
Global Occurrence, Geology and Characteristics of Hydrothermal-Origin Kaolin Deposits

1
Department of Geological Engineering, Istanbul Technical University, 34469 Istanbul, Turkey
2
Department of Geology, University of Georgia, Athens, GA 30602, USA
3
Department of Chemical & Chemical Processing Technologies, Konya Technical University, 42250 Konya, Turkey
*
Author to whom correspondence should be addressed.
Minerals 2024, 14(4), 353; https://doi.org/10.3390/min14040353
Submission received: 12 March 2024
/
Accepted: 20 March 2024
/
Published: 28 March 2024
(This article belongs to the Section Mineral Deposits)
Abstract
:Kaolin-group minerals occur in nature as the result of high-sulfidation acid sulfate, sulfur-poor HCl-, HF- and H2CO3-rich acidic fluid-related hydrothermal alterations and in situ geochemical weathering. These minerals possess different crystallographic and chemical properties that determine their application areas, mainly in the ceramic and paper industries, and as nanocomposite materials. The physicochemical properties of hydrothermal kaolin deposits are the result of the type of parent rock, the effect of the regional tectonism-associated magmatism, and the chemical features of hydrothermal fluids that interact with the deep basement rocks. However, understanding these geothermal systems is one of the most challenging issues due to the rich mineralogical assemblages, complex geochemistry and isotopic data of hydrothermal alteration zones. This study evaluates the formation of hydrothermal-origin kaolin-group minerals by considering their characteristics of hydrothermal alteration, isotopic compositions and differences in characteristic properties of low- and high-sulfidation occurrences; this paper also addresses mineralogical and structural differences between hypogene and supergene kaolin formations, and kaolin–alunite–pyrophyllite association, and it provides examples of worldwide occurrences. The study of the mineralogical assemblages, geochemistry and isotopic data of the hydrothermal alteration zones is one of the most challenging subjects in terms of gaining a detailed understanding of the geothermal systems. Silicification processes are subsequent to late-stage alteration after the completion of kaolinization processes, erasing existing hydrothermal mineralogical and geochemical traces and making interpretation difficult. In the early stages involving magmatic–hydrothermal-origin acidic geothermal fluids, the latter comes from the disproportionation of SO2 (+H2O) and H2S oxidation to H2SO4 in hydrothermal environments. In the later stages, due to spatial and temporal changes over time in the chemistry of geothermal fluids, the system comes to have a more alkali–chloride composition, with neutral pH waters frequently saturated with amorphous silica which characteristically precipitate as siliceous sinter deposits containing large amounts of opal-A.
1. Introduction
Humankind have used kaolin since the early ages to make ceramic wares, which have existed in every phase of our life. Archeologists have discovered that glazed pottery-type ceramics were first made in Anatolia in 6000 BC. These initial ceramic products spread to the Aegean Islands and then to other Mediterranean countries after Anatolia and Egypt. In this way, a key industry was born. The oldest known kaolinite-rich ceramic artefact is dated as early as 28,000 BCE (Before Common Era) during the Paleolithic period [1]. During the Shang (1700–1027 BC) dynasty period, the first fired glazed ceramics were produced in China. At Erlitou site in Yanshi, Henan province, fired ceramics appeared in the 13th–17th centuries [2]. The kaolin group is characterized by one tetrahedral and one octahedral (1:1) sheet and a dioctahedral structure. Kaolin groups can have mineralogical differences depending on their formation environment and which parent rocks they are derived from, and this property controls which industrial area they are used in, such as ceramics, porcelain, paper and plastics. Detailed studies of clays in the regolith (critical) zone, their characterization, the formation of clay minerals, biological factors, redox reactions and clay sequences have been previously discussed [3]. Kaolinite is soft, earthy and usually a white-to-cream color, but its color may change to white in yellowish and reddish kaolinite-rich rocks containing goethite, hematite and ferrihydrite and depending on other impurities contained in the rock. The kaolin-group minerals include dioctahedral minerals kaolinite (Al2Si2O5(OH)4), dickite, nacrite and halloysite (Al2Si2O5(OH)4·2H2O).
The physicochemical properties of kaolinite-rich rocks depend on crystal structures, together with other accessory minerals, such as illite, smectite, quartz and alunite-group minerals. Kaolin-group minerals are formed as (1) hydrothermal alterations in volcanic rocks and tuffs, (2) a weathering (in situ residual) product from mostly Fe-poor granites, rhyolites, andesitic tuffs or even gneisses, (3) sedimentary kaolins which are eroded from a source area, transported and deposited in a continental or coastal environment and (4) mixed types of kaolin deposits (supergene alteration in hydrothermal and/or sedimentary deposits) [4,5]. All economic sedimentary kaolin deposits are formed from in situ weathered (and deferrated) fine-grained sediments that often have detrital kaolinite components, and these formation processes are the combination of (2) and (3). The supergene origin of worldwide kaolin deposits has been discussed in detail in a previous study [6]. In situ and hydrothermal kaolin, called primary kaolin, is formed by processes involving surface and underground waters or hydrothermal fluids. Sedimentary kaolin, called secondary kaolin, is kaolinized after deposition and subsequently weathered by the presence of organic acids; nitrification and acidification take important roles during these processes. Dickite is a high-temperature mineral [7] and nacrite forms as a minor mineral along the fault and shear zones under high stress conditions where geothermal fluid temperature is >80 °C. Halloysite occurs, as both hydrated and dehydrated types, in saprolites as a weathering product in tropical soil profiles and as a hydrothermal mineral next to limestone blocks which provide both a geochemical buffer system and drainage systems that are suitable for discharging the mineral [4,8,9]. The most common parent minerals for the occurrence of kaolin-group minerals are glass-rich volcanic materials (such as tephra, unconsolidated ash and lapilli tuff), pumice or even perlite, and rarely muscovite or micas which are much more resistant to hydrolysis compared to feldspars and felsic rocks [10]. The alteration in K- and Na-feldspars to become kaolin-group minerals is explained by a hydrolysis reaction:
2KAlSi3O8 + 9H2O +2H+ → 2K+ + 4H4SiO4 + Al2Si2O5(OH)4
Due to the amorphous structure of tuffs, which are glassy felsic volcanic rocks, and their porosities, they have a greater tendency to undergo hydrothermal alteration compared to other crystalline materials in nature. For this reason, siliceous tuffs are the major parent rocks of kaolin minerals in almost all geological environments under acidic conditions. High-sulfidation alteration is characterized by alunite + kaolinite + quartz +/− pyrite assemblage, which results from chemical leaching by the fluids concentrated in H2SO4. According to [11,12], these processes are created in three principal geologic environments: (1) by atmospheric oxidation of sulfides in the supergene environment, (2) by atmospheric oxidation at the water table in the steam-heated (oxidation H2S; H2S2− + H2S6+O4 = So + S4+O2 + 2H2O) environment of H2S released from deep sources and (3) by the disproportionation of magmatic SO2 to H2S (SO2 disproportionates according to the reaction 4S4+O2 + 4H2O = 3H2S6+O4 + H2S2+) and H2SO4 in magmatic hydrothermal environments (condensing magmatic vapor; H2S > SO2) and in magmatic steam environments (expanding magmatic vapor; SO2 > H2S) in silicic and andesitic volcanic terranes.
Acidic hydrothermal waters are formed in two ways: (1) rising gases from deep boiling reservoirs that are mixed with circulating oxygenated meteoric water (steam is heated) in near-surface conditions [13] or (2) condensation of hydrothermal H2S in surficial oxygenated groundwater (steam condensates) [14]. These fumaroles are originally dissolved under high temperature–pressure conditions in the deep reservoir and due to the oxidation of chloride by the alkali–chloride process at neutral pH, carrying dissolved silica, are separated from the fluids at a shallow depth. Fumaroles-rich hydrothermal waters are generally found in the vicinity of upflow areas above the water table at shallow depths [13]. For example, in the Kanlaon Volcano (Mambucal), Philippines, geothermal waters also have acidic, oxidized and sulfate-rich hot springs. The temperatures of these dilute acid–sulfate fluids range from 66° to 95 °C, and they are low in Cl−, rich in SO42−, characterized by an elevated SO42−/Cl− ratio of ~15 and have pH values between 3 and 6.7 [15]. Kaolinite, halloysite, cristobalite and alunite are the characteristic mineral assemblages of advanced argillic alteration formed by acid–sulfate hydrothermal fluids.
Alkali–chloride waters are typical of the deep and high-temperature geothermal fluids which exhibit large-flow springs in the field. Argillic–propylitic alteration exhibits silica (amorphous silica, cristobalite and quartz) albite, adularia, illite, chlorite, zeolites, pyrite, pyrrhotite and base-metal sulfides. The subsurface silicification and silica sinter are found to be discharge features, which indicate the presence of fossil hot springs and geysers activities, and the temperature of waters is about ~200 °C [13].
The aim of this review is to focus on high-sulfidation hydrothermal alterations, detailing evaluations of genesis and isotopic composition, and distinguishing high- and low-sulfidation processes, morphological diversity and mineralogical and geochemical properties, and to compare different schools of thoughts. The close association between kaolin-group minerals, silica sinter deposits and silicification processes will be discussed with special reference to the distinction among hydrothermal alteration zones and the comparison of worldwide famous kaolin deposits.
2. Mineralogy and Chemistry of Kaolin-Group Minerals
Kaolin minerals have a triclinic crystal system and consist of a tetrahedral (T–SiO4) sheet and oxygen ions bonded to an octahedral (O–Al(OH)3) sheet. The structural formula for kaolin is Al2Si2O5(OH)4 and it theoretically contains SiO2 46.54%, Al2O3 39.50% and H2O 13.96%. There are polytypes of kaolin-group minerals: kaolinite, dickite, nacrite, 7 Å halloysite and 10 Å halloysite. The regularly distributed cation site vacancies in the octahedral sheet and the distortions caused by these vacancies with respect to the bonding of the octahedral and tetrahedral sheets appear to be the dominant structural feature of these minerals [16]. Kaolinite forms book-shaped triclinic flat crystals, whereas dickite, although of monoclinic symmetry, exhibits rhomboidal prisms. Kaolinite consists of a layer stacking sequences which are a one-layer polymorph in which the vacancy is located at the B position. Dickite is formed by regular alternation which is a two-layer polymorph of the stacking sequence of the B- and C-type layers. The dimensions of the dickite unit cell are smaller than those of the two kaolinite unit cells that are superposed along the c∗ direction. Nacrite is a monoclinic polymorph with two layers [17]. The distinguishing of kaolin-group minerals using XRD and other crystallographic data is listed in previous works [6,16,18]. Detailed information on the mineralogy and crystallographic structure of most clay minerals for those readers who are interested can be found in the textbooks of [3,19,20,21].
Dickite, as a high-temperature mineral, is less common than kaolinite and it occurs alongside pyrophyllite as a high-temperature mineral and also in porphyry environments, thus in subvolcanic porphyries. The grain size of dickite is larger than that of kaolinite, and dickite is known to have a wide stability field in the field of diagenesis and metamorphism, and is also commonly found near to fault zones where the hydrothermal fluid temperature is the maximum [9,22,23]. Ref. [24] also suggested that in the kaolinite–dickite conversion process, there is a dissolution–reprecipitation process. Because dickite and nacrite are restricted to hydrothermal settings, nacrite developed into a singular occurrence as euhedral, pseudo-hexagonal or elongated crystals and it has been identified as filling very thin, irregular veins associated with quartz and dolomite in the Betic Cordilleras, Spain [23]. During burial diagenesis, the kaolinite-to-dickite transition in sandstones occurs with increasing depth (and temperature) over the interval from 3000 m to 5000 m in the North Sea, corresponding to a model temperature range between 100 °C and 165 °C [25]. Moreover, the replacement of pre-existing kaolinite by dickite was observed with increasing depth of sandstones; at 1.4–2.7 km below the seafloor (100 °C), sandstones predominantly contain kaolinite, whereas deeper than 3.2 km (130 °C), mainly dickite is present. The transformation of kaolinite to dickite occurs at 3.1 km (120 °C) below the sea floor on the Norwegian continental shelf [26]. Nacrite and kaolinite occurrences from the alteration in Permian ignimbrites, Rochlitz, Germany, developed as a result of a hypogene alteration in rhyolites, rhyolitic tuffs, and are often interpreted as a subtype of high-temperature- and high-sulfidation-type argillitization [27,28].
The hydrated halloysite 10 Å, typically characterized by a tubular structure, accommodates interlayer water and irreversibly dehydrates even at room temperature in a couple of weeks to a 7 Å structure [29,30,31]. Halloysite is a fast-forming metastable precursor to kaolinite. Hydrothermal alteration processes and the micromorphological characterization of the kaolinite samples of the Turplu halloysite mine, Turkey, using FE-SEM revealed booklet-shaped structures (Figure 1a,b), alunite rhombohedral (Figure 1c), which suggests steam-heated genetic origin [32] and presents excellent examples of halloysite tubular structures (Figure 1d). Halloysite presents a hollow open-ended tubular micromorphology (Figure 1e) and spherical-shaped crystal growth of halloysite on the edges of kaolinite flakes (Figure 1f), with tubular internal diameters of 10–20 nm and outer diameters of 110–220 nm [33]. The globular cluster microstructures appear to represent an original growth habit as the result of in situ crystallization from a solution phase, and these globular clusters show diameters of 80–200 nm with unrolled particles adjacent to each other (Figure 1f). Possible spherical growth of halloysite on the edges of kaolinite flakes and altered feldspar grains, developing bending crystals and curved surfaces, may have formed through the rapid dissolution and continuous distribution of crystal dislocations [34,35,36]. Pseudo-spherical or spheroidal halloysite is related to the saturation state of solutions. Due to the high dissolution rate of volcanic glass, the solution in contact with the glass is likely to be highly supersaturated [35].
Ref. [37] explained that cylindrical forms of halloysite are typically smaller in both diameter and length than prismatic forms and prismatic halloysite is a common morphology found in many samples. Ref. [38] proposed that tubular halloysite initially forms as the hydrated type with the pseudo-mirror plane of the kaolinite layers perpendicular to the tube axis, and later dehydrates and finally transforms into a prismatic halloysite. Some kaolinite crystals occur as curved flakes with dimensions between 2000 and 4000 nm. Tubular halloysite has three types of surfaces: the internal lumen surface, covered by Al–OH groups; the external siloxane surface, with a negative surface charge; and the interlayer surface, blocked by hydrogen bonding [39]. The TEM observations show that almost all halloysites have a hollow interior (Figure 2). End views of the tubular particles by the replica method (at −180 °C) are shown for samples dominated by 10 Å halloysites and 7 Å halloysites in Figure 3. End views reveal hydrated, ring-shaped tube diameters measuring ~300 nm. Dehydrated spiral-shaped halloysite measures 500 nm. Tube diameters appear widened due to the replica technique, which provides ~20 nm thick platinum and carbon coatings on the clay surface. Some crevices exist in the inner part of the dehydrated halloysite, while other tubes become segmented into units of considerable thickness upon dehydration. The mechanism for outer diameter expansion (from 300 nm to 500 nm or 60% change) is related to the rolled structure of halloysite tubes, the cleavage surfaces of which lose the attractive force between the layers. During the dehydration of rolled tubular halloysite, due to the releasing of 2H2O molecules, the interior bonding breaks free and the tubes expand. Ref. [40] similarly described the expansion mechanism of dehydrated halloysite tubes as being like book pages rolled up by a hand and then expanding upon relaxation of the grip (Figure 3). As a summary, Figure 4 shows a schematic representation of acid–sulfate alteration processes of the various crystal habits under hydrothermal conditions.
3. Mineral Stability and Thermodynamics
The first thermodynamic approach of the Al2O3-SiO2-H2O system at 25 °C and in 1 atm conditions was created for the physicochemical processes of sedimentary kaolin deposits by [41,42]. The stability diagrams of mineral pair associations, such as gibbsite–kaolinite, kaolinite–montmorillonite, amorphous silica–kaolinite and montmorillonite–amorphous silica were prepared based on field observations of natural occurrences [41]. As a natural phenomenon, amorphous silica controls high silica levels, and kaolinite forms at intermediate and gibbsite at low silica levels. The stability of these minerals is controlled by pH vs. the activity of aluminum ion relations. The formation of gibbsite, kaolinite, montmorillonite and amorphous silica appears to be controlled by a combination of the rate of kinetics reactions and solution equilibria. The rate of dissolution of unstable silicates appears to control the H4SiO4 level and the most stable silicates at the H4SiO4 level appear to precipitate in response to solution equilibria [41]. From a thermodynamic point of view, only quartz and diaspore are stable minerals in the stability diagram. Moreover, when kaolinite becomes more stable than montmorillonite, it represents the lowest aluminum hydroxide potential until the silica activity decreases to the activity of H4SiO4 of about 4.7. In the low-silica environment above the activity of H4SiO4 of 4.7, the most stable mineral of the group is gibbsite.
The thermodynamic properties of the formation of kaolin-group minerals at 298 °K have been determined: kaolinite 24,120.2 ± 6.6 kJ/mol, dickite 24,107.6 ± 5.7 kJ/mol, nacrite 24,104.0 ± 7.6 kJ/mol and halloysite 24,097.5 ± 5.6 kJ/mol [43]. Due to small differences between the values, they do not change significantly with pressure and temperature throughout their formation range. For this reason, due to the enthalpy values of the reactions, halloysite, nacrite and dickite are metastable phases with respect to kaolinite and must be interpreted in terms of chemical reaction rates. In addition, experimental thermodynamic studies have revealed that the kaolinite-to-dickite transformation reaction can be performed in acid solutions at temperatures ranging from 150° to 300 °C under vapor-saturated conditions [44]. One of the important conclusions that they reached is that kaolinite is metastable relative to dickite at temperatures of at least 350 °C and many hydrothermal systems are controlled by the kinetics of this reaction. In addition, the kaolinite-to-dickite transformation in natural systems is an irreversible process due to crystallographic orientation, as seen in the Düvertepe kaolin region [9]. Observations of active hydrothermal systems in Japan confirm that dickite forms in hydrothermally altered rocks and veins at higher temperatures (150–270 °C) in deeper, altered zones [45]. At these temperatures, ref. [44] supported this idea and explained that dickite precipitates directly from supersaturated solutions without the transient formation of kaolinite. The stability fields of K-feldspar, white mica, pyrophyllite and kaolin-group minerals are mostly controlled by the activity ratio of K+/H+ and temperature [46]. The polytype transition of kaolinite to dickite in active geothermal systems occurs at about 200 °C [47] and these observations were confirmed by previous experimental studies [44]. Zonal arrangements of paragenesis in the three types of hydrothermal alteration systems are shown based on chemical properties in solutions (Table 1).
