest below, the water molecules occupy space between the X ions along the c-axis, then the maximum number of water molecules per unit cell would be two. Of course, the molecules might also occupy hydroxide or fluoride vacancies, which could result in a maximum mpuc value of greater than two.
Third, structural water in CCaApOH and CCaApF can be removed by heating, but is only partially replaced by immersion of the dehydrated apatite in water. Fourth, the extent of rehydration is strongly increased by an increase in temperature and less strongly by an increase in immersion time. Fifth, crystallite size is increased by thermally-induced dehydration, probably as a result of the effect of temperature in decreasing crystal strain and increasing the stability of the crystallites. Crystallite size also is increased by rehydration, likely a result of increased time and/or temperature as well as the diffusion-stimulated dissolution/reprecipitation process for crystal growth.
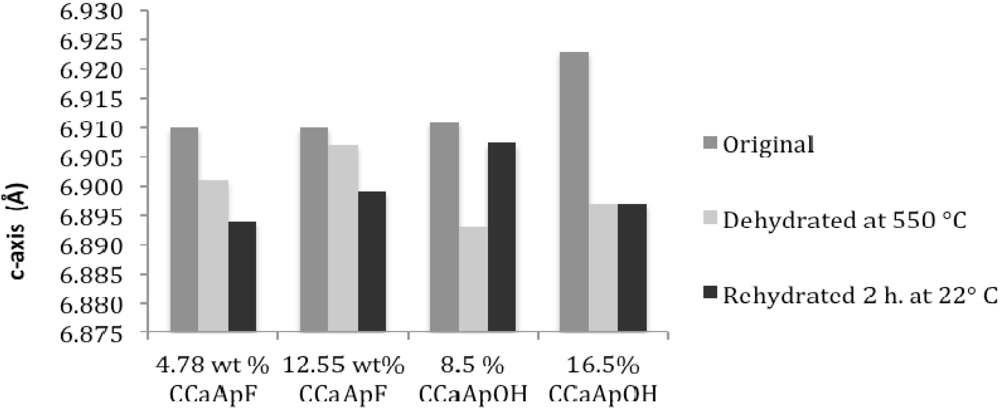
Sixth, both dehydration and rehydration lead to a generally small (<0.02 Å) increase in a-axis and a decrease in c-axis value relative to the lengths of the axes in the original hydrated apatite. The slightly longer a-axis of tooth enamel compared to precipitated hydroxylapatite has been attributed to the presence of lattice water [
5,
23]. Holcomb and Young observed a contraction in the a-axis of about 0.014 Å at about 300 °C, which they attributed to loss of structural water [
23]. LeGeros
et al. [
5] found a decrease in the a-axis of a synthetic carbonated hydroxylapatite upon heating from 200 °C to 400 °C, which they inferred was due to water loss. This result was followed by a significant increase in the a-axis during heating to 800 °C, which they attributed to loss of carbonate. Their work also shows that after heating to 550 °C the a-axis is longer than the length at 200 °C [
5]. Hence, the larger values obtained by us for the a-axis after heating to 550 °C are in agreement with the findings of LeGeros
et al. [
5]. The larger value for the a-axis after heating to 550 °C is likely due in part to thermal expansion of the lattice [
6,
19]. Although the removal of a species from a structure would normally suggest a decrease in “size” of the structure, it is possible that the presence of water in the c-axis channels (see below) provides for increased attraction of ions to these H-bonding, dipolar water molecules. Such attraction in turn may produce anisotropic contraction leading to a slight decrease in the a-axis and an increase in the c-axis relative to the anhydrous apatite. It should be noted that there is more variability in the a- and c-axis data after rehydration at 22 °C for 2 h.