Experimental studies in simulated laboratory conditions at various pH values (2 to 14) and temperatures (200 to 240 °C) revealed that there was no influence of temperature on defect density but pH is important [49]. From pH 2 to 8, kaolinite formed, with increasing pH > 8, pseudo-boehmite and kaolinite started to form, and above pH of 10, kaolinite and hydrated nepheline and paragonite formed. In addition, a continuous series was observed from a low-defect kaolinite at the most acidic final pH to a high-defect kaolinite at the most basic final pH. Ref. [49] also found out that increasing pH values cause an increase in both cation adsorption and elongation in kaolinite particles.
High-temperature experimental studies to better understand equilibrium thermodynamic models are more reliable for describing kaolinite dissolution and nucleation processes. One of these reliable studies indicated that the hydrothermal precipitation of kaolinite starts from amorphous gel-like aluminosilicates, with a Si/Al ratio from 1.8 to 0.76; temperatures of 150, 175, 200, 225 and 250 °C were examined [50] and it was found that kaolinite crystallinity is dependent on time, temperature and the Si/Al ratio of the starting material. It is also known that the rate of kaolinite crystallization increases with higher compared to lower temperatures. During the first stage of crystallization, a kaolinite-like structure forms due to much faster reaction rates (82 ± 5 kJ/mol), and at the second stage, hexagonal-outlined platy crystals form with average activation energies of 71 ± 5 kJ/mol [50].
4. Stable Isotope Studies
The term “isotopes” is derived from Greek, meaning equal places, and the number of protons in nuclei are the same but the number of neutrons are different. The higher mass numbers (the sum of the number of protons and neutrons in the nucleus) indicate heavier isotopes (more neutrons), and therefore, the density of water 1H216O is lighter than 2H216O (D216O) and 1H218O at 20 °C. The partitioning of isotopes between phases, components, aqueous species or organisms with different isotope ratios is called “isotope fractionation” and there is also equilibrium isotope fractionation, as well as two types of kinetic isotope fractionations: reaction kinetics (the breaking of bonds) and transport kinetics (diffusion). If two components or aqueous species are in isotopic equilibrium, they are controlled by energy levels; components with a stronger bond strength tend to concentrate the heavier isotopes [51]. Isotopic fractionation can be measured by isotope analysis and ratios of isotopes, which are an important tool in understanding geochemical and biological systems. In this concept, stable isotope studies have played an important role in advancing the understanding of the origin of mineralization systems and the sources of H, O, S and C in the evolution of geochemical processes. In particular, H and O isotopic studies of meteoric and geothermal waters have a distinct advantage because the sources of aqueous solutions can be better determined. According to [52], supergene clays can be distinguished from hypogene clays; supergene kaolinites are generally richer in O18 and depleted in D relative to hypogene clays from a given porphyry environment. Refs. [53,54] conducted stable isotope geochemistry studies on clay minerals, shales and ocean sediments. Ref. [52] critically reviewed existing data, discussed discrepancies and made some new suggestions for equilibrium fractionation factors considering all existing data on clay minerals by proposing equilibrium H- and O-isotope fractionation equations between water and individual clay minerals. The original isotopic composition of clay minerals does not change unless they are affected by diagenetic or metamorphic processes.
Stable isotope studies on the active hydrothermal system in Valles Caldera, New Mexico, presented that there was little or no change between the isotopic composition of the meteoric water entering the hydrothermal system and that of hydrothermal waters flushed out on the surface after deep circulation [55]. The δ18O values of altered volcanic rocks are about 5.5‰ lower than those of unaltered rocks at the edge of the caldera. These measured data corroborate with the idea of [51] as described above. In addition, the δ18O values are about 7‰ lowered in altered volcanic rocks at the center of the caldera. In addition, these findings have concluded that the water/rock ratio is very large in the Valles’ hydrothermal activity. The Jemez volcanic field, New Mexico, presents two types of mineral assemblages. First, active and fossil acid–sulfate processes produce kaolinite–alunite alterations which occur along the caldera ring fracture and within the resurgent dome, and second, the chlorite–sericite alterations are mostly formed in subsurface rocks [56]. The isotope data of water and kaolinite show that the interaction temperatures are between 60 and 280 °C, which is a very large interval, in the Valles Caldera. Recent studies revealed that the present-day meteoric waters are not in chemical equilibrium with kaolinite. They have probably formed from earlier chemically and isotopically different hydrothermal fluids, most probably from a steam-heated environment.
Kaolinite and dickite δ18O values from the Cretaceous shales of the Gibraltar Strait suggest that an increase in the water isotopic composition and smectite dissolution almost occurred at the same time and consequently triggered the occurrences of I/S mixed-layers and quartz [23]. The minimum formation temperatures of 62 °C for kaolinite and 86–96 °C for dickite can be calculated, indicating that the depth of burial was about 2–3 km. But dickite progressively replaced kaolinite within the depth of 2500–5000 m in sandstone-–hale reservoirs of the North Sea [25] and the transition occurred in the Rotliegend sandstones at about 130 °C.
The origin of kaolinization in the surficial conditions of the Ploemeur Variscan leucogranite, Armorican Massif, NW France, is debatable [57]. It is more likely that low-temperature hydrothermal fluids slightly altered near-surface rocks that may have led to an initiation of the alteration process. Some quartz veins may be evidence of late-stage hydrothermal events at temperatures of the order of 100–150 °C (δ18OQtz = 23 ± 1‰). By the first stage of partial alteration, surface rocks became more open to surface weathering conditions; this is a kind of “preparation of ground process”. This event probably was favored for the development of subsequent supergene alteration, the overprinting process and the increasing permeability, like similar processes proposed for the two-step processes of kaolinization in Cornwall, UK [58]. Cornwall’s granites are two-mica granites, meaning they contain muscovite and biotite, plus quartz, K-feldspar and plagioclase (alkali granites). Similar processes of late-stage fracture related to occurrences of six parallel silicified veins inside kaolin bodies have been observed (Figure 3 in [33]), which indicates changes in pH and in the chemistry of geothermal fluids from early-stage slightly acidic solutions to late-stage near-neutral pH alkali–chloride geothermal activities. The genesis of Cornwall kaolinites, formed in a tropical-to-warm temperate climate over a time period extending from 300–275 Ma (late Carboniferous–Permian), in altered granites is also debatable, because there is some evidence of low-temperature supergene and/or deep hydrothermal origin [58].
Another mixed-type example of kaolin deposit is that similar to the Armorican Massif in France whereby kaolins are formed by in situ alterations of common silicate minerals in the Cape Peninsula Granite, South Africa [59]. The calculated isotopic model temperature of the kaolinite indicates formation at around 20 °C in equilibrium with water that had isotopic ratios slightly lower than present-day meteoric water. Various kaolin deposits of the Cape Peninsula region for the bulk <38 µm fraction present δD (−60 to −50‰) and δ18O (18.0 to 20.2‰) values. Isotopic data also suggest that a slightly cooler and wetter climate rather than a tropical climate was dominant at the time of kaolinization during the Quaternary. The low-temperature weathering origin of the kaolinite is supported by the fractionation factor (αnonOH/OH) which is determined for a single kaolinite sample. Only, the illite did not form in equilibrium with the kaolinite but possibly formed during late-stage magmatic–hydrothermal processes through fluid interaction with the granite at higher temperatures [59]. The isotopic composition of ambient meteoric waters suggests that kaolinite formed by low-temperature meteoric weathering. As a result, early-stage hydrothermal illite and then late- and main-stage supergene kaolin occurred after emplacement of the pluton.
The δ18O, δD and δ34S values and trace element data were used for the acid–sulfate steam-heated alteration of andesitic tuffs in order to elucidate the genesis of halloysite and alunite deposits in the Biga Peninsula, Turkey [60]. The range of δD values of halloysite samples from Turplu is from −58.4 to −68.6‰ and the δ18O values are in the range between 16.7 and 18.1‰. In addition, pyrite and alunite samples from the same mine have δ34S values of 0.6/1.8% and 4.8/7.9‰, respectively (Figure 5). These positive δ34S values indicate magmatic origin. Sulfur gases (H2S-SO2) of hypogene origin rose from deep magmatic sources, where they were dissolved in hydrothermal fluids and carried throughout the fracture systems to the surface where they encountered cool and oxygenated groundwater.
Alunite (KAl3(SO4)2(OH)6) and halloysite associations were frequently seen in upwelling hydrothermal waters containing major H2S and SO2 gases. The occurrence of H2SO4 in this hydrothermal system enhanced the acidity of geothermal fluids, thus enhancing advanced argillic alteration. Hypogene alunite samples also have high (1%–2.5%) P2O5 contents, suggesting deep chemical leaching of phosphorus from a magmatic origin. The P-bearing analogue is crandallite (CaAl3(PO4)(PO3OH)(OH)6), with a rare earth element substituting Ca and forming fiorencite (CeAl3(PO4)2(OH)6); intermediate compositions also occur, such as woodhouseite (CaAl3(PO4)(SO4)(OH)6), which commonly contains Sr, Ba and Pb. Mineralogical and chemical zonation suggests hypogene alunite, which provides information on changes in fluid composition over time, and the detailed electron microprobe analyses of P-bearing alunite are listed [60].
The Tolfa district, Italy, and Marysvale, Utah, are good examples of steam-heated acid–sulfate environments above adularia–sericite-type epithermal systems [11]. Such acid–sulfate alteration zones represent characteristic vertical zoning systems which occur over adularia–sericite-type bases and precious metal ore deposits. The δ34S values of most of the alunites of steam-heated origin suggest the same as the pioneer H2S and δD values similar to those of local meteoric water, and isotope studies of alunites present reasonable temperature ranges from 90 to 160 °C [11].
In the Blanquita kaolin mine, Province of Río Negro, Argentina, kaolinites range from +3.6 to +9.2‰ for O and from −74 to −103‰ for H and the calculated isotopic composition of the geothermal fluid is in equilibrium with kaolinite at 350 °C [62]. These values are compatible and isotopically equilibrated with a mixture of high-temperature siliceous magma or magmatic hydrothermal fluids and meteoric waters.
The Karacayır kaolin deposit in western Turkey is hosted by rhyolite and andesite of the Miocene volcanics, and by the Paleozoic mica schists [63]. Kaolinite is mostly concentrated together with accessory minerals in the center but outwards and upwards from the deposit, there is relatively more smectite, illite, chlorite and Fe-oxide minerals. The Karacayır kaolinite and smectite have δ18O and δD values ranging from 11.6 to 20.4‰ and from –79 to –112‰, respectively. The isotopic model formation temperatures were determined for the Karacayır kaolinite (61.6–131.7 °C) and smectite (61.2–148.9 °C). The negative δ34S values for pyrite (–3.4‰), chalcopyrite (–0.2‰) and gypsum (–0.8‰) reflect formation under the influence of a mixture of hydrothermal activity and a minor amount of biologic activity, and isotope equilibrium temperatures are in the range of 80–125 °C, calculated from pyrite–chalcopyrite pairs, suggesting an epithermal alteration process. Hypogene mineralization is characterized by the sulfides (pyrite and chalcopyrite) and sulfates (gypsum). The negative δ34S value for pyrite suggests that local meteoric water partially mixed with sulfur-bearing geothermal fluids during the last stages of hydrothermal activity or the input of biogenic activities, such as the dissolution of sulfur which possibly came from coal deposits in groundwater circulation in the vicinity of the kaolin deposit [60,64]. In addition, negative δ34S values indicate that the sulfate/sulfide ratio decreased ƒO2 with the escape of sulfur-bearing gases from the melt and the consequent depletion in the δ34S values [65]. Also, ref. [63] reported that the low isotopic values of gypsum near the top of the deposit may suggest that gypsum forms rapidly relative to the alteration of the pyrite-bearing host rocks and recent oxidation [66]. If gypsum, as in the first example, is heavier than its precursor sulfide, there is an indication of some degree of biogenic sulfate reduction before gypsum precipitation.
5. Hypogene vs. Supergene Origin of Kaolinization
Hypogene processes have been commonly used by economic geologists and they refer to ore deposits, associated gangue and alteration products that were formed by the presence of ascending high-temperature solutions. Supergene processes apply to the rock weathering near the surface conditions by descending meteoric waters. In many geothermal areas, it has been shown that meteoric–hydrothermal solutions may be ascending from a few km deep. The physical–chemical conditions of the occurrences of kaolin deposits are the subject of scientific debate on whether kaolinization has a supergene or hypogene origin and what is their structural impact on the phyllosilicates. Primary kaolin deposits may form in situ from granitic, siliceous volcanic and pegmatitic rocks through weathering (supergene kaolins) or hydrothermal activity (hypogene kaolins) [4,67]. Several methods have been proposed to distinguish between supergene and hypogene kaolin deposits, such as the following:
- Chemical compositions of kaolin [22];
Supergene or hypogene origins of kaolin deposits are more associated with regional tectonics, the position of the water table and the episodes of magmatism that control the circulation patterns of geothermal fluids, pH, the chemistry of the fluids and paleoclimatic conditions. In addition, the depth of low- or high-temperature geothermal reservoirs, which control kaolinization processes in active graben structure or fracture-dominated active or fossil hydrothermal fields, will affect the influence of the origin of kaolin deposits and the surrounding rocks. High-temperature fumaroles and acid springs are common surface expressions in steam-heated environments associated with degassing processes and high sulfidation (HS); they are mostly formed in stratovolcano settings at a depth leading to porphyry mineralization and intermediate sulfidation (IS) [76]. Hot spring epithermal-style mineral systems are a continuous spectrum from surface chemical precipitates to deep veins and fissure fillings (detailed process information is given in Table 2). The determination of hypogene and supergene alteration criteria of kaolin deposits is listed in Table 3.
The highwalls in some kaolin deposits exhibit the shape of the kaolinization processes and the stages of associated mineralization, as well as the influence of regional tectonics on the structural geology and chemistry of geothermal fluids, which better show in funnel-shaped structures, volcanic geomorphologies and silicified veins and are associated with fault zones. Generally, deep ascending geothermal fluids which cause fracture-dominated vein-type or funnel-shaped kaolin mineralization have more potential to create relatively pure kaolin deposits. However, it is not possible to distinguish all kaolin bodies as supergene or hypogene; many deposits are overprinted above hypogene activities.
In the high-sulfidation gold mineralization from the Amazon Craton of Brazil, it has been presented that the decreasing temperature of hydrothermal fluids is indicated by the diaspore–pyrophyllite–kaolinite association [77]. The high-sulfidation occurrences mostly consist of intense silicified veins, zones and silica sinter deposits, which are called the “silica cap”, that are covered by Fe-rich siliceous rocks. Strong advanced argillic alterations mostly present with high-temperature hydrothermal breccias in near-surface conditions and their mineral assemblages are pyrophyllite, quartz, pyrite, andalusite and diaspore, which frames alunite-rich zones with vuggy silica, that characterize strong acid sulfate leaching. The advanced argillic alteration grades with increasing temperature downward and especially outward to intermediate argillic (clay minerals: kaolinite, smectite and illite), sericitic–phyllic (fine-grained white mica, quartz, sericite and pyrite,) and propylitic (epidote, chlorite, quartz, pyrite and altering feldspars, biotite and amphibole) alterations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 2.
Comparison of the geological features of high sulfidation vs. low sulfidation (a = abundant, m = minor and vm = very minor) [61,78,79,80,81,82,83,84,85].
| High Sulfidation (HS) Acid Sulfate Type or Alunite–Kaolinite Type | Low Sulfidation (LS) Adularia–Sericite Type | |
|---|---|---|
| Based on Alteration and Mineralogy | High sulfur/metal ratio (enargite, gold, luzonite and covellite) | Low sulfur/metal ratio (sphalerite, galena, tetrahedrite, chalcopyrite, cinnabar and stibnite) |
| Silicic, advanced argillic alteration, and dominated by alunite and pyrophyllite at deeper levels | Quartz-dominant, sericite, intermediate argillic and chloritic alteration with adularia, +/− calcite, epidote and smectite | |
| Hydrothermal Fluids | Formed by acid pH solutions, S-rich, oxidized overprinting of descending fluids, probably saline initially, a predominantly magmatic origin, 4%–10% NaCl and variable | Formed by near-neutral pH, S-poor and reduced hypogene fluids, low salinity, gas-rich (CO2 and H2S), a predominantly meteoric origin and <1% NaCl |
| Genetically Related Volcanic Rocks | Mainly andesite–rhyodacite and their subvolcanic intrusive equivalents | Andesite, rhyodacite and rhyolite |
| Deposit Form | Disseminated, breccia and veinlets, dominant replacement: common stockworks, minor veins, massive sulfide replacement pods and lenses, stockwork and vuggy breccias. Irregular deposit shapes determined by host rock permeability and ore-controlling fracture systems. | Open-space veins are dominant, stockwork is common, and dissemination and replacement are minor; brecciation and cross-cutting veinlets with subhedral quartz crystals |
| Alteration Zone | Aerially extensive and visually prominent; advanced argillic (zonation: quartz, alunite, pyrophyllite, kaolinite, illite, halloysite, dickite and chlorite) | Commonly restricted and visually subtle; adularia–sericite (zonation: quartz/chalcedony, calcite, adularia, sericite and chlorite) |
| Tectonic Setting | Extensional and transtensional settings, commonly in volcano–plutonic continent-margin, oceanic arcs and back-arcs. In zones with high-level magmatic emplacements. | Back-arc environments, granite replacement contacts, above volcano-sedimentary strata, and biotite-rich gneiss |
| Silica Sinter Mass | Dominant cover at the top of altered zones from late-stage alkaline chloride solutions, depression, and focus on upflow | From near-neutral pH, commonly along fracture zones, only at surface, <200 °C, rapid cooling fluid, and boiling at depth |
| Geologic Setting | Subvolcanic to volcanic in calderas, flow-dome complexes, and rarely maars and other volcanic structures. Genetically related to epithermal porphyry copper systems | Skarn zones, volcanic–carbonate contacts, and hydrothermal breccia at depth indicates a zone of intense boiling |
| Host Rock Types | Pyroclastic, flow rocks, andesite, dacite, rhyodacite, Permeable intervolcanic units, and domes | Andesite, ignimbrites, volcanic cones, shoshonitic rocks, domes and diatremes |
| Weathering | Weathered rocks contain limonite (jarosite–goethite–hematite) and a groundmass of kaolinite and quartz. Fine-grained supergene alunite veins and nodules are common. | Iron cap-rocks are common, fault associated and large alteration zones, and limonitization is common |
| Associated Deposit Types | Porphyry Cu, Mo, Au, Ag and epithermal Au-Ag | Mn-bearing calcite and rhombic adularia crystals |
| Ore Controls | Hydrothermal breccia and diatremes, fault-controlled breccias around upflow and outflow areas, and permeable lithologies | Base metal-enriched at depth and higher concentration; hydrothermal fluids show a vertical evolution |
| Sulfur Species | Oxidized sulfur species (SO2, SO42− and H2SO4) in ore fluid/vapor; atmospheric oxidation of fine-grained sulfide within surficial weathering zone | Reduced sulfur species (HS− and H2S) in ore fluid/vapor; condensation of high-T magmatic vapor with HCl + SO2 ascending |
| Quartz Gangue | Fine-grained, massive, mainly of replacement origin; residual, slaggy (vuggy) quartz commonly hosts ore, banded veins, hydrothermal breccias, fine-grained quartz, of steam-heated origin, above water table | Chalcedony and/or quartz displaying crustiform–colloform banding, bladed quartz, cockade and carbonate-replacement textures; open-space filling bladed calcite, massive chalcedony, and comb-banded quartz, 80%–90% SiO2 |
| Carbonate Gangue | Absent | Ubiquitous, commonly manganoan |
| Other Gangue | Barite widespread with ore; native sulfur commonly fills open spaces | Barite and/or fluorite present locally; barite commonly above ore |
| Sulfide Abundance | 10–90 vol.%, mainly fine-grained, partly laminated pyrite | 1–20 vol.%, but typically < 5 vol.%, predominantly pyrite |
| Metals Present | Cu, Au and As (to a lesser degree: lesser Ag, Bi, Te and Pb) | Au and Ag (to a lesser degree: As, Sb, Se, Hg, Zn, Pb and Cu) |
| Frequency and abundance of ore and gangue minerals | ||
| Ubiquitous | Pyrite (a) Quartz (a) Enargite–luzonite (+/−) | Pyrite (a) Quartz (a) |
| Common | Chalcopyrite (m), kaolinite–dickite (m), alunite (m), illite (m), covellite (m), barite (m), native gold (vm), tellurides (vm), diaspore (vm), tennantite (+/−), tetrahedrite (+/−), sphalerite (+/−), galena (+/−) and pyrophyllite (+/−) | Illite (a), smectite (m), native gold (vm) chalcopyrite (vm), tetrahedrite (vm), arsenopyrite (m), tellurides (vm), pyrargrite (vm), chalcedony (+/−), adularia (+/−), electrum (+/−), calcite (+/−) and sphalerite (+/−) |
| Uncommon or Rare | Chalcedony (m), smectite (m), electrum (vm), selenides (vm), pyrargyrite (vm), arsenopyrite (vm), cinnabar (vm) and stibnite (vm) | Selenides (vm), stibnite (vm) cinnabar (vm), enargite–Luzonite(vm), tennantite (vm), covellite (vm), barite (vm) and kaolinite (vm) |
| Absent Except as Overprint | Calcite Adularia | Pyrophyllite Diaspore Alunite |
| World’s Famous Examples | Nansatsu, Japan Wheaton Mountain, Yukon Mt. McIntosh/Hushamu, British Columbia El Indio, Chile Summitville, Colorado Goldfield and Paradise, Nevada Temora, New South Wales, Australia Pueblo Viejo, Dominica Chinkuashih, Taiwan Rodalquilar, Spain Lepanto and Nalesbitan, Philippines Lagunas Norte, Peru | Apacheta, Peru Mule Canyon, Nevada Emperor Gold, Vatukoula, Fiji Broadlands–Ohaaki, New Zealand Hishikari, Japan Axi, Xinjiang, China Osilo, Sardinia, Italy Pongkor, Indonesia Nazareno, Peru Omu Camp, Hokkaido, Japan Golden Cross, New Zealand River Reef Zone, Watuputih Hill, Indonesia |
| Distinguishing Criteria | Hypogene Kaolin Occurrence | Supergene Kaolin Occurrence |
|---|---|---|
| Altered mineral association | Kaolinite, alunite, halloysite, quartz, natroalunite, iron oxide, sulfide minerals, montmorillonite and illite | Halloysite, kaolinite, alunite, natroalunite, illite, montmorillonite and gibbsite |
| Kaolin type | Kaolinite, dickite ± halloysite | Halloysite and kaolinite |
| Kaolinite crystal habits | Euhedral, equidimensional, blocky, booklet stacks and vermiculate booklets | Anhedral, randomly distributed and randomly scattered crystals |
| Halloysite crystal habits | Well-formed tubes; spherical | Booklet with bent ends, long and thin tubes, and spherical |
| Alunite crystal habits | Pseudo-hexagonal, euhedral < 10 µm with dissolution pits, a formation temperature 200–350 °C | Semi-euhedral < 5 µm or earthy, could occur down to 60 °C, commonly with Jarosite |
| Quartz crystal habits | Euhedral 30 µm, vuggy silica and spherical quartz | Euhedral |
| Kaolinite, quartz and alunite association zones | Quartz enrichment along the fracture zone, alunite and dickite or kaolinite around the quartz veins, kaolinite increases away from fault zone, and pyrite underlies kaolinite. Advanced stage of formation of kaolinite crystals. | Kaolin (kaolinite and halloysite) and the quartz accompanying kaolinite or halloysite may contain small amounts of alunite. Well-formed with dissolution pits; early stage of halloysite tubes. |
| Massive alunite body + opal-CT + quartz association zones | Widely distributed alteration minerals; apatite enrichment in alunite nodules | Alunite–jarosite mineralization; oxidation of primary sulfides during weathering |
| Massive silicified masses and siliceous veins along fault zones | Abundant within fault zone, >50 m thick deposits at main fault zone, silica sinter deposition and fracture filling | Poor in quartz content; scattering and fracture filling |
| Geologic setting | Hydrothermal environments and related to steam-heated process; acid–sulfate alteration zones | Influenced by semi-arid and tropical climates. P-bearing bauxites were emplaced under subtropical to tropical climates, with soil profile development. |
| (Ba + Sr) vs. (Ce + Y + La) | Enrichment in Ce + Y + La content; decreasing in Ba + Sr content | Enrichment in Ba + Sr content; decreasing in Ce + Y + La content |
| δ18O vs. δD of kaolin | δ18O enrichment; slight δD depletion | δ18O enrichment; δD enrichment |
| δ34S of alunite and pyrite | δ34S enrichment or close to magmatic S values, 16‰ to 31‰ larger than that of associated pyrite | Mostly δ34S depletion due to sulfur-reducing bacteria |
All mineral assemblages, geologic settings, geothermal fluids, weathering, sulfur species, rock types and various aspects of associated kaolin + Au deposits with high-sulfidation (HS) vs. low-sulfidation (LS) properties are summarized in Table 2. HS epithermal deposits originate from magmatic fluids which segregate into two phases during transportation. In the first phase, HS deposits are the products of the interaction of volatile magmatic fluids with groundwater, so there are fast-moving and less dense geothermal fluids. In the second phase, LS epithermal mineralization forms under the conditions of slower-moving and denser magmatic fluids and mineralization occurs as a result of groundwater dilution (lowering acid ions and pH) and the cooling and lowering of ƒO2.
According to some of the proposed epithermal models, the major difference between HS and LS deposits originates from the chemistry of fluids that causes differences in the ore body form and mineralogical assemblages; there are also partial mineralogical overlaps that may occur in some deposits and their mineral zonings of deposits differ significantly. There are not too many places on the earth which exhibit the co-occurrence of both HS- and LS-type deposits in the same volcanic field. The Seongsan hydrothermal system is one of these rare and unique places that shows the occurrence of early-stage high sulfidation of hydrothermal alunite–kaolin deposits and later-stage low (intermediate)-sulfidation episodes of epithermal Au-Ag deposits in the same geothermal field, which is closely related to silicic volcanism in the Cretaceous Haenam volcanic field, South Korea [69]. These HS and LS types of hydrothermal systems show a close genetic relationship temporally and spatially, and this presents a textbook example of the changes in pH, chemistry and temperature of geothermal fluids and depth of geothermal reservoirs through time as the results of progressively active regional tectonism and magmatism. The Cretaceous (81 Ma) hydrothermal system was initiated with the volcano–tectonic extensional system of the southwestern part of the South Korea peninsula. During the post-volcanic activities, ascending acidic and oxidized hydrothermal fluids originated from shallow deep siliceous igneous rocks which caused acid–sulfate high-sulfidation-type Au-Ag mineralization, and as a result of the HS, alunite–kaolin deposits formed in the early stage at shallow depths and were overprinted in the late stage by phyllic alteration. As the hydrothermal system evolved to later stages (~75 Ma), the ascending hydrothermal fluids were mixed with meteoric waters and became diluted, resulting in the formation of low-sulfidation-type Au–Ag deposits throughout open-fracture systems surrounded by hot springs due to the influence of phyllic alteration. The distribution of gold mineralization within an acid sulfate alteration zone are is to the quantity of the dense fluids available, which determines the extent of mixing of the metal-bearing dense fluid with groundwater.
Well-known magmatic-origin HS processes are located in the Nansatsu district of southern Kyushu, Japan [88,89]. Their distinguishing feature is the vuggy silica and more massive silicified rocks at the core where the host rock is initially permeable and enveloped in a very narrow (1–3 m) clay alteration zone at the rim of the deposit. Silica leached from andesitic lava and pyroclastic flows by acid chloride–sulfate waters, and these probably formed when magmatic vapors containing HCl and SO2 (+H2SO4) condensed into meteoric water [89]. Kaolinite–alunite–(pyrophyllite–diaspore) alteration products overlying the silica zone and propylitic and illite–smectite alterations were surrounding the silica and silicified zones. The Nansatsu-type gold deposits were found to be irregularly disseminated in the silica core, which was probably formed about 200–300 m below the paleosurface. Generally, the pH of geothermal fluids under the influence of the mixture of meteoric waters affects the rock as follows: at pH 2, vuggy silica, at 2–3, kaolinite–illite, at 3–5, illite-smectite and above 5, propylite alterations occurred in the Nansatsu deposits [88]. Hypogene alunite of the Nansatsu district formed from a mixture of magmatic fluid (δ18O = 7 ± 2‰; δD = −25 ± 5‰) with local present-day meteoric waters (δ18O = −7.3‰; δD = −48‰) [89]. In contrast, the clays indicate values of δ18O = 6‰–8‰ from local meteoric water values, probably due to water–rock interaction. These observations match very well with the genetic model of low-salinity magmatic vapor plumes and surrounding groundwater interaction in the porphyry system model [90,91].
Another important example of a multistage high-sulfidation epithermal system is the giant Pascua Au-Ag-Cu deposit in Chile and Argentina [92]. The hydrothermal activity produced advanced argillic and vuggy silica alterations and hypogene alunite. The advanced argillic alteration, extensive vuggy silica and high-sulfidation mineralization are the results of high-level hydrothermal systems and interaction of the wall rocks with acidic vapors developed above a porphyry stock.
Every REE and all trace elements have different mobility and differential resistance against weathering impacts on these hydrothermal deposits, so some trace elements are diagnostic for weathering, such as HREE, Cr and Ti, but weathering depends primarily on the protolith. The genetic distinction between alunite and aluminum–phosphate–sulfate (APS) minerals containing hypogene and supergene kaolinization can be further examined on the basis of geochemical approaches using plots of some trace elements [22,27,93]. But differential element mobility depends on the mineral phases and their stability. Thus, all geochemical diagrams cannot be used to distinguish hydrothermal from supergene kaolins, as the temperature of alterations is not the crucial factor for the element’s concentration. The enrichment of S, Ba and Sr relative to protolith is common in hydrothermal-origin kaolin deposits, whereas Cr, Nb, Ti and La are mainly concentrated during surface weathering [22]. These researchers found that hypogene kaolin deposits in Peru are enriched in S and P due to the presence of alunite and assumed that the source of P is in phosphate-bearing minerals, which are the products of magma differentiation. As a result, the chemistry of deep ascending geothermal fluids changed over time and these variations were reflective of the mineral assemblages. At the same time, Zr and Ti are very immobile under near-atmospheric conditions, so if the concentration of the unaltered protolith is known, they can be used as the conservative elements to indicate the stages of rock weathering [94,95]. Three different Ti-bearing phases, including anatase, rutile, and a poorly defined nanocrystalline form, were reported from the Huber formation of the East Georgia kaolin deposit [96].
Ref. [62] studied the Blanquita kaolin mine emplaced in rhyolitic tuffs (Rio Negro Province, Argentina), which mineralogically is characterized by the presence of dickite, kaolinite, alunite and pyrophyllite, whereas in the Equivocada mine, kaolinite is accompanied by dickite and traces of alunite. The trace element contents and ratios of kaolin samples supported that hypogene-origin kaolin deposits occurred under the influence of hydrothermal alteration of the enclosing rhyolites.
The regional tectonism and magmatism of the environments, where kaolin occurred, are of particular importance because they control the exploration strategy and the exploitation of kaolin deposits [5]. The definitions of the terms supergene (exogenic) or hypogene (endogenic) explain the origin of kaolin, but the key important point is to understand the origin of geothermal waters, i.e., where they are originally coming from. Mineral associations of kaolins will also help to elucidate their origins, but the paleotemperatures of geothermal reservoirs cannot be used as an indicator of its supergene or hypogene origin (Figure 5). Ref. [97] reported isotopic compositions of clay minerals from porphyry copper deposits at Santa Rita, New Mexico, and initially demonstrated that supergene clays can be distinguished from hypogene clays; supergene kaolinites are generally richer in 18O relative to hypogene clays from a given deposit.
The Patagonia Province of Argentina has many kaolin deposits of hypogene or supergene origins [98]. The origin of these deposits has been identified based on the δ18O and δD isotopic composition of kaolinite and/or dickite, and statistical multivariate methods were used to model the behavior of immobile trace elements during the alteration. The principal component method was used to elucidate the characteristic variables of each deposit and correlate them with others, and [98] concluded that the first component indicates that Fe2O3, Y, Rb, U and HREE are more abundant in the supergene deposits, whereas Sr, Pb, S and V are more abundant in the hypogene deposits. The second component shows that S, P2O5 and LREE are enriched in the hydrothermal deposits, whereas Zr is more abundant in those formed under weathering conditions because Zr is a very immobile element in surface conditions.
S isotopes provide valuable information about the source of ore-forming hydrothermal fluids and their physical and chemical evolutions, such as changing pH through time, redox conditions and temperature. Thermodynamic models can be used for predicting a range of geologically realistic hydrothermal fluids, and S isotopic compositions are controlled by the ratio of oxidized (SO42−) to reduced (H2S) species and reaction kinetics [99]. This ratio is strongly affected by changing temperature, fO2 and pH, and the SO42−/H2S ratio can change significantly during cooling. Thermodynamic models are compatible with the S isotope evolution of porphyry and high-sulfidation (acidic; δS34sulfate) low-temperature hydrothermal fluids. The S isotopes of low-sulfidation (δS34sulfide) low-temperature hydrothermal fluids do not fit with thermodynamic models, and the mixing of seawater with biogenic sources naturally causes disequilibrium between the reduced and oxidized S species. The S isotopic fractionation between the coexisting pyrite and sulfate minerals indicates an equilibrium temperature of 380 ± 50 °C [100]. If two-phase (liquid–vapor) conditions existed at the time of sulfate and sulfide mineral deposition, the formation pressure would correspond to 130 to 200 bar. At high temperatures (380 ± 50 °C), ascending deep magmatic gases mixing with geothermal waters cause the redox reaction that is related to the occurrence of alunite.
4SO2 + 4H2O → 3H2SO4 + H2S
Under extreme conditions, the presence of HCl and H2SO4 in high-temperature ascending solutions reflects the intense high-sulfidation alteration leading to the formation of silica sinter, alunite-group and kaolin-group mineralization [101].
One of the unique examples of active hydrothermal fields is located in the Kamchatka peninsula. The Karymsky Volcanic Centre of the Kamchatka active volcanic terrane consists of two different Akademii Nauk and Karymsky hydrothermal systems. The Akademii Nauk is a typical high-temperature boiling system, with Na-Cl waters (TDS ~1 g/L), low gas content (CO2-N2) and deep calculated temperatures of ~200 °C. In contrast, the springs of the Karymsky system have lower temperatures (up to 42 °C) and strong gas bubbling, TDS ~2.5 g/L, and are enriched with HCO3− and SO42−, with Mg2+ being the main cation [102]. In mature hydrothermal systems, light sulfur isotopic composition between δ34S = −3.4‰ and +1.2‰ implies that the origin of sulfates is linked to surficial oxidation of H2S or the mixing of meteoric water, and this system is represented by neutral Na + K–chloride (bicarbonate) fluids. But in immature hydrothermal systems, there is a higher sulfur isotopic composition (δ34S = +8.2‰), which could either result from the disproportionation of magmatic SO2 or from the hydrolysis of elemental sulfur at high temperatures between 100 and 350 °C [15]. In addition, it is hypothesized that isotopic re-equilibration occurs as long as the aqueous solution is acidic [103].
6. Genesis of Kaolin Deposits
Kaolin deposits are part of the group of industrial raw materials and important sources for ceramic and paper industries. The genesis of kaolin-group minerals is commonly compared with the epithermal gold deposits due to the high-sulfidation process and economic importance. The well-known world kaolin mining districts in terms of both volume, quality and production quantity are in Georgia, USA, the Rio Capim basin, northern Brazil, Cornwall, UK, Bavaria and Saxony, Germany, and the Maoming basin of Guangdong Province, southern China. Moreover, kaolin production is reported in some other countries such as the Czech Republic, Turkey, Ukraine, Spain, South Korea, Malaysia, Vietnam, Bulgaria, Iran, South Africa, Colombia, and including ball clay, Australia.
The transformation of feldspars and arkosic sediments to kaolinite results from weathering or hydrothermal alteration, chemical leaching and partial precipitation of silica according to the equation below:
2KAlSi3O8 + 3H2O → Al2Si2O5(OH)4 + 4SiO2 + 2KOH
As the results of the dissolution of feldspars and kaolin crystallization, geothermal fluids become enriched with Al3+, as well as H4SiO4 after consuming H+, as follows:
2(K,Na)AlSi3O8 + 9H2O + 2H+ → Al2Si2O5(OH)4 + 4H4SiO4 + 2(K,Na)+
The removal of alkalines from feldspar and volcanic tuffs, which is the most suitable rock type for alteration due to its amorphous structure, in the unsaturated zone continues until the ascending percolating geothermal fluids become supersaturated with kaolinite. The formation of vermiform book-shaped kaolinite occurs as a result of in situ forming of kaolinite. The morphologies of kaolinite crystals have genetic links with the environments of occurrence, which are controlled by the parent rocks, pH and concentration of ions in the circulating fluids [104].