Two simple models can be envisaged to account for these observations. The first involves the formation of the apatite structure in a two-step process. In the first step the structure begins to assemble by ion aggregation, and portions of the hexagonal structure form with water molecules in the channels defined by the Ca(2) cations. In the presence of carbonate ions, the second step involves replacement of phosphate ions by carbonate ions (in type-B substitution) and evolution of the structure. In the carbonated apatite structure, the accommodation of water molecules does not require widening of the channels in the concomitant removal of Ca(2) and hydroxyl ions. Rather, the water molecules are present in the nascent structure that is formed before substitution occurs. A second model involves simultaneous incorporation of water and carbonate into a poorly ordered phase. This model does not suffer from the kinetically unlikely replacement of carbonate for phosphate necessary in the first model. Both models rationalize the lack of relationship between carbonate and water content. Because a total of two molecules of water per unit cell can fit between the monovalent anions in the channels this model also accounts for the nearly constant value of mpuc of close to two.
Dehydration requires an increase in kinetic energy sufficient to evacuate water molecules from the channels, which presumably exit primarily at basal crystallite terminations along the c-axis [
19,
23]. With a c-axis length of ca 7 Å, only about 7 to 10 unit cells stacked along the c-axis provide structures of the appropriate length for a newly formed crystallite. For water molecules toward the interior of the crystallite, the energy required for expulsion will be greater than for those closer to the terminations. It is therefore expected that removal of all of the structural water would be difficult and that water expulsion would occur over a range of temperatures.
Because water is confined in the solid apatite and translationally free in the surrounding liquid water, removal of the structural water molecules from the solid is an entropically favored process. Moreover, dehydration is an enthalpically unfavorable process (∆H > 0) because of the stronger hydrogen-bonding and charge-dipole interactions within the apatite relative to those in bulk water. Because the temperature dependence of the equilibrium between structural