The most common ore classifications have been achieved by genetic means by economic geologists. Ref. [4] proposed more field observations related to the genetic classification of kaolin: (a) hydrothermal, (b) residual (in situ weathering), (c) mixed hydrothermal and residual, and (d) sedimentary deposits. The hydrothermal and residual kaolin occurrences are classed as primary and the sedimentary occurrences as secondary because transported and re-deposited kaolin occurs commonly in shallow lacustrine or paleo-shoreline environments. Primary kaolin deposits imply in situ occurrence by the alteration of volcanic tuffs, gneisses, granitic/rhyolitic and arkosic sedimentary rocks. Sedimentary kaolins are deposited in sedimentary basins as lense beds under the influence of groundwater movements and organic acids coming from a swamp environment. Ref. [105] subdivided kaolin deposits into three genetic groups: (1) residual kaolin deposits/primary deposits, which are products of rock weathering, (2) hydrothermal kaolin deposits/primary deposits and (3) sedimentary kaolin and kaolinitic clays/secondary deposits in lakes or coastal regions. The weathering process includes parts of the sedimentary processes and other processes, such as soil development, hydrology, geomorphology and climatology. However, ref. [5] criticized this classification, because some kaolin deposits have weathering overprinting or multiple processes. Under these conditions, he suggested that it is very difficult to give a genetic name, such as primary weathering, residual deposit, supergene alteration or pre-primary hypogene deposit. To avoid these deficiencies, refs. [27,28] summarized many commodity groups and proposed a new “Chessboard classification scheme of minerals deposits”. However, this scientific classification has a low practical use rate by field geologists. It covers various subject matters and interdisciplinary approaches from all branches of geosciences such as geology, mineralogy, pedology and hydrology [5]. For this reason, the researchers decided to follow [4] classification from an economic geologist’s point of view as it is more suitable for the field applications.
7. Hydrothermal Kaolin Deposits
Hydrothermal systems are commonly found in convergent margins, which are orogenic belts with a high geothermal gradient, or divergent margins or intracontinental rifts, which are non-orogenic areas [106]. The thermodynamic behavior of seawater, groundwater or meteoric waters infiltrating underground through cracks and/or permeable zones in these regions with high heat flow, in other words, thermal convection currents, forms geothermal systems [14,107]. High heat flux may be associated with active or pre-existing magmatic activity or it may develop with tectonic uplift. In conclusion, hydrothermal systems can be related to compressive fluid flows in the depth zone of the crust [70,106]. There is a well-known chemical rule that elements dissolve quickly in acidic hydrothermal solutions as temperature (>300 °C) and pressure increase, and these features favor the migration of dissolved elements in hydrothermal systems, whereas at cooler temperatures, such as where geothermal solutions are mixed with paleo-groundwater and the carrying capacity of the elements is diminished, some saturated dissolved elements may precipitate in surface conditions [101]. Under extreme conditions, the presence of HCl and H2SO4 in the geothermal fluids leads to the precipitation of residual silica, alunite, kaolinite-group minerals and anhydrite.
The temperatures of the fluids in magma-related hydrothermal systems can be changed depending on space and time and vary between 40 and 500 °C [106,108,109,110]. Hydrothermal fluid-related deposits are distinguished from each other based on their characteristic properties such as rock types, mineralogical composition, textural properties, the thickness of deposit, and biological content [111,112,113,114]. In hydrothermal systems, there are three types of main hydrothermal springs defined as being carbonate-rich, alkaline chloride and acid–sulfate. (1) Carbonate-rich fluids often produce travertine (CaCO3) deposits [115,116]; (2) alkali chloride fluids with neutral pH waters are mostly saturated with amorphous silica, which typically precipitates sinter deposits that are characteristics of opal-A [117]; and (3) acid–sulfate fluids are generally low in chlorides and high in sulfate and pH which are related to H2S oxidation to H2SO4. The chemical compositions of geothermal waters from the volcanic fields of Japan and the Kuril Islands show that usually, volcanic activity is marked by an increase in the SO4/Cl ratio during or after the eruptive event (phreatic or phreatic–magmatic). These volcanic activities depend on hydrogeological conditions of the Kuril Islands that control the appearance of the acid sulfate chloride springs [118].
It has been proposed that the water–rock interactions are complex processes that take place over time, especially in shallow geothermal systems, that usually result in the production of intermingled alkali chloride and acid spring deposits [119,120,121,122,123,124]. Acid–sulfate thermal fluids are produced by the condensation of steam and vapors as they rise through cracks and crevices at temperatures below 400 °C [12,106]. These waters are characterized by low pH (<4) and low chloride ratios (SO42−/Cl− = 2–10) as the result of the H2S + 4H2O = H2SO4 + 4H2 oxidation reaction. The formation of acid–sulfate thermal waters in fossil and active geothermal areas shows regional diversity and variability depending on the chemical composition of volcanic rocks, which geothermal waters pass through, temperature, chemical composition, pH of solutions and water/rock ratio. Small changes in the characteristic features of these solutions cause many changes in clay minerals and secondary minerals [17].
Additionally, the processes of hydrothermal alteration present with alteration veins and mineral zonation, which are most commonly observed in porphyry Cu-Au systems (Figure 6). These alteration zones are the natural products of water–rock interactions which transport heat and chemical constituents at different pH to serve a series of evolving physicochemical reactions. During the hydrothermal fluids and surface rocks reactions, the changes in thermodynamic conditions such as temperature, pressure, oxygen fugacity and sulfur fugacity control the precipitation of precious elements partly underlying kaolinization [125]. Depending on the mineral assemblage of wall rocks and the pH of geothermal fluids, alteration patterns exhibit temperature gradients in mineral assemblages. These physicochemical gradients of altered mineral assemblages are summarized in a previous study [126]. The chemistry and pH of hydrothermal fluids are controlled by the type of magmatic rocks and fluid/vapor compositions, in which geothermal waters pass through. The metaluminous magmas produce potassic and sericitic alterations upon cooling with high K/Na contents (such as granites) (path A; Figure 6). The peraluminous magmas or magmas with low K/Na content produce advanced argillic alteration upon cooling (path B; Figure 6). High water/rock ratios generate advanced argillic alteration with the condensation of vapor (path C; Figure 6).
The evolution of hydrothermal alterations was classified by using the model of a porphyry system and a group of mineral assemblages in ore bodies and the adjacent rocks [127]. The typically observed temporal evolution in porphyry ores is from early-stage, high-temperature biotite ±K-feldspar assemblages (potassic alteration) to muscovite ±chlorite assemblages (sericitic alteration) to low-temperature, clay-bearing assemblages (advanced argillic and intermediate argillic alteration), which is consistent with progressively greater acidity and higher fluid-to-rock ratios in fluids, prior to their eventual neutralization. In this late stage of advanced argillic alteration processes, kaolin and alunite mineralizations are formed and superimposed on ore and potassic alterations, where advanced argillic alteration (especially as quartz + alunite) is preserved spatially above ore.
7.1. Alunite–Kaolinite–Halloysite–Pyrophyllite Association
The alunite–kaolinite–halloysite–pyrophyllite association is common in many kaolin quarries, especially in Japan, around the world. For this reason, it has been explained in detail under a separate heading. The processes of advanced argillic–propylitic alteration are the results of hydrothermal fluid and rock interactions that are controlled by a heat source, pH and the chemical composition of solutions evolving from a series of physicochemical reactions. These intermediate–high temperature reaction series are characterized by wide alteration haloes and mineral zonation in the vicinity of upflow areas, such as those observed in porphyry Cu-Au systems [127]. Depending on the mineral assemblage of wall–rocks, the pH and the chemistry of geothermal fluids, and temperature gradients, the final products of alteration patterns between fluids and wall–rock composition can be varied. The products of physicochemical conditions between temperature gradients and alteration mineral assemblages in porphyry systems at high temperatures and epithermal systems at low temperatures are summarized in Figure 6.
The alunite supergroup (containing more than 40 minerals) has the general formula AB3(XO4)2(OH)6, where A is a monovalent or divalent large ion (K, Na, NH4, H3O, Ca, Ba, Pb, Bi or REE2+), B is trivalent Al3+ or Fe3+ REE3+ and X is S6+, As5+ and P5+ (SO4, AsO4 and PO4) [128,129]. Most alunite (KAl3(SO4)2(OH)6) samples are enriched in REE contents with respect to the parent rocks. Alunite is of particular interest among all hydrothermal minerals, as it has an open-formula structure that allows for the replacement of many cations and anions and also forms in several of the hydrothermal environments. The REE enrichments of alunite nodules (4–6 cm) can be changed depending on the chemical composition of the magmatic rocks in which geothermal fluids pass through, the pH and the temperature conditions of halloysite deposits. The enrichment of trace elements and REEs relative to the whole rock is larger in alunite than halloysite, which suggests that there are differences in the temporal and spatial distributions of pH and the chemistry of geothermal fluids among alunite and halloysite deposits [9,60]. The occurrence of halloysite always requires adjacent highly permeable drainage and buffer rocks, such as limestone, for it to form, otherwise kaolinite forms. The pH buffering capacity of limestone is more important. It will neutralize the acidic Al- and Si-rich fluids, and thus halloysite will precipitate and excess silica must leave the system. A good example of high-temperature hydrothermal deposits of kaolin in which nacrite, dickite and kaolinite are intergrown occurs in the foothills of the Sierra Madre Occidental, Nayarit, Mexico [130]. Similar observations have been made for dickite mineralization along the fault zone in the Düvertepe kaolin district, Turkey. Geologically, nacrite has been reported as a valid kaolin–mineral indicator of high temperature. Dickite is known not only from high-temperature environments but also as an authigenic mineral in the pores of deeply buried diagenetic sandstone and in hollow chert nodules from a later Carboniferous (Pennsylvanian) conglomerate derived from underlying earlier Carboniferous (Mississippian) rocks, 358.9–323.2 Ma, in Missouri [130].
The stability of alunite is also related to aqueous SO42− sulfate as well as the K+ and H+ concentrations [131]. The experimental studies revealed that an increase in the activity products of H+ and SO42− causes thermodynamic shifting in the stability fields from K-feldspar to white mica to kaolin-group minerals and finally to alunite as pH decreases to about 2.5, which results in intense chemical leaching and only silicification occurs. If the K+ concentration is high, white mica (muscovite and illite) can coprecipitate with alunite [131], as reported based on field observations from the Düvertepe kaolin district [9].
In high-temperature acid sulfate–chloride hydrothermal systems, the mixing of boiling geothermal water with groundwater in near-surface conditions and the intermittent input of high-temperature acid components from a deep source commonly occur as natural phenomena. As the result of these changes in a deep source, the chemistry of fluids is subject to continuous changes in temperature, pH, cation and anion compositions, salinity and so on, leading to variation in the crystal chemistry of alunite and immiscibility between Na– and K–alunite at low temperatures. Apatite is present as a primary mineral in some host andesitic rocks at deep sources, and is possibly released during the initial stage of acid leaching and carried out with the variable PO4/SO4 ratios in the fluid to the surface. Both apatite and alunite minerals have open-crystal formula structures suitable for ion exchange processes. For this reason, some alunite minerals include a significant amount of apatite or phosphate ions in their formula structure, up to 37 wt.% P2O5 in the microenvironment [60]. Back-scattered electron composition images show a typical growth banding texture in alunite, like plagioclase zonation, which is similar to timewise annual growth rings of the trees, and can be used to understand the development stages of the hydrothermal system [132]. These variations in textural zonation and the compositional changes in the crystal chemistry of alunite originate from the spatial and temporal changes in the magmatic hydrothermal system.
Solubility diagrams are useful tools for understanding and interpreting chemical reactions and (alunite–kaolinite–halloysite) mineral assemblages. These thermodynamic diagrams of hydrous alumina and aluminum silicate minerals based on log(Al3+) and log(H4SiO4) relations are provided for gibbsite, kaolinite and pyrophyllite–diaspore at 200 °C and 300 °C and at pH of 2.3, 3 and 4 [133]. Under hydrothermal conditions, as long as the dissolution reaction continues between geothermal fluids and K-feldspar, circulating fluids become saturated with respect to a group of some elements, and these saturated elements begin to precipitate naturally when the P-T relations decrease in near-surface conditions. The remaining solutions change their compositions as a result of precipitation [133]. As a natural phenomenon, hydrothermal fluids circulate continuously on their migrating pathways and a series of reactions occur as a function of migration distance. As a result of the continuation of hydrothermal reactions, the remaining solutions attain saturation with respect to other minerals and different hydrothermal minerals are precipitated in turn over time. Based on experimental studies, metastable boehmite is formed around 200 °C, while diaspore is formed at higher temperatures [134]. Ref. [135] found that muscovite/illite changed to pyrophyllite through kaolinite, and the alteration occurred in two steps, from sericite to kaolinite at 260 °C and from kaolinite to pyrophyllite at 270 °C. In contrast, they also observed that natural kaolinite was not changed to pyrophyllite at 270 °C, but this was possible in a two-step reaction chain from sericite to kaolinite and then to pyrophyllite. The rate of experimental reaction in each step is dependent on the reaction temperature and chemical composition of the solution.
The sequences of precipitation of minerals are as follows: aluminum hydroxide → kaolinite or pyrophyllite → silica mineral + kaolinite or pyrophyllite in weakly acidic solutions and silicate mineral → silica mineral + kaolinite or pyrophyllite is expected in strongly acidic solutions. These mineral transformations occur at 300 °C and in pH 3 conditions as follows: boehmite → kaolinite → amorphous silica + kaolinite or diaspore → pyrophyllite → α-quartz + pyrophyllite [133]. Sulfur-rich fumaroles are commonly found in the Japanese volcanism and are sometimes exploited for kaolinite and halloysite.
Alunite- and kaolinite-group minerals and pyrophyllite (Al2Si4O10(OH)2) associations in high-temperature hydrothermal systems have been studied in detail using core drilling samples in Japan. Pyrophyllite occurs at medium-grade metamorphic belts or is commonly related to Quaternary or Pliocene volcanism. Ref. [136] gives one of the best upper stability limits of kaolinite (+quartz) at 390 ± 10 °C and 2 kbar, and these low-grade (greenschist facies) conditions depend on fluid composition (XCO2):
The Pötürge area is located on the presently active tectonic zone in the Malatya, Turkey. The Pütürge Fault is a left-lateral strike-slip fault with a northwest dipping plane and extends toward the Sivrice–Doganyol Fault, which is part of the main East Anatolian Fault Zone. Metamorphic-hosted pyrophyllite occurrences are common in the shear zones, mostly as lens shapes along minor faults in the Pötürge area. Based on fluid inclusions in quartz (121.3–172.6 °C) and stable isotope studies, it was estimated that alteration to pyrophyllite and kaolinite occurred at temperatures of 150 and 100 °C, respectively [137]. The occurrence of pyrophyllite and dickite is probably related to the interactions of heated meteoric water and metamorphic rocks [137]. Pyrophyllite occurrences are found within kyanite gneisses overlying the mylonitic granitic gneisses. Similar to metamorphic rocks (disthene-bearing kyanite gneiss), they are overprinted by an acid high-temperature alteration in Turkey. The major pyrophyllite occurrences are located in North Carolina, USA, which is part of the Carolina Slate Belt that is characterized by high-alumina minerals [138].
The mineral stability temperature of pyrophyllite ranges from 275 to 350 °C; however, the formation temperature of pyrophyllite depends on H2O content and other fluid species, e.g., CH4 and CO2 [139]. In addition, silica-supersaturated fluids could result in the precipitation of pyrophyllite instead of kaolin up to 100 °C during the hydrothermal alteration [140,141]. Thus, the presence of hydrothermal quartz veins including CO2-rich fluids and the existing SO2 for the formation of alunite suggest that pyrophyllite occurs under relatively low-temperature conditions [137]. Similar results have been obtained based on fluid-inclusion studies on the quartz that were determined at <245 °C [142]. The dissolution of mainly feldspars, mica and tuffs under acidic conditions suggests that alunites are formed and excess silica is transported away from the system:
3(K,Na)AlSi3O8 + 2SO2 + 22H2O = (K,Na)Al3(SO4)2(OH)6 + 9H4SiO4 + 2(K,Na)OH
Mineral dissolution in the Al–silicates is incongruent with the gradual loss of SiO2 to the hydrothermal fluids. Thus, gradual and continuous silica-leaching and silica-fixation reactions are chemically controlled by the pH of fluids in mineral assemblages [140]. Dickite is a high-temperature mineral and rarely forms on the extension of pyrophyllite fibers as an alteration and hydrolysis product:
Ref. [143] explained alteration mineral zonation in high-temperature geothermal fields from the deep center to the surface rim as (a) a pyrophyllite zone at the center, (b) a corundum, diaspore and pyrophyllite zone, (c) an alunite–kaolin zone, (d) a silicification zone impregnated with Fe-minerals, (e) a silicification zone, and (f) weakly altered wall rock from Shokozan mine, Hiroshima Prefecture, Japan. Due to subduction tectonics and island-arc volcanism of Japan, Quaternary volcanism and their regional tectonic activities created suitable environments for the circulations of acidic hydrothermal fluids (pH < 4) that caused deep alteration of the volcanic formations [143]. In low-pH conditions, the solubility of aluminum and silicon has reverse relations in organic and inorganic systems. In the inorganic system, aluminum becomes more soluble than silicon, and consequently, silica forms porous deposits (vuggy quartz) in the deep hydrothermal systems. The first altered zone along the fracture systems near vuggy quartz occurrences is formed of kaolin-group minerals. As long as the concentration of H2SO4 diminishes, the altered rocks contain smectites on the outside, further away from the vents or fault zones, and both pH values and the temperature of hydrothermal fluids decrease. The acid leaching of cations in the very acid domain by the oxidation of H2S serves to precipitate alunite in steam-heated environments associated with halloysite or/and kaolinite.
One of the unique hydrothermal alteration areas in Japan is the Mitsuishi district which is characterized by the alteration zone from the center to the margin: Quartz zone → Roseki zone (pyrophyllite, pyrophyllite–sericite, sericite and/or kaolinite zones) → K-feldspar–sericite zone → albite zone [144]. This Roseki zone is overlain by a weakly propylitized zone, which is composed mainly of plagioclase, quartz, chlorite and calcite, containing small amounts of alkali–feldspar, albite, sericite, biotite and pyrite. The fluid-inclusion studies using euhedral quartz crystals from the Quartz Zone revealed that the hydrothermal solution was between 300 and 350 °C. Thermochemical consideration, fluid-inclusion studies, chemical compositions of altered rocks, and mineralogy and element mobility calculations present that the hydrothermal solutions initially ascending through the column of the original rocks were strongly acidic, in a temperature range of 300–350 °C and oversaturated with SiO2. As the geothermal solutions infiltrated through the column, pH increased from 4.5 to 6.9 after the occurrence of the formation of Quartz zone.