water and bulk water is controlled by the enthalpy change for the dehydration process, an increase in temperature should increase the amount of water displaced during dehydration. For the reverse process of rehydration, an increase in temperature should decrease the amount of water replaced into the structure of the apatite. However, rehydration appears to be mainly a rate-driven process that benefits primarily from the kinetic energy of the bulk water molecules and their rate of diffusion into and through the apatite structure.
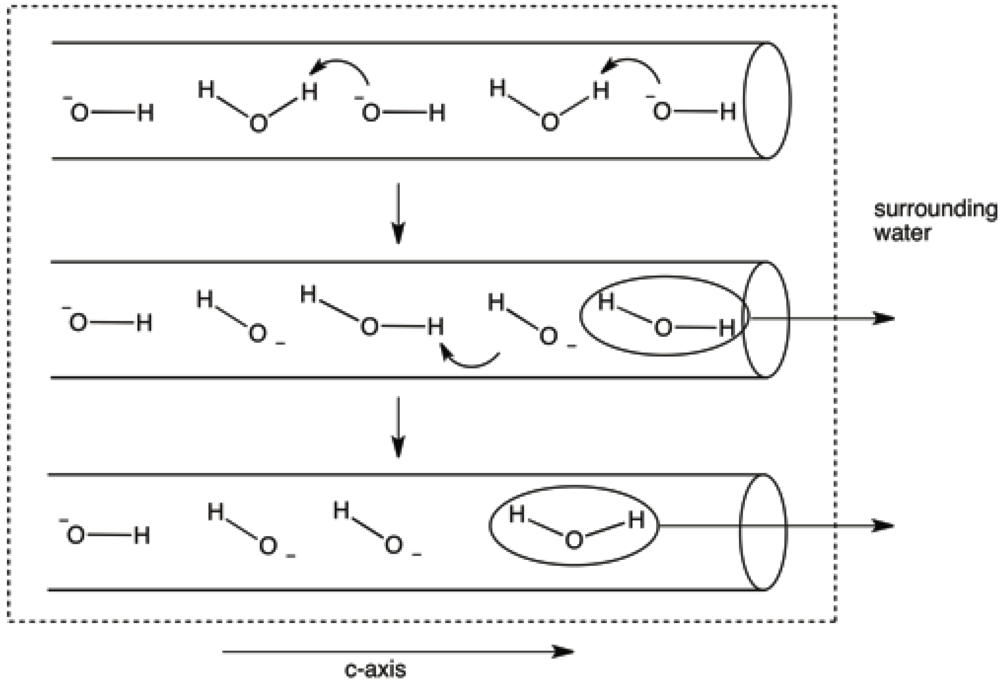
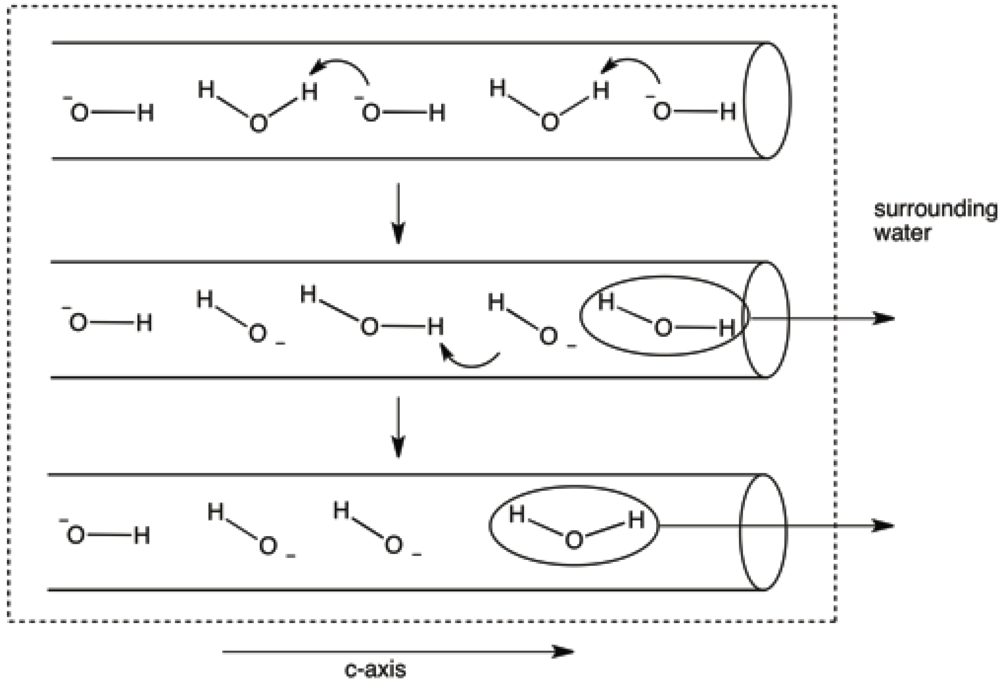
It seems likely that water would enter, and leave, the structure through termination sites of the c-axes. One could further speculate that the passage of water molecules (both during dehydration and rehydration) through the c-axis channels might occur, especially at higher temperatures, by transfer of a proton from a water molecule to a neighboring hydroxide ion (see
Figure 14), resulting in expulsion of water molecules at the basal termini and an increase in hydroxide ions in the channel. Eventually, repulsion of proximate hydroxide ions and attractions to other ions would result in movement of the ions to their lowest energy lattice positions, and dehydration would be complete. This mechanism would be consistent with the well-known observation that the hydroxide ion concentration in carbonated and non-stoichiometric apatites increases as a function of temperature [
4,
23,
30,
31,
32,
33,
34,
35,
36].
Figure 14.
Model invoking proton transfers from water molecules to hydroxide ions in the c-axis channels. This model eliminates the necessity for translation of water molecules past hydroxide ions as the former exit the c-axis terminus.
Figure 14.
Model invoking proton transfers from water molecules to hydroxide ions in the c-axis channels. This model eliminates the necessity for translation of water molecules past hydroxide ions as the former exit the c-axis terminus.
Other researchers have proposed that the lattice water molecules occupy hydroxide vacancies [
5,
11,
37,
38,
39]. The processes of both dehydration and rehydration therefore increase diffusion along the c-axis channels possibly stimulating recrystallization. These changes are also consistent with our observations of changes in peak widths in XRD patterns.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}