Studies on pyrophyllite in the Kuroko Deposits of Japan showed that high dissolved SiO2 concentrations caused the direct precipitation of microcrystalline quartz associated with nacrite and pyrophyllite [145]. The stability relationship between kaolinite and pyrophyllite [140] reveals that kaolinite forms at a lower temperature than pyrophyllite due to the presence of dissolved SiO2 (Figure 7). Ref. [45] supported this idea and noted that kaolin minerals formed at 100 to 200 °C and pyrophyllite formed at temperatures above 230 °C in Japan’s Ohtake geothermal system.
The spatial and temporal relations of the acid sulfate and other types of alterations of high-temperature hydrothermal systems in the Kamikita area, Japan, are well documented especially using a large number of core drilling data [154]. Four types of alteration are distinguished based on mineral zones and each zone is illustrated in Figure 8. The parent rocks are the Miocene massive and brecciated basaltic, andesitic and dacitic lava flows and volcaniclastic rocks. Alteration type I is composed of mica, amphibole, zeolite, epidote, chlorite-group and smectite-group minerals and represent the western part of the Kamikita area. In this zone, it is thought that the temperature ranges from >300 °C in the biotite–actinolite zone to <100 °C in the smectite zone. In addition, the transformation from smectite to chlorite to form corrensite takes place between 100 and 200 °C. Alteration type II represents the central area, and the K-feldspar zone is in the lower part, a mixed-layer illite/smectite mineral zone is in the upper part and an illite/chlorite zone is in the middle part. Abundant barite was identified in core samples. The presence of abundant regularly interstratified illite/smectite (R1) (tosudite and rectorite) alteration indicates a temperature of ~100 °C, but the whole alteration type-II zone formed at approximately 100–300 °C. The whole range of alteration type III is located in the eastern part and is composed of laumontite and wairakite zones, which is characteristic of propylitic alteration, and this zone probably occurred at temperatures of 200–300 °C. Alteration type IV is distributed in the central part and characterized by the acid sulfate alteration minerals, such as pyrophyllite–diaspore and the mixed-layer I/S zone observed from the center to the margin (Figure 8). The type-IV acid sulfate alteration temperature was changed in the range of 200–300 °C. The alunite zone is observed in core samples of the central part and comprised of mainly alunite, quartz, dickite, topaz, zunyite and pyrite [154]. The distribution of alunite shows a repeated variation at 200 m intervals associated with dickite and quartz.
It is proposed that these variations in the amount of alunite could be related to pH fluctuations. At lower pH, the fluids favor the precipitation of alunite by the presence of K+, but alunite reacts with quartz to form dickite in higher pH fluids [154,155]. Alunite usually shows heterogenous compositional zoning at the microscale within a single grain; these properties are well documented using electron microprobe data (Table 4 in [60]) and these compositional differences are common regardless of different occurrences. Based on EDS analyses of alunite in the K-Na-2Ca diagram from the Kamikita area [155], the K/(K + Na) ratio ranges from 0.1 to 0.9 and the Ca content is less than 0.3. All of these studies indicate as a natural phenomenon that the temperature, pressure, pH, ƒO2, ƒCO2 and chemical composition of high-temperature hydrothermal fluids temporally and spatially change as the active tectonism and subduction volcanism continue in almost all geothermal systems.
Active geothermal fields are the surface exposures of deep circulated geothermal waters originating from the emplacement of magmatic intrusions and regional tectonism [17]. Due to these major geologic activities, using the hydrothermal system model of the Philippines [164], advanced argillic alteration formed over the top of the porphyry system, which consists of HS processes, indicating that quartz–alunite and intermediate parts are the epithermal ore deposits.
7.2. Silica Sinter and Silicification Processes
The above-mentioned deposits refer to mineral associations formed in relation to the acidic conditions of the geothermal fluids. However, water–rock interactions are complex processes, especially in shallow geothermal systems, which can show a transformation from an acid–sulfate active solution to an alkali–chloride solution over time and vice versa [119,122,123]. Sinter deposits are the surface products of geothermal systems that are oversaturated in silica and are precipitated from near-neutral pH alkali–chloride fluids. This process is commonly observed in many hydrothermal kaolin deposits as the result of the alkalization of pH and the silica enrichment of geothermal fluids after the kaolinization processes, and this process represents the last product of epithermal mineralization. The active faulting at Mangatete, Taupo Volcanic Zone, New Zealand, is a good example of fascinating hydrothermal alteration environments exhibiting many different products of recent volcanism [114,165]. Additionally, silica sinter deposits may host disseminations of Au, As, Sb and Ag mineralization in near-surface conditions, whereas Au, Ag and As are precipitated as a results of boiling at depth, characterized by hydrothermal brecciation [70]. Veins and stockworks with sulfides (Cu, Pb and Zn) are in the lower levels and Au, Ag, As, Sb and Tl are in the upper levels of the hydrothermal systems due to mineral stability conditions.
Chemical silica deposits which are observed in the form of beds or mounds in the field are also quite resistant to erosion. In this way, silica caps protect underlying kaolin deposits against erosion and they can have topographical highs and small hills [126,157,166,167,168]. These sinter deposits develop near the surface and above the water table in subaerial volcanic terrains where the heat input of volcanic activity and subterranean magmas is sufficient to cause hydrothermal circulation of groundwater from deep reservoirs with minor magmatic input [126,169,170]. The cooling near-neutral-pH alkali–chloride thermal fluids at the surface, from ~100 °C to ambient temperatures, contain metastable silica that becomes insoluble and precipitate as opal-A. Silica diagenesis continues through a series of polymorphs from opal-A, upon a change in pH and referred to as silica residues, to opal-CT/-C and then to microcrystalline quartz, which takes more than 20,000 years, resulting in massive, thick, highly silicified rocks, which groundwater infiltrates after the discharging of alkali chloride liquids [117,146,157,171,172,173,174]. Fluctuations in groundwater tables cause erosion or the rapid cooling of boiling geothermal fluids, in which dissolved silica was saturated at depth with respect to quartz and in near-surface conditions became supersaturated with respect to amorphous silica, which results in silica deposition, either as chalcedony at less than 200 °C or amorphous silica at 100 to 150 °C [109,175]. Fluid-inclusion studies on quartz from wall rock alteration zones showed that the homogenization temperatures range from 100 to > 500 °C [82,176]. Silica-rich solutions in hot springs and surrounding pools may contain the unicellular cyanobacterium Synechococcus elongatus and without these microorganisms, continuous silica input from the hot springs leads to well-laminated silica sinters [177]. So, thick massive silica sinters and highly silicified rocks, including “vuggy silica” textured rocks, are the products of large-scale continuous silica input in relatively short episodes [178,179,180].
Strong acidic solutions create high silica activity [181,182] and amorphous silica precipitates with alunite and kaolin-group minerals at 500 bars and 600 °C, at about 1000 mg/kg (0.0294 M) [183]. Two processes, geochemical and adiabatic cooling, are working hand-in-hand here; First, when the temperature of geothermal fluids drops due to mixing groundwater, known as decompressional boiling, silica-supersaturated fluids cannot carry high silica concentrations that cause very rapid amorphous silica precipitation in fracture zones and on the surface. Second, this rise in the boiling-temperature solutions is a very fast adiabatic process and there is no time for heat transfer to the wall rock, so silica cannot precipitate during ascending. Silica concentrates in the residual solutions after steam separation, which are in equilibrium with quartz at 210 °C in reservoir conditions, yield saturated solutions with respect to amorphous silica at 100 °C, and at both temperatures, the silica concentration is ~380 ppm [182]. Later, a sequence of silica polymorphs occurs, ultimately resulting in microcrystalline quartz [146,159,171]
Ref. [126] classified sinters as follows: (1) the different lithofacies of sinter deposits, from proximal apron (65–100 °C), through the middle (45–65 °C) and distal apron (<45 °C), to geothermally influenced marsh environments (tepid to ambient) (Cady and Farmer, 1996); (2) varied breccias commonly associated with sinters; and (3) some silicified features that may be misidentified as siliceous sinters, known collectively as pseudo-sinters. The recurring gradient of distinctive, environmentally dependent facies assemblages of the proximal, middle and distal apron and geothermally influenced marsh areas may extend laterally for several meters, up to hundreds of meters, the extent depending on fluid volume, flow rate, seasonality, paleoslope, etc. Within and between the facies assemblages of sinters, individual facies may gradationally transform into one another. This transition between lithofacies may occur over small spatial scales, depending on several processes including changes in thermal fluid flow rates, relative fluid discharge volumes, pool depths and fluid temperature, as well as local topography [126].
Determination of the lithofacies characteristics of fossil silica sinters and paleoenvironmental and paleogeothermal fluid properties is achieved by comparing them with modern-day analogues. Preserved sinter deposits, lithofacies relations with respect to hot spring discharge temperature gradient, and biota living in the thermal fluids and in surrounding pools are shown in a simplified model of thermal spring deposits [126]. The concentrations of major ionic species in geothermal fluids have higher dissolved solute contents in higher temperatures [157]. Temperatures at depth and the dissolved silica vs. pH relation after cooling through steam formation from natural springs from different geothermal systems are shown in Table 4. In addition, Table 4 shows that silica dissolved at depth depends on the temperature, pressure and pH properties of the geothermal fluids of the reservoir, as well as the chemical properties and mineralogical structure of the rocks which the fluids pass through. Also, it is experimentally documented that silica solubility at 500 bars of pressure increases from 20 ppm containing 0% NaCl at 400 °C to 40 ppm, 23 wt.% NaCl, at 470 °C in geothermal waters [184]. These observations elucidate why high-temperature geothermal fields have more silicified zones or silica sinter deposits where springs and geyserite emerge on the surface.
Apart from sinter precipitation on the surface, another effective process in epithermal processes is silicification. The silicification process initiates in phreatic zones of pre-existing deposits, including the oversaturation and remobilization of silica in the micropores of the surrounding rocks. Many polymorphs of quartz can be observed as a result of this process, which is described as the chemical precipitation of silica-oversaturated waters in the voids, fractures and cracks in the rocks. The reason for this is the temporal evolution of the precipitated quartz polymorphs. In this process, firstly, blankets of opal begin to invert to chalcedony over time below the vadose zone [76,169], and finally, in time, chalcedony inverts to quartz [109]. A laterally developed silicification zone has been observed around the epithermal veins in the bedrock, which extend radially over large distances (hundreds of meters) [9] due to the widespread presence of high-porosity and -permeability bedrock (i.e., tuffs, andesites and volcano-clastics), and these porous rocks are easily silicified in their shallow levels [168]. Silicified rocks occur in the form of mounds in a very wide area due to their resistance to erosion [87].
8. Examples of Worldwide Kaolin Deposits
8.1. Kaolin–Au Mineralization, Turkey
Since the Middle Miocene, western Turkey has caused an N-S extension due to the E-W compression that has resulted in crustal thinning and, consequently [185], increases in the geothermal gradient which heated the post-arc basin due to enhanced asthenospheric flow as a result of slab tearing below western Anatolia [186]. Early Miocene volcanism, characterized by the deposition of calc–alkaline tuffs, ashes and pyroclastic rocks, has been modified in this extensional tectonic regime [187,188]. These conditions were favorable for the mobilization and concentration of Au and for acidic hydrothermal alteration in the shallowest part of the crust due to alkaline magmas formed in the sub-back-arc metasomatized lithospheric mantle [189,190]. All of these geological features provided suitable environments for the deposition of Au and kaolin-group minerals in western Anatolia. Miocene precious ore deposits in western Turkey are known to be formed by the following process: Cinarpinar porphyry Cu-Au-Mo system, the porphyry Mo-Cu Pinarbaşi system in the Menderes massif [191], the low-sulfidation Kısacık, Küçükdere and Ovacık Au-Ag deposits, and the IS to LS Efemcukuru Au deposit [192,193]. The richness of the ore minerals makes all of these quarries different from other mines.
The Kışladağ Au deposit is located in Uşak in western Turkey, and it was the first porphyry-type gold mineralization discovered in Turkey [194]. The Kışladağ porphyry Au deposit is related to monzonite-intrusive and subvolcanic rocks of the Beydağı volcanic complex that includes andesites, latites, trachytes, dacites, rhyodacites and rare basalts invaded the pre-Cretaceous Menderes metamorphic rocks [195,196]. Based on their mineralogical assemblages of alterations and chemical compositions, these rocks have been subdivided [197] as follows: (i) Advanced argillic alteration is represented by quartz–alunite and dickite–pyrophyllite–pyrite mineral assemblages [196]. (ii) Argillic alteration is dominated by the development of kaolinite, halloysite and smectite [196,198]. The argillic alteration overprints all other alteration assemblages. In the transition from deep porphyry-type mineralization to shallow epithermal-type mineralization deposits, kaolinite is commonly observed in the argillic alteration zones [194].
Efemçukuru is another vein-type epithermal gold deposit that comprises two mineralized NW-trending quartz–rhodochrosite veins and is associated with kaolinization in Ovacik, Aegean Region of Turkey. Six main stages of veining were identified by [192] based on mineralogy, textures and cross-cutting relationships. These are as follows: Early veins of quartz, chlorite and calc–silicates (stage I) are cut by two stages of brecciated and banded veins of quartz, rhodochrosite, rhodonite and pyrite (II, III). Later veins with disseminated (IV) and massive base-metal sulfides (V) are cut from previous phases, and subsequently from late quartz–carbonate veinlets (VI). Gold occurs as electrum and with pyrite and galena in stages III, IV and V. Belonging to this deposit is the alteration of rocks that host the Kestanebeleni and Kokarpınar veins that can be classified into three main types: (1) calc–silicate alteration: pre-epithermal, (2) interlayered illite–smectite alteration and (3) halloysite–kaolinite alteration [192]. The calc–silicate alteration zone broadly follows the NW-SE fault structural trends within a 150–200 m halo on the surface and is most intense in the center of the Kokarpınar region. On the other hand, the illite–smectite and kaolinite–halloysite trends occur only within 75 m of the epithermal veins and rhyolite dikes, respectively. These minerals occur proximal to rhyolite dikes and fault zones in the Efemçukuru host rocks and also kaolinite and halloysite alter from primary feldspars in rhyolite dikes [192]. Heavily comminuted fault zone cores contain abundant kaolinite and their lack of spatial and temporal overlap with illite, smectite, chlorite or the calc–silicate minerals indicate a supergene origin. Kaolinite and halloysite as supergene alterations are common in many LS and IS epithermal systems, indicating that early, intermediate and late sulfidations gradually changed temporally and spatially due to active regional tectonism and volcanism of the Aegean Region. The vertical and horizontal distribution of kaolinite and halloysite mineralization shows the dominant control of the structure of the fluids along the fault-related zones in Efemçukuru, Turkey.
8.2. Çanakkale and Düvertepe Kaolins, Turkey
Industrial mineral deposits in the Biga Peninsula in Turkey include varying contents of kaolin-group minerals; alunite and quartz have attracted great interest due to the raw material demands of the ceramic industry [9,33,60]. All of these quarries are distinguished by the presence of rich kaolin formations, as well as large amounts of alunite and silica caps in the upper zones of the quarries. Additionally, their ore metal content is very low. The Düvertepe is one of the most important kaolin districts and is located at the end of the Simav Graben in western Turkey. Kaolin in the Düvertepe region was formed within Miocene rhyolite–rhyodacites and tuffs and also N-S extending developed in accordance with tectonism in the Simav graben. Upward fan-shaped silicification, which is located above the kaolin zones, suggests that adiabatic solutions follow the fracture zones and mark the upflowed areas of compressional boiling fluids [9]. The Çanakkale province is another well-known industrial mineral resource area in the Biga Peninsula. Acid–sulfate-type ascending hydrothermal fluids caused argillic alteration zonation around andesitic lavas and tuffs. The first zone includes massive rich quartz (massive silicified rocks), the second is a mixture of rich quartz + alunite + kaolinite/dickite/halloysite, the third one is rich in alunite + kaolin/dickite/halloysite ± rare quartz, and the fourth zone is enriched with kaolinite ± rare alunite ± rare quartz. Toward the andesitic tuffs, it grades into a kaolinite ± feldspar ± illite ± montmorillonite zone. These kaolin deposits reflect the hypogene–supergene transition quite well with the change in the mineral content and crystallization degree [33]. This type of mineral zoning was proposed for the Kütahya kaolin-group deposits in Turkey [199] that were formed through hydrothermal alteration of products of the Neogene volcanism. The proposed alteration zonation outward from the main kaolin deposits is as follows: A) inner zone, kaolinite > smectite + illite + opal-CT + feldspar; B) middle zone, feldspar + kaolinite + quartz + smectite + illite; C) outward zone, quartz + feldspar + volcanic glass. Different polymorphs of kaolin-group minerals were observed but the particle size was changed corresponding to the temperature of formation. The best examples of traces of hydrothermal evolutions in the rock records with changes in the pH and chemistry of hydrothermal fluids through time and space from highly acidic, acidic, and neutral to alkali chloride solutions can be seen on the geological map [9]. Because there are mainly kaolin and alunite deposits, the presence of very minor bentonite mineralization and very thick silica sinter deposits (Table 3) is reported in the Düvertepe region [9]. The epigenetic model for the Düvertepe kaolin district, including kaolin-group, alunite and silica sinter deposits, in the west of Simav Graben is summarized in Figure 9.
The most important formations of the kaolin deposits occurred along the subdivision of the North Anatolian Fault, the NE–SW-trending Çan-Etili-Bayramiç fault, and the zone located within the Çan Volcanics in the Etili area, Çanakkale [87]. Three different mineral assemblages were observed in three deposits: (A) the Bahadırlı quarry; kaolinite + alunite ± quartz ± smectite ± plagioclase ± K-feldspar; (B) the Caltıkara quarry: kaolinite + quartz + alunite ± Fe-oxide and ore minerals; and (C) the Duman quarry: hanging wall block of kaolinite + quartz + plagioclase ± smectite and a footwall block of kaolinite ± alunite ± smectite ± quartz ± plagioclase ± K-feldspar ± gypsum. The footwall blocks reflect the mineralogical, chemical and isotopic properties dominated by the effect of acid–sulfate hydrothermal fluids at the base. However, the mineralogical composition, geochemical properties and 34S isotope characterization of the upper zones of the footwall block reveal the hypogene–supergene transition with the effect of the alkali–chloride fluids. These three kaolin deposits located in the same host rock are very close to each other along the Çan-Etili-Bayramiç fault zone but experienced different stages and degrees of hydrothermal alteration processes (Table 3). Each deposit was presumably affected by the groundwater and meteoric waters differentially heated by two different hydrothermal processes in different episodes [87]. In the first stage, highly acidic and oxidizing conditions prevailed, as shown by the presence of alunite and kaolinite, an indicator of high-temperature mineralization, formed in relatively deep epithermal zones. During the second stage, kaolinite, smectite and halloysite were formed.
Most hydrothermal kaolin deposits exhibit partly silicified veins, fault-related silicified zones within the kaolin body or silica sinter deposits on the surface where geothermal fluids emerge (Table 3). The Bodurlar deposit of the Çanakkale district shows an example of late-stage six silicified fracture zones, which occur within the same wide flower-like fault zone that passes through the kaolin deposit along which ascending hydrothermal fluids are believed to have moved. Kaolinization forms within massive white- and purple-colored tuffs with plagioclase and biotite fragments in the ash matrix. In nature, amorphous materials, such as volcanic ashes and tuffs, have a greater tendency to undergo acid sulfate alteration compared to crystalline materials. For this reason, kaolinization of volcanic tuffs can take place much more easily than weathered granites or other silicate group minerals. The physicochemical reactions are generally multistage; therefore, in many cases, due to the limitations in the reaction time of metasomatic processes, these hydrothermal reactions have not always reached to final equilibrium among different mineral phases. As the result of incomplete chemical reactions, the mineral assemblages, the physicochemical properties and the quality of kaolin deposits are very heterogenous.
Kaolinite crystals have well-formed hexagonal vermiform book shapes, alunite crystals are idiomorphic rhombohedral forms, and needle- and tubular-shaped halloysites are common in the alunite facies in the Düvertepe and Sarıbeyli–Sığırlı deposits. Common coexistence of halloysite and alunite in hydrothermal acid–sulfate alterations was interpreted as the presence of H2SO4 which enhanced the acidity in hydrothermal solutions that led to advanced argillic alteration o the Biga Peninsula [60]. Well-ordered hexagonal vermiform kaolinite grows in the range from 325 to 375 °C depending on the pressure [136,201]. Kaolinite and dickite minerals were observed together in the high-temperature zones in these deposits. A lack of pyrophyllite and dickite indicates hypogene kaolinization that develops in the distal zone at a temperature below 300 °C. The other mineral associations besides kaolin-group minerals facilitate the correlation and understanding of the epithermal-type mineralization that occurs during advanced argillic alteration.
Differential element mobility in hydrothermal kaolin deposits has been observed during acid sulfate alteration of the Biga Peninsula [9,33,202]. The Sarıbeyli-Sığırlı kaolin deposits have high Ba + Sr concentrations above 2000 ppm and low Ce + Y + La concentrations (<150 ppm). These elements are largely sourced from K-feldspars, which undergo dissolution in the initially undersaturated geothermal solutions. Using the classification scheme of [22], the majority of Sarıbeyli-Sığırlı samples indicate the hypogene area and the Bodurlar samples indicate the mixed and supergene areas [33]. Similar observations were made in the Düvertepe kaolin region which contains the coupled elements (Cr + Nb) at about 8–50 ppm, which suggests hypogene origin. This phenomenon better explains Cr + Nb enrichment in supergene environments.
The coexistence of alunite with kaolinite requires pH < 4 and this kind of acidic solution is typical of hypogene environments [22]. Also, at pH < 4, Ti and Fe can be dissolved and are removed from the system; therefore, Ti + Fe content is low in hypogene environments. Trace element composition of the Düvertepe samples fit in the range of hypogene environments [9]. Ba and Sr can substitute for each other and often occur at elevated levels in hypogene deposits. Light REEs are considerably enriched in supergene kaolinization [22,203]. This is expected due to the solubility behavior of LREEs under hot and acidic conditions. The Düvertepe kaolins are between supergene and hypogene alteration fields with an influence of supergene overprint after hypogene alteration. Direct comparison between the Simav and Peruvian data sets is partially limited by differences in host rocks and the absence of copper oxide minerals in the Simav area [22].
8.3. Halloysite Deposits, Turkey
The Biga Peninsula is considered the unique area for halloysite–alunite deposits in the world and is located in the Turplu, Kızıldam, Kırıklar, Taban and Soğucak quarries of the Balıkesir and Çanakkale districts [60]. Hypogene halloysite mineralization was controlled by low-pH geothermal water, fluid temperatures below 100 °C near the surface, good permeability, and karstic environments for the best drainage system, which is necessary for the migration of Si and other excess cations. The occurrence of hydrated halloysite in areas with a high-moisture regime is critically important and it requires permanently humid or water-saturated environments not only during the occurrence but also for the preservation, and it must be stored in water caps in laboratory conditions. Otherwise, hydrated halloysite (10 Å) turns into dehydrated halloysite (7 Å), which is an irreversible reaction.
The Taban halloysite quarries, which are located at 900 m above sea level, 20 km NE of the town of Yenice, consist of three separate NNE-trending occurrences called lower, middle and upper quarries, and the fault zone cuts through the halloysite deposits in the same direction [204]. The halloysite occurrences in the lower quarry are located within a fault zone in a narrow valley at 750 m elevation and are bounded by partially pyrite-bearing altered andesites on the east side and karstic limestones on the west. Halloysite, located in a fault zone with approximately NW trending in the middle quarry at an elevation of 800 m, is bounded by altered volcanics in the east and appears to be embedded in a limestone pit in the western block. There is a scattered Mn-bearing halloysite zone between halloysite and limestone, and limestone blocks are surrounded by a thin manganese concentric sheath, which indicates high Eh > 0.4, pH > 8 and pO2 > 1 in an oxidizing environment. At about 300 m NNW of the middle quarry, the upper quarry shows a similar near-horizontal halloysite occurrence (Figure 10).
Karst limestones are suitable for the penetration of hydrothermal solutions formed in reservoir rock environments, as well as in the natural environment, as a result of the dissolution effects of andesitic tuffs with acidic solutions which are required for halloysite precipitation by changing pH, establishing a geochemical balance. Large dissolution and decomposition traces are observed at the surfaces of limestones, as well as residues on the carbonate blocks due to acidic leaching coated with manganese in the halloysite deposit. In this way, the occurrences of the Gönen-Balya halloysites are similar to the Utah-Dragon mine [205] in terms of the geological model that occurred next to or on the karstic limestones.
The Soğucak halloysite deposit appears to have formed in association with the limestones within the lower Miocene andesitic pyroclastic and andesitic lavas sequence. The halloysite deposit is located in the south of Yenice town; the N-S trend is nearly 45–50 m wide, 125–130 m long and reaches a thickness of 12–15 m [206]. It is located adjacent to the limestone blocks of different dimensions within the deposit. The Soğucak halloysite deposit contains halloysite, kaolinite, smectite, illite, alunite, jacobsite, pyrocroite, hematite, goethite, birnessite and pyrite and alunite minerals also accompany the composition. Oxidation of pyrite is the source of Fe stains on the halloysite-rich rocks, and Fe and Mn are transported by hydrothermal ferrous mafic in volcanics of H2S in solutions along the faults. The observed iron and manganese minerals in the halloysite deposits are mostly concentrated at the contacts between limestones and halloysites. The Fe- and Mn-bearing minerals have developed by moving from the rim to the core of limestone blocks, because they can only precipitate in basic environments.
Turplu quarry is characterized by the presence of rich halloysite minerals. XRD analysis shows that the deposits comprise a mineral assemblage that mainly includes 10 Å and 7 Å halloysite, but also feldspars, quartz, goethite, gibbsite, alunite, smectite and illite. The XRD analysis and field observations also reveal pyrite, realgar, cinnabar, orpiment, azurite, bornite, chalcopyrite, psilomelane, hematite, gypsum and other clays that are sometimes associated with halloysite and alunite mineralization. Pyrite is mostly concentrated within the fault clays with small amounts of chalcopyrite. As and Cu sulfides are irregularly distributed in very small discontinuous bands in the clay body. Hand-specimen and XRD studies of the unaltered tuffs reveal that they consist of devitrified glass and pumice fragments, with small phenocrysts of quartz, feldspars and biotite. Other unaltered volcanic flows contain variable abundances of plagioclase, orthopyroxene, hornblende and biotite phenocrysts. Halloysite samples from the deposit follow an almost linear Zr vs. TiO2 trend and suggest that this is possibly inherited from the altered host rock. Mineralogical, chemical and morphological analysis of the Turplu halloysites indicate that their origins result from three general periods of occurrence. These stages include (1) the period of initial deposition of volcanic tuffs and andesites on top of a weathered karstic carbonate terrain, (2) a subsequent period of extensional and strike-slip faulting events and contemporaneous hydrothermal fluid circulation through the faults (i.e., hypogene alteration) and (3) a present-day near-surface weathering period (i.e., supergene alteration) associated with continued faulting. Halloysite forms from andesitic tuffs and devitrified glasses through a dissolution–precipitation mechanism at low pH (2–3) in boiling mud-pool environments. The changes in pH through time and space during hydrothermal activity are strongly controlled by the proposed dissolution–precipitation processes, and this epithermal genetic model is summarized in Figure 11.
8.4. Halloysite Deposits, New Zealand
The Maungaparerua halloysite deposit is located on the further end of the North Island of New Zealand and it was formed by the alteration of host rhyolitic rocks [207]. The Miocene and Pliocene Parahaki volcanics, composed of dacites and rhyolites, are characterized by hosting a wide variety of halloysite minerals. The 40Ar/39Ar dating of the rhyolite dome complex is 10.1 ± 0.03 Ma and bounded by a series of hydrothermally altered basalt flows, siltstone and rhyolitic tuffs [208]. Deep intense supergene weathering is the result of the humid climate superimposed on the hydrothermally altered halloysite deposit. Mineralogical assemblage contains about 50% quartz + fine amorphous silica (cristobalite) and 50% of a mix of halloysite, kaolinite and allophane with a small amount of plagioclase feldspar. The rhyolite dome shape overburdens the morphology and covers the soft halloysite clays, and the upper part, with an average of 15 m, of the halloysite alteration deposit consists of relatively soft clay and underneath this zone, the soft clay becomes hard and dense as a result of overprinting of intense surficial weathering. A genetic model to explain these alteration sequences was proposed [209]. Feldspar alters to amorphous oxides, then amorphous oxides react to form allophane + silica minerals + iron oxides. Based on increasing crystallinity, these mineral transportations occurred as follows: β-allophane → α-allophane → halloysite → kaolinite.
Based on 40Ar/39Ar dating, the emplacement of the rhyolite dome complex was 3.7 ± 0.04 Ma (Early Pliocene) and O and H isotopic compositions of halloysite samples indicate supergene rather than hydrothermal origin; also, this finding is consistent with weathered clay profiles in halloysite-rich zones [208]. However, field studies show the presence of earlier hydrothermal alteration in the form of silicified rhyolite 800 m to the west of the “Southern Area” and kaolinite–pyrite alteration in the adjacent basalt. The authors interpreted that these observations indicate the beginning of the initial processes of hydrothermal alterations, which is considered “the preparation of ground mass”, superimposed with deep weathering of sanidine-rich rhyolite under water-saturated subtropical conditions, which is the dominant process for the development of a halloysite-rich weathering zone which extends up to 24 m deep from the surface in the Maungaparerua clay deposit.
At the Matauri Bay halloysite deposit, north of Maungaparerua, a sanidine rhyolite dome (40Ar/39Ar dated at 10.1 ± 0.03 Ma) intrudes an older basalt and is overlain by alluvial sediments and a younger basalt (4.0 ± 0.7 Ma) [210]. In the Kerikeri-Matauri Bay district, volcanic alkali rhyolite was extruded as domes and cooled rapidly, with fine-grained feldspar subsequently highly altered to a halloysite-rich clay layer [211] between 10 and 30 m thick [205]. Other halloysite deposits are present in different rhyolite domes in the district and the largest is the Maungaparerua dome covering 140 ha. The δ18O values of halloysite samples range from 23.7 to 33.7‰ and δD values range from −51.2 to −57.5‰, suggesting dominant influences of supergene effects over hydrothermal alteration, and that the activities of young tectonism and magmatism cannot be ruled out. Due to geographic locations, paleo-climatic effects and similarities in volcanic activities, most of New Zealand’s halloysite deposits have genetic similarities and the dominance of halloysite over kaolinite was favored by supergene weathering during the late Miocene-Pliocene subtropical weathering regime [210].
Another economic halloysite deposit is reported in the Quaternary pumiceous rhyolitic tephra deposits of ~0.93 Ma (Te Puna tephra) and ~0.27 Ma (Te Ranga tephra) in the Tauranga area, eastern North Island [36]. The proposed genetic changes are solubilized volcanic glass + plagioclase → halloysite spheroids → halloysite tubes → halloysite plates → halloysite books. In contrast to other deposits, there is no occurrence of kaolinite in Tauranga; it is more likely that low permeability largely causes the deposits to remain in a locally wet environment, which is extremely important for the hydrated metastable halloysite books that have not transformed to kaolinite.
8.5. Halloysite Deposits, Mexico
The occurrence of supergene and hypogene halloysite in a porphyry epithermal system Au (Cu-Mo) (pyrite + marcasite + enargite), which is structurally and lithologically controlled by matrix-rich breccia pipes, appears at Cerro la Mina, Chiapas, southeastern Mexico [212]. This region is known as a tectonically active and complex region along the Chiapanecan volcanic arc, and is also located at the junction of the North American, Caribbean and Cocos plates. The Quaternary stratigraphy of the Cerro la Mina area presents with pyroclastic flows at 1.04 ± 0.04 Ma (U-Pb, zircon) and volcaniclastic matrix-rich breccias that were invaded by monzodiorites, alkali intrusions and feldspar–biotite–phyric trachyandesites dome cross-cuts, which are interpreted to be a volcanic–hydrothermal breccia pipe [213]. The eruption of El Chichón volcano in 1998 ejected anhydrite-bearing trachyandesite pyroclastic material from the active volcano-hydrothermal system. A boiling crater spring discharged Cl-rich and S-poor geothermal water at almost neutral pH (up to 6.7) with an isotopic composition close to that of subduction-type magmatic water (δD −15‰; δ18O +6.5‰) [161].
Ref. [213] classified the Cerro la Mina magmatic hydrothermal alteration systems, which are composed of (A) early porphyry-style potassic and late-potassic alteration which occurred after the formation of the breccia pipe, including quartz + K-feldspar ± biotite and pyrite, (B) phyllic alteration composed of quartz, muscovite, illite, illite–smectite, chlorite, calcite, gypsum and tourmaline, and is associated with pyrite ± chalcopyrite ± molybdenite found in veins, and (C) advanced argillic–argillic alteration overprinted potassic and phyllic alteration. The advanced argillic–argillic zone includes low-temperature (<110 °C) hypogene halloysite + kaolinite occurrences extending from 800 to 250 m and is deeper than higher-temperature (>120 °C) quartz + dickite ± kaolinite ± pyrophyllite ± alunite that occur from 250 m to the present-day surface. Below the 250 m zone, argillic alteration has produced a halloysite–kaolinite–rich zone. These zones also typically contain pyrite, marcasite and enargite, and traces of galena and sphalerite. Halloysite association with gypsum and jarosite has been attributed to a supergene origin [212] and this mineralogical paragenesis distinguishes Mexican quarries from others, whereas halloysite occurring with quartz, alunite, dickite, kaolinite and pyrite is proposed as being of hypogene origin. In the Cerro la Mina volcanic–hydrothermal system, the roughly 800 m deep halloysite zone suggests that higher-temperature potassic alteration was overprinted by extensive lower-temperature advanced argillic alteration, which implies significant temperature changes over time, possibly due to regional uplift and exhumation.
8.6. Kaolin Deposits, Italy
Sardinia island is very famous due to major kaolin deposits in Italy and these deposits are the products of hydrothermal alteration of the Tertiary rhyolite ignimbrites and andesitic basaltic lavas [4]. In addition, ref. [214] reported in more detailed studies that massive andesites, dacites and mildly welded rhyodacitic ignimbrites are the precursors of kaolin in north-western Sardinia. The dominant kaolin-group mineral is well-crystallized kaolinite with minor amounts of dickite, halloysite and allophane. These kaolin deposits from the andesites contain pyrite and jarosite, and the deposits which formed from the dacites contain up to 5% alunite. Several fluid-dominated hydrothermal systems control the local-scale structural framework, morphology and composition of the kaolin deposits that are linked to the NNW-SSE-trending normal faults which formed during the extension regime of the regional tectonics of the Corsica and Sardinia system. Based on the morphological properties, attitude, fracturing and porosity of the parent rocks, four different types of kaolin deposits are recognized; these are bedform, mushroom, fault parallel and funnel. The remarkable funnel morphology was generated by repeated hydrothermal eruptions, which is an indicator of ascending geothermal fluids.
8.7. Kaolin Deposits, Japan
The Kohdachi kaolin deposit is located to the north of Hiroshima and originates from the hydrothermal alteration of mainly fine to medium-grained biotite–muscovite granite. The kaolin deposit is ~250 m in length and ~100 m in width, extending from NE to SW, and is overlain by a sand and mud bed of the Tertiary age and a gravel bed of the Quaternary age [215]. The Kohdachi deposit contains three main alteration zones: (i) halloysite with a weak silicification zone, (ii) a halloysite–kaolinite zone and (iii) a kaolinite zone in a W-E sequence. Based on SEM observations, large dissolution pits were observed on the surface of quartz crystals from the kaolin zone. Silicification and dissolution pits on the surface of quartz crystals in different areas of the deposit indicate a changing pH of geothermal fluids over time. Silicification, (i) the precipitation of Fe-oxides at the border of zones and (ii) the dissolution of quartz in different areas of the deposit indicate a variable pH within a distance of <100 m. Based on H isotope data, the formation temperature of the Kohdachi kaolinite may be roughly estimated at 70–150 °C and this suggests that it is formed by hydrothermal alteration [216]. Typically, the halloysite zone is more dominant in the central part, and the kaolinite zone is the margin of the kaolinized granitic rocks. Two different clay veins were found in the kaolin body and the surrounding rocks: (i) white veins consisting of kaolinite and/or halloysite and (ii) pale green veins composed of kaolinite and a small amount of illite mineral. Weak silicification and euhedral quartz crystals were observed in the halloysite zone, and the amounts of quartz in the halloysite zone were slightly greater than in the kaolinite zone [215]. This observation indicates that the pH of geothermal solutions was lower during the occurrence of halloysites compared to the formation of kaolinites.
The Toyoha mine in the Green Tuff region of Hokkaido, Japan, consists of many epithermal vein-type deposits which are associated with kaolinization; this is characterized by the major alteration of Miocene volcanic rocks of acidic to intermediate compositions [217]. Many minerals are identified in the Toyoha mine, such as lead–zinc vein-type deposits, but the principal minerals are pyrite, sphalerite, galena, quartz, carbonates and chlorite. Some secondary origin minerals are kaolinite, caryopilite, goethite, native silver and szmikite (MnSO4H2O). This quarry differs from the quarry described above in its chaotic mineral composition, with the presence of both ore minerals, carbonates and secondary mineral assemblages including kaolin. The textures of ores from the Toyoha deposits strongly suggest that most of the ores have suffered from extensive alteration after deposition. Late-stage kaolinite sometimes fills the interspaces of these sphalerites, which indicates the changes in the pH regime through time and space due to the influences of active island-arc volcanism and regional tectonism in Japan. The geothermal gradient in the mining area is very steep, about 20 °C/100 m, and hot springs are often encountered in the mine. The textural evidence and the presence of hot springs suggest that secondary alterations of ores are quite common in the deposit [217]. Based on mineralogical assemblages, it is assumed that the temperature of formation of the Toyoha deposits was in a range from 150 to 250 °C.
8.8. Kaolin Deposits, China
One of the largest kaolin deposits of China is in the Dongguanxia mine located in Fujian province and it is formed in the upper part of the biotite granite pluton of Mesozoic magmatism. These igneous activities affected the folded basement terrain of southeastern China [218]. The kaolin bodies are lenticular or bedlike-shaped and some others show funnel or trough-like geometry with a maximum depth of 162 m. Regional tectonics and the shape of the kaolin body indicate that ascending geothermal fluids enabled the occurrences of many kaolin deposits. The kaolin deposits in the Zhanjiang area in Guangdong province are characterized by a mixture of stacking disorder kaolinite and tubular halloysite with some illite. The kaolinization process was reported as the result of alteration and weathering of Jurassic biotite–granites which invaded Cambrian sediments [218]. The Mengson halloysite deposit is a good example of hydrothermal alteration of granite and feldspar–quartz assemblages without mafic minerals in the Yunnan province. The Longyan kaolin is a mixture of halloysite, kaolinite and others, and Guangxi province is the major source of halloysite production.
The most significant and the best-quality kaolin deposits in China are located near Suzhou in Jungsu province [219] and primary resources have been derived from the alteration of Mesozoic granitic rocks and associated volcanics. As the result of multi-stage emplacements of granitic rocks and cyclic intrusions of Jurassic volcanism, intermediate composition, low-pH and low-temperature hydrothermal alterations played a key role during kaolinization. The thickness of the kaolin zone ranges from 5 to 30 m and the maximum thickness reaches 80 m [4]. The mineral assemblage is kaolinite and halloysite with a minor amount of alunite, pyrite, sericite, quartz and chalcedony [211]. This quarry is distinguished from other quarries in the region by the hard-white alunite nodules found in the clay body.
The Longmen kaolin deposit is located in the western margin of Ganxi volcanics, Jiangxi Province, China [220]. The host rocks are rhyolitic crystal-vitric tuff and minor lapilli tuff of the Late Jurassic Ehuling Formation, and hydrothermal kaolinite deposit, together with some Mo, Cu, Pb and Zn mineralization, which is characterized by a higher metal content compared to other quarries and consists of kaolinite, dickite and pyrophyllite with minor amounts of quartz, sericite and pyrite. The cross-cuttings of the N-E- and N-W-trending fault zones occur in the kaolin mining area, along which small late-stage dykes, such as felsites, sillite, and quartz dykes, appear. The ore bodies consist of mostly vein or vein-like structures, and occur in the fault-related altered rocks. Based on mineral assemblages, the formation temperature of the kaolin deposit falls within the range of 270–350 °C.
8.9. Kaolin Deposits, Mexico
The Guanajuato mining district is located in the central part of the Guanajuato Mountain Range; there are both Ag and Au deposits from low-sulfidation quartz and calcite-rich epithermal veins from a kaolin-production point of view. Hydrothermal kaolin deposits extend about 3 km long and the area is located in the west of Guanajuato. The kaolin deposits occurred through the hydrothermal alteration of rhyolite flow-breccia and probably welded tuffs [4]. The most refractory clay is comprised chiefly of kaolinite and finely crystalline quartz. The giant silica sinter, “silica gossan”, deposits have been deposited as surface covers over the fractures; such Fe-rich silicifications, “silica cap”, are fairly common over the Mexican hydrothermal clay deposits [221]. Partial desilication of parent rocks yields kaolinite with a parallel orientation with the chloritic schist, amorphous silica and fine-grained quartz bands intermixed with kaolinite. Another famous kaolin deposit is near San Luis Potosi [222].
Another kaolin deposit is located in San Luis Potosi and the products of hydrothermal alteration, which is controlled by N-S-trending fault zones, are of the Tertiary rhyolite, flow-breccia and welded tuffs [221]. Late-stage silicification processes resulted in massive colloform opal and silica sinter deposits that were discontinuously underlain by the kaolinization (±halloysite) exposures approximately 5 km long, indicating a change in the chemical compositions of hydrothermal fluids. In addition, the high-temperature hydrothermal origin of nacrite, dickite and kaolinite mineralization occurred in the foothills of the Sierra Madre Occidental, Nayarit [130].
9. Conclusion and Keys Points for Future Prospects
The main emerging points are as follows from this paper:
- The most important point in examining the kaolin formations proposed in this study is that the data should be evaluated together rather than obtained separately. In this study, the inter-evaluation of the data was achieved by examining the important kaolin formations in different literature sources and re-interpreting the tables and figures by combining them. In this way, it was easier to determine hypogene, supergene, high-sulfidation and low-sulfidation types by comparison, as well as to determine the formation environments of altered mineral associations and to interpret their origins.
- The dominant process in the formation of kaolin and alunite-group minerals was an early-stage acid–sulfate hydrothermal alteration of tuffs and other volcanic rocks. This was followed by a later stage where alkali–chloride fluids are responsible for the silicification and silica sinter deposition within the volcanic–hydrothermal system. Sinter deposits and silicifications were found at the top and adjacent to altered zones, where the focus mechanism was upflow. High sulfidation was controlled by the fault systems and these steam-heated environments included sulfide-enriched vapors, initially saline (4%–10% NaCl), and then oxidized overprinting by S-rich descending magmatic fluids, and finally groundwaters mixed to varying degrees in the vadose zone.
- Although kaolinization processes are widely observed all over the world, whether they occur under hydrothermal alteration or weathering conditions, they vary considerably in terms of mineral paragenesis and formation morphology. The difference in mineral paragenesis can manifest itself in the form of kaolin-group polymorphs, and also these kaolin-group minerals are often associated with alunite-group minerals, silica, ore minerals, other clay minerals, carbonates, sulfates, etc. The reason for this large mineral assemblage should be regarded as the host rock mineralogical composition and porosity, the composition of the active solution, and the chemical character and physical conditions such as temperature and pressure.
- Considering all of these processes, we can collect information about many parameters, such as mineral associations, host rock chemistry and composition, composition of the active hydrothermal solutions, origin and determination of high-sulfidation and low-sulfidation types, and hypogene and supergene characterization during the evaluation of any kaolin formation. At this stage, in addition to mineralogical and petrographic descriptions, detailed chemical analyses and isotopic examinations should be frequently used.
- The occurrence and differentiation of conditions for halloysite and kaolinite formation are still subject to debate. Due to the high permeability and porosity of karst, limestones conditions were suitable for the penetration of acid sulfate hydrothermal solutions that result in the dissolution of shallow andesitic tuffs and volcanic rocks in steam-heated environments. Field observations suggest that the close associations of halloysite and alunite (±apatite) play a role in hypogene processes. As a result of large dissolution and decomposition on the surfaces of karstic limestones, traces of thin Mn- and Fe-crusts surrounding the embedded limestone blocks both inside and underlying the karstic limestone beds of the halloysite quarry were observed. We attribute this to changing pH, which increases within the local geochemically balanced environment. As a result, it is possible to hypothesize that the forming of halloysite started following the steam-heated shallow hypogene acid sulfate alteration process of volcanic rocks. Later, it was under the influence of supergene overprinting adjacent to and/or underlying karstic limestones beds which provide better drainage and act as a geochemical buffer. Based on SEM morphology studies, halloysite was probably formed in low-temperature (30–40 °C) conditions and a continuous moisture-rich environment.
- The main morphologies of halloysite (tubular, spheroidal and platy) and the Fe content suggest that a role is played by structural Fe in the determination of its micromorphology. The occurrences of halloysite morphologies are exhibited in specific geologic features, which were not observed for other kaolin minerals. Halloysite deposits were found in association with karstic limestone contacts next to the deposit. Otherwise, kaolinite morphologies would take place.
Author Contributions
Conceptualization, Ö.I.E. and H.Ü.E.; methodology, Ö.I.E.; software, H.Ü.E.; data curation, Ö.I.E. and H.Ü.E.; writing—original draft preparation, Ö.I.E. and H.Ü.E.; writing—review and editing, Ö.I.E. and H.Ü.E.; visualization, H.Ü.E.; supervision, Ö.I.E.; project administration, Ö.I.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data are contained within the article.
Acknowledgments
The authors express their deepest gratitude to Hans Albert Gilg for his critical review and constructive comments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schroeder, P.A.; Erickson, G. Kaolin: From ancient porcelains to nanocomposites. Elements 2014, 10, 177–182. [Google Scholar] [CrossRef]
- Keller, W.D.; Cheng, H.; Johans, W.D.; Meng, C.S. Kaolin from the original Kaoling (Gaoling) mine locality, Kiangsi Province, China. Clays Clay Miner. 1980, 28, 97–104. [Google Scholar] [CrossRef]
- Schroeder, P.A. Clays in the Critical Zone; Cambridge University Press: Cambridge, UK, 2018; 246p. [Google Scholar]
- Murray, H.H. Kaolin minerals: Their genesis and occurrences. In Hydrous Phyllosilicates (Exclusive of Micas); Bailey, S.W., Ed.; Reviews in Mineralogy; Mineralogical Society of America: Washington, DC, USA, 1988; Volume 19, pp. 67–89. [Google Scholar]
- Dill, H.D. Kaolin: Soil, rock and ore from the mineral to the magmatic, sedimentary and metamorphic environments. Earth Sci. Rev. 2016, 161, 16–129. [Google Scholar] [CrossRef]
- Ece, O.I.; Ercan, H.U. Supergene origin of kaolin deposits. In Supergene Mineral Deposits; Butt, C., Bowell, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2024; in press. [Google Scholar]
- Bailey, S.W. Hydrous Phyllosilicates: (Exclusive of Micas); Walter de Gruyter GmbH & Co KG: Berlin, Germany, 2018; Volume 19. [Google Scholar]
- Dominguez, E.; Iglesias, C.; Dondi, M. The geology and mineralogy of a range of kaolins from the Santa Cruz and Chubut Provinces, Patagonia (Argentina). Appl. Clay Sci. 2008, 40, 124–142. [Google Scholar] [CrossRef]
- Ece, O.I.; Ekinci, B.; Schroeder, P.A.; Crowe, D.; Esenli, F. Origin of the Duvertepe kaolin-alunite deposits in Simav Graben, Turkey: Timing and styles of hydrothermal mineralization. J. Volcanol. Geotherm. Res. 2013, 255, 57–78. [Google Scholar] [CrossRef]
- Psyrillos, A.; Howe, C.H.; Manning, D.A.C.; Burley, S.D. Geological controls on kaolin particle shape and consequences for mineral processing. Clay Miner. 1999, 34, 193–208. [Google Scholar] [CrossRef]
- Rye, R.O.; Bethke, P.M.; Wasserman, M.D. The stable isotope geochemistry of acid-sulfate alteration. Econ. Geol. 1992, 87, 225–262. [Google Scholar] [CrossRef]
- Rye, R.O. A review of the stable-isotope geochemistry of sulfate minerals in selected igneous environments and related hydrothermal systems. Chem. Geol. 2005, 215, 5–36. [Google Scholar] [CrossRef]
- Nicholson, K. Geothermal Fluids: Chemistry and Exploration Techniques; Springer: Berlin/Heidelberg, Germany, 1993; 268p. [Google Scholar]
- Mazot, A.; Bernard, A.; Fischer, T.; Inguaggiato, S.; Sutawidjaja, I.S. Chemical evolution of thermal waters and changes in the hydrothermal system of Papandayan volcano (West Java, Indonesia) after the November 2002 eruption. J. Volcanol. Geotherm. Res. 2008, 178, 276–286. [Google Scholar] [CrossRef]
- Maximo, R.P.R.; Bernard, A.; Maussen, K.; Joiris, E.; Rebadulla, R.R.R. Geochemical studies of thermal waters from Kanlaon Volcano, Negros Island, Philippines. J. Volcanol. Geotherm. Res. 1999, 374, 39–51. [Google Scholar] [CrossRef]
- Moore, D.M.; Reynolds, R.C. X-ray Diffraction and the Identification and Analysis of Clay Minerals; Oxford University Press: Oxford, UK, 1997; 378p. [Google Scholar]
- Meunier, A. Clays; Springer: Berlin/Heidelberg, Germany, 2005; 486p. [Google Scholar]
- Dixon, J.B. Kaolin and serpentine group minerals. In Mineral in Soil Environments; Dixon, J.B., Weed, S.B., Eds.; Soil Science Society of America (SSSA): Madison, WI, USA, 1989; Volume 1, pp. 467–525. [Google Scholar]
- Weaver, C.E. Clays, Muds, and Shales; Development in Sedimentology; Elsevier: Amsterdam, The Netherlands, 1989; Volume 44, 799p. [Google Scholar]
- Brindley, G.W.; Brown, G. Crystal Structures of Clay minerals and Their X-ray Identification; Mineralogical Society: London, UK, 1980; 486p. [Google Scholar]
- Bergaya, F.; Theng, B.K.G.; Lagaly, G. Handbook of Clay Science; Development in Clay Science; Elsevier Ltd.: Amsterdam, The Netherlands, 2006; Volume 1, 1234p. [Google Scholar]
- Dill, H.G.; Bosse, H.R.; Henning, K.H.; Fricke, A.; Ahrend, H. Mineralogical and chemical variations in hypogene and supergene kaolin deposits in a mobile fold belt—The Central Andes of northwestern Peru. Miner. Depos. 1997, 32, 149–163. [Google Scholar] [CrossRef]
- Ruiz Cruz, M.D.; Reyes, E. Kaolinite and dickite formation during shale diagenesis: Isotopic data. Appl. Geochem. 1998, 13, 95–104. [Google Scholar] [CrossRef]
- McAuley, G.E.; Burley, S.D.; Fallick, A.E.; Kusznir, N.J. Palaeohydrodynamic fluid flow regimes during diagenesis of the Brent Group in the Hutton–NW Hutton reservoirs: Constraints from oxygen isotope studies of authigenic kaolin and reverse flexural modeling. Clay Miner. 1994, 29, 609–626. [Google Scholar] [CrossRef]
- Beaufort, D.; Cassagnabere, A.; Petit, S.; Lanson, B.; Berger, G.; Lacharpagne, J.C.; Johansen, H. Kaolinite-to dickite reaction in sandstone reservoirs. Clay Miner. 1998, 33, 297–316. [Google Scholar] [CrossRef]
- Ehrenberg, S.N.; Aagaard, P.; Wilson, M.J.; Fraser, A.R.; Duthie, D.M.L. Depth-dependent transformation of kaolinite to dickite in sandstones of the Norwegian continental shell. Clay Miner. 1993, 28, 325–352. [Google Scholar] [CrossRef]
- Dill, H.G. The “chessboard” classification scheme of mineral deposits: Mineralogy and geology from aluminum to zirconium. Earth Sci. Rev. 2010, 100, 1–420. [Google Scholar] [CrossRef]
- Dill, H.G. Authigenic heavy minerals a clue to unravel supergene and hypogene alteration of marine and continental sediments of Triassic to Cretaceous age (SE Germany). Sediment. Geol. 2010, 228, 61–76. [Google Scholar] [CrossRef]
- Bailey, S.W. Halloysite—A critical assessment. Sci. Geol. 1990, 86, 89–98. [Google Scholar]
- Joussein, E.; Petit, S.; Churchman, J.; Theng, B.; Righi, D.; Delvaux, B. Halloysite clay minerals—A review. Clay Miner. 2005, 40, 383–426. [Google Scholar] [CrossRef]
- Ece, O.I.; Schroeder, P.A. Clay mineralogy and chemistry of halloysite and alunite deposits in the Turplu Area, Balikesir, Turkey. Clays Clay Miner. 2007, 55, 18–35. [Google Scholar] [CrossRef]
- Itaya, T.; Arribas, A., Jr.; Okada, T. Argon release systematics of hypogene and supergene alunite based on progressive heating experiments from 100 to 1000 °C. Geochim. Cosmochim. Acta 1996, 60, 4525–4535. [Google Scholar] [CrossRef]
- Ercan, H.U.; Ece, O.I.; Schroeder, P.A.; Karacik, Z. Differentiating styles of alteration within kaolin-alunite hydrothermal deposits of Canakkale, NW Turkey. Clays Clay Miner. 2016, 64, 245–274. [Google Scholar] [CrossRef]
- Cravero, F.; Churchman, G.J. The origin of spheroidal halloysites: A review of the literature. Clay Miner. 2016, 51, 417–427. [Google Scholar] [CrossRef]
- Cravero, F.; Maiza, P.J.; Marfil, S.A. Halloysite in Argentinian deposits: Origin and textural constraints. Clay Miner. 2012, 47, 329–340. [Google Scholar] [CrossRef]
- Cunningham, M.J.; Loew, D.J.; Wyatt, J.B.; Moon, V.G.; Jock Churchman, G. Discovery of halloysite books in altered silicic Quaternary tephras, northern New Zealand. Clay Miner. 2016, 51, 351–372. [Google Scholar] [CrossRef]
- Hillier, S.; Brydson, R.; Delbos, E.; Fraser, A.; Gray, N.; Pendlowski, H.; Phillips, I.; Robertson, J.; Wilson, I. Correlations among the mineralogical and physical properties of halloysite nanotubes (HNTs). Clay Miner. 2016, 51, 325–350. [Google Scholar] [CrossRef]
- Kogure, T.; Mori, K.; Drits, V.A.; Takai, Y. Structure of prismatic halloysite. Am. Mineral. 2013, 98, 1008–1016. [Google Scholar] [CrossRef]
- Yuan, P.; Tan, D.; Annabi-Bergaya, F. Properties and applications of halloysite nanotubes: Recent research advances and future prospects. Appl. Clay Sci. 2015, 112–113, 75–93. [Google Scholar] [CrossRef]
- Kohyama, N.; Fukushima, K.; Fukami, A. Observation of the hydrated form of tubular halloysite by an electron microscope equipped with an environmental cell. Clays Clay Miner. 1978, 26, 25–40. [Google Scholar] [CrossRef]
- Kittrick, J.A. Soil minerals in the Al2O3-SiO2-H2O system and a theory of their formation. Clays Clay Miner. 1969, 17, 157–167. [Google Scholar] [CrossRef]
- Kittrick, J.A. Precipitation of kaolinite at 25 °C and 1 Atm. Clays Clay Miner. 1970, 18, 261–267. [Google Scholar] [CrossRef]
- De Ligny, D.; Navrotsky, A. Energetics of kaolin polymorphs. Am. Mineral. 1999, 84, 506–516. [Google Scholar] [CrossRef]
- Zotov, A.; Mukhamet-Galeev, A.; Schott, J. An experimental study of kaolinite and dickite relative stability at 150–300 °C and the thermodynamic properties of dickite. Am. Mineral. 1998, 83, 516–524. [Google Scholar] [CrossRef]
- Hayashi, M. Hydrothermal alteration in the Ohtake geothermal area, Kyushu. J. Jpn. Geotherm. Energy Assoc. 1973, 10, 9–46. [Google Scholar]
- Hemley, J.J. Some mineralogical equilibria in the system K2O-Al2O3-SiO2-H2O. Am. J. Sci. 1959, 257, 241–270. [Google Scholar] [CrossRef]
- Reyes, A.G. Petrology of Philippine geothermal system and the application of alteration mineralogy to their assessment. J. Volcanol. Geotherm. Res. 1990, 43, 279–304. [Google Scholar] [CrossRef]
- Utada, M. Hydrothermal alteration related to igneous acidity in Cretaceous and Neogene formations of Japan. Min. Geol. Jpn. Spec. Issue 1980, 12, 79–92. [Google Scholar]
- Fialips, C.I.; Petit, S.; Decarreau, A.; Beaufort, D. Influence of synthesis pH on kaolinite “crystallinity” and surface properties. Clays Clay Miner. 2000, 48, 173–184. [Google Scholar] [CrossRef]
- Huertas, F.J.; Fiore, S.; Huertas, F.; Linares, J. Experimental study of the hydrothermal formation of kaolinite. Chem. Geol. 1999, 156, 171–190. [Google Scholar] [CrossRef]
- O’Neil, J.R.; Johnson, C.M.; White, L.D.; Roedder, E. The origin of fluids in the salt beds of the Delaware Basin, New Mexico and Texas. Appl. Geochem. 1986, 1, 265–271. [Google Scholar] [CrossRef]
- Sheppard, S.M.F.; Gilg, H.A. Stable isotope geochemistry of clay minerals. Clay Miner. 1996, 31, 1–24. [Google Scholar] [CrossRef]
- Savin, S.M.; Epstein, S. The oxygen and hydrogen isotope geochemistry of clay minerals. Geochim. Cosmochim. Acta 1970, 34, 25–42. [Google Scholar] [CrossRef]
- Savin, S.M.; Epstein, S. The oxygen and hydrogen isotope geochemistry of ocean sediments and shales. Geochim. Cosmochim. Acta 1970, 34, 43–63. [Google Scholar] [CrossRef]
- Lambert, S.J.; Epstein, S. Stable isotope investigations of an active geothermal system in Valles Caldera, Jemez Mountains, New Mexico. J. Volcanol. Geotherm Res. 1980, 8, 111–129. [Google Scholar] [CrossRef]
- Woldegabriel, G. Hydrothermal alteration in the Valles caldera ring fracture zone and core hole VC-1: Evidence for multiple hydrothermal systems. J. Volcanol. Geotherm. Res. 1990, 40, 105–122. [Google Scholar] [CrossRef]
- Boulvais, P.; Valet, J.M.; Esteoule-Choux, J.; Fourcade, S.; Martineau, F. Origin of kaolinitization in Brittany (NW France) with emphasis on deposits over granite: Stable isotopes (O, H) constraints. Chem. Geol. 2000, 168, 211–223. [Google Scholar] [CrossRef]
- Sheppard, S.M.F. The Cornubian batholith, SW England: D/H and 18O/16O studies of kaolinite and other alteration minerals. J. Geol. Soc. 1977, 133, 573–591. [Google Scholar] [CrossRef]
- Harris, C.; Compton, J.S.; Bevington, S.A. Oxygen and hydrogen isotope composition of kaolinite deposits, Cape Peninsula, South Africa; low-temperature, meteoric origin. Econ. Geol. 1999, 94, 1353–1366. [Google Scholar] [CrossRef]
- Ece, O.I.; Schroeder, P.A.; Smilley, M.J.; Wampler, J.M. Acid-sulphate hydrothermal alteration of andesitic tuffs and genesis of halloysite and alunite deposits in the Biga Peninsula, Turkey. Clay Miner. 2008, 43, 281–315. [Google Scholar] [CrossRef]
- Heald, P.; Foley, N.K.; Hayba, D.O. Comparative anatomy of volcanic-hosted epithermal deposits: Acid-sulfate and adularia-sericite types. Econ. Geol. 1987, 82, 1–26. [Google Scholar] [CrossRef]
- Marfil, S.A.; Maiza, P.J.; Cardellach, E.; Corbella, M. Origin of kaolin deposits in the “Los Menucos” area, Rio Negro Province, Argentina. Clay Miner. 2005, 40, 283–293. [Google Scholar] [CrossRef]
- Kadir, S.; Erkoyun, H. Genesis of the hydrothermal Karaçayır kaolinite deposit in Miocene volcanics and Palaeozoic metamorphic rocks of the Uşak-Güre Basin, western Turkey. Turk. J. Earth Sci. 2013, 22, 444–468. [Google Scholar] [CrossRef]
- Szynkiewicz, A.; Moore, C.H.; Glamoclija, M.; Pratt, L.M. Sulfur isotope signatures in gypsiferous sediments of the Estancia and Tularosa Basins as indicators of sulfate sources, hydrological processes, and microbial activity. Geochim. Cosmochim. Acta 2009, 73, 6162–6186. [Google Scholar] [CrossRef]
- Sakai, H.; Casadevallt, J.; Moore, J.G. Chemistry and isotope ratios of sulfur in basalts and volcanic gases at Kilauea volcano, Hawaii. Geochim. Cosmochim. Acta 1982, 46, 729–738. [Google Scholar] [CrossRef]
- Cunningham, C.G.; Rye, R.O.; Rockwell, B.W.; Kunk, M.J.; Councell, T.B. Supergene destruction of a hydrothermal replacement alunite deposit at Big Rock Candy Mountain, Utah; mineralogy, spectroscopic remote sensing, stable isotope, and argon age evidences. Chem. Geol. 2005, 215, 317–337. [Google Scholar] [CrossRef]
- Murray, H.H.; Keller, W.D. Kaolins, kaolins and kaolins. In Kaolin Genesis and Utilization; Murray, H.H., Bundy, W., Harvey, C., Eds.; The Clay Mineral Society: Boulder, CO, USA, 1993; pp. 1–14. [Google Scholar]
- Dill, H.G.; Kus, J.; Dohrmann, R.; Tsoy, Y. Supergene and hypogene alteration in the dual-use kaolin-bearing coal deposit Angren, SE Uzbekistan. Int. J. Coal Geol. 2008, 75, 225–240. [Google Scholar] [CrossRef]
- Lee, G.; Koh, S.; Pirajno, F. Evolution of hydrothermal fluids of HS and LS type epithermal Au-Ag deposits in the Seongsan hydrothermal system of the Cretaceous Haenam volcanic field, South Korea. Ore Geol. Rev. 2014, 61, 33–51. [Google Scholar] [CrossRef]
- Pirajno, F. Subaerial hot springs and near-surface hydrothermal mineral systems past and present, and possible extraterrestrial analogues. Geosci. Front. 2020, 11, 1549–1569. [Google Scholar] [CrossRef]
- Keller, W.D. Scan electron micrographs of kaolins collected from diverse environments of origin—I. Clays Clay Miner. 1976, 24, 107–113. [Google Scholar] [CrossRef]
- Keller, W.D. Scan electron micrographs of kaolins collected from diverse environments of origin—II. Clays Clay Miner. 1976, 24, 114–117. [Google Scholar] [CrossRef]
- Keller, W.D. Scan electron micrographs of the kaolinization process including examples from the Bohemian Massif. Schriftenr. Geol. Wiss. 1978, 11, 89–108. [Google Scholar]
- Marumo, K.; Matsuhisa, Y.; Nagasawa, K. Hydrogen and oxygen isotopic composition of kaolin minerals in Japan. Dev. Sedimentol. 1982, 35, 315–320. [Google Scholar]
- Gilg, H.A.; Hülmeyer, S.; Miller, H.; Sheppard, S.M.F. Supergene origin of the Lastarria kaolin deposit, South-Central Chile, and paleoclimatic implications. Clays Clay Miner. 1999, 47, 201–211. [Google Scholar] [CrossRef]
- Sillitoe, R.H. Epithermal paleosurfaces. Miner. Depos. 2015, 50, 767–793. [Google Scholar] [CrossRef]
- Juliani, C.; Rye, R.O.; Nunes, C.M.D.; Snee, L.W.; Silva, R.H.C.; Monteiro, L.V.S.; Bettencourt, J.S.; Neumann, R.; Neto, A.A. Paleoproterozoic high-sulfidation mineralization in the Tapajos gold province, Amazonian Craton, Brazil: Geology, mineralogy, alunite argon age, and stable-isotope constraints. Chem. Geol. 2005, 215, 95–125. [Google Scholar] [CrossRef]
- Panteleyev, A. Epithermal Au-Ag-Cu: High Sulfidation. 2005. Available online: https://emrlibrary.gov.yk.ca/ygs/open_files/2005/2005-5/h04_high_sulphidation_epithermal_au_ag_cu.pdf (accessed on 6 April 2021).
- Hedenquist, J.W. Volcanic-related hydrothermal systems in the Circum-Pacific Basin and their potential for mineralization. Min. Geol. 1987, 37, 347–364. [Google Scholar]
- Hedenquist, J.W.; Browne, P.R.L. The evolution of the Waiotapu geothermal system, New Zealand, based on the chemical and isotopic composition of its fluids, minerals and rocks. Geochim. Cosmochim. Acta 1989, 53, 2235–2257. [Google Scholar] [CrossRef]
- Berger, B.R.; Bonham, H.F., Jr. Epithermal gold-silver deposits in the western United States: Time-space products of evolving plutonic, volcanic, and tectonic environments. J. Geochem. Explor. 1990, 36, 103–142. [Google Scholar] [CrossRef]
- Henley, R.W.; Berger, B.R. Magmatic-vapor expansion and the formation of high-sulfidation gold deposits: Chemical controls on alteration and mineralization. Ore Geol. Rev. 2011, 39, 63–74. [Google Scholar] [CrossRef]
- Hedenquist, J.W.; Watanabe, Y.; Arribas, A. Hypogene alunite from the El Salvador district, Chile, indicates potential for a blind porphyry copper center. Econ. Geol. 2020, 115, 231–239. [Google Scholar] [CrossRef]
- Hedenquist, J.W.; Arribas, A. The tops and bottoms of high-sulfidation epithermal ore deposits. In Proceedings of the 5th Biennial Meeting of the Society for Geology Applied to Mineral Deposits, London, UK, 22–25 August 1999. [Google Scholar]
- Arribas, A. Characteristics of high-sulfidation epithermal deposits, and their relation to magmatic fluid. In Magmas, Fluids, and Ore Deposits; Thompson, J.F.H., Ed.; Mineralogical Association of Canada Short Course; Mineralogical Association of Canada: Quebec, QC, Canada, 1995; Volume 23, Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.459.114&rep=rep1&type=pdf (accessed on 22 March 2022).
- Simeone, R.; Dilles, J.H.; Padalino, G.; Palomba, M. Mineralogical and stable isotope studies of kaolin deposits: Shallow epithermal systems of western Sardinia, Italy. Econ. Geol. 2005, 100, 115–130. [Google Scholar] [CrossRef]
- Ercan, H.Ü.; Ece, Ö.I.; Schroeder, P.A.; Gülmez, F. Characteristics and evolution of the Etili silica sinter epithermal deposits, Çanakkale-Turkey: Relation to alkali-chloride vs acid-sulphate fluids. Ore Geol. Rev. 2022, 142, 104726. [Google Scholar] [CrossRef]
- White, N.C. High sulfidation epithermal gold deposits: Characteristics and a model for their origin. Geol. Surv. Jpn. Rep. 1991, 277, 9–20. [Google Scholar]
- Hedenquist, J.W.; Matsuhisa, Y.; Izawa, E.; White, N.C.; Giggenbach, W.F.; Aoki, M. Geology, geochemistry and origin of high sulfidation Cu-Au mineralization in the Nansatsu District, Japan. Econ. Geol. 1994, 89, 1–30. [Google Scholar] [CrossRef]
- Henley, R.W.; McNabb, A. Magmatic vapour plumes and ground-water interaction in porphyry copper emplacement. Econ. Geol. 1978, 73, 1–20. [Google Scholar] [CrossRef]
- Fournier, R.O. Conceptual models of brine evolution in magmatic-hydrothermal systems. In Volcanism in Hawaii; USGS Professional Paper 1350; Hawaiian Volcano Observatory: Hilo, HI, USA, 1987; pp. 1487–1505. [Google Scholar]
- Chouinard, A.; Williams-Jones, A.; Leonardson, R.W.; Hodgson, C.J.; Silva, P.; Tellez, C.; Vega, J.; Rojas, F. Geology and genesis of the multistage high-sulfidation epithermal Pascua Au-Ag-Cu deposit, Chile and Argentina. Econ. Geol. 2005, 100, 463–490. [Google Scholar] [CrossRef]
- Dill, H.G. The geology of aluminium phosphates and sulphates of the alunite group minerals: A review. Earth-Sci. Rev. 2001, 53, 35–93. [Google Scholar] [CrossRef]
- Railsback, B. Earth scientist’s periodic table of the elements and their ions. Geology 2003, 31, 737–740. [Google Scholar] [CrossRef]
- Yau, Y.C.; Peacor, D.R.; Essene, E.J. Authigenic anatase and titanite in shales from the Salton Sea Geothermal Field, California. Neues Jahrb. Mineral. Monatsh. 1987, 19, 441–452. [Google Scholar]
- Schroeder, P.A.; Shiflet, J. Ti-bearing phases in an east Georgia kaolin deposit. Clays Clay Miner. 2000, 48, 151–158. [Google Scholar] [CrossRef]
- Sheppard, S.M.F.; Nielsen, R.L.; Taylor, H.P. Oxygen and hydrogen isotope ratios of clay minerals from porphyry copper deposits. Econ. Geol. 1969, 64, 755–777. [Google Scholar] [CrossRef]
- Cravero, F.; Marfil, S.A.; Maiza, P.J. Statistical analysis of geochemical data: A tool for discriminating between kaolin deposits of hypogene and supergene origin, Patagonia, Argentina. Clay Miner. 2010, 45, 183–196. [Google Scholar] [CrossRef]
- Hutchison, W.; Finch, A.A.; Boyce, A.J. The sulfur isotopic evolution of magmatic-hydrothermal fluids: Insights into ore-forming processes. Geochim. Cosmochim. Acta 2020, 288, 176–198. [Google Scholar] [CrossRef]
- Ohmoto, H.; Rye, R.O. Isotopes of sulfur and carbon. In Geochemistry of Hydrothermal Ore Deposits, 2nd ed.; Barnes, H.L., Ed.; Wiley Interscience: New York, NY, USA, 1979; pp. 509–567. [Google Scholar]
- Hedenquist, J.W.; Simmons, S.F.; Giggenbach, W.F.; Eldridge, S.C. White Island, New Zealand, volcanic-hydrothermal system represents the geochemical environment of high-sulfidation Cu and Au ore deposition. Geology 1993, 21, 731–734. [Google Scholar] [CrossRef]
- Taran, Y.; Kalacheva, E.; Inguaggiato, S.; Cardellini, C.; Karpov, G. Hydrothermal systems of the Karymsky Volcanic Centre, Kamchatka: Geochemistry, time evolution and solute fluxes. J. Volcanol. Geotherm. Res. 2017, 346, 28–39. [Google Scholar] [CrossRef]
- Marini, L.; Moretti, R.; Accornero, M. Sulfur Isotopes in Magmatic-Hydrothermal Systems, Melts, and Magmas. Rev. Mineral. Geochem. 2011, 73, 423–492. [Google Scholar] [CrossRef]
- Keller, W.D. Morphology of kaolinite weathered from a non-feldspathic mica-phyllite. Clay Miner. 1981, 16, 289–295. [Google Scholar] [CrossRef]
- Lorenz, W.; Gwosdz, W. Bewertungskriterien für Industrieminerale, Steine Und Erden: Teil 1 Tone; Geologisches Jahrbuch Reihe H; Bundesanstalt für Geowissenschaften und Rohstoffe: Hanover, Germany, 1997; Volume 2, pp. 1–108. (In German) [Google Scholar]
- Pirajno, F. Hydrothermal Processes and Mineral Systems; Springer: Berlin/Heidelberg, Germany, 2009; 1250p. [Google Scholar]
- Henley, R.D.; Ellis, A.J. Geothermal systems ancient and modern: A geochemical review. Earth-Sci. Rev. 1983, 19, 1–50. [Google Scholar] [CrossRef]
- Cooke, D.R.; Simmons, S.F. Characteristics and genesis of epithermal gold deposits. Soc. Econ. Geol. Rev. 2000, 13, 221–244. [Google Scholar]
- Hedenquist, J.W. Exploration for epithermal gold deposits. Soc. Econ. Geol. 2000, 13, 245–277. [Google Scholar]
- Simon, A.C.; Frank, M.R.; Pettke, T.; Candela, P.A.; Piccoli, P.M.; Heinrich, C.A. Gold partitioning in melt-vapor-brine systems. Geochim. Cosmochim. Acta 2005, 69, 3321–3335. [Google Scholar] [CrossRef]
- Takeno, N. Thermal and geochemical structure of the Uenotai geothermal system, Japan. Geothermics 2000, 29, 257–277. [Google Scholar] [CrossRef]
- Jones, B.; Renaut, R.W. Hot spring and geyser sinters: The integrated product of precipitation, replacement and deposition. Can. J. Earth Sci. 2003, 40, 1549–1569. [Google Scholar] [CrossRef]
- Lynne, B.Y.; Campbell, K.A. Diagenetic transformations (opal-A to quartz) of low and mid-temperature microbial textures in siliceous hot spring deposits, Taupo Volcanic Zone, New Zealand. Can. J. Earth Sci. 2003, 40, 1679–1696. [Google Scholar] [CrossRef]
- Drake, H.; Heim, C.; Roberts, N.M.; Zack, T.; Tillberg, M.; Broman Åström, M.E. Isotopic evidence for microbial production and consumption of methane in the upper continental crust throughout the Phanerozoic eon. Earth Planet. Sci. Lett. 2017, 470, 108–118. [Google Scholar] [CrossRef]
- Fouke, B.W.; Farmer, J.D.; Des Marais, D.J.; Pratt, L.; Sturchio, N.C.; Burns, P.C.; Mykell, K.; Discipulo, M.K. Depositional Facies and Aqueous-Solid Geochemistry of Travertine-Depositing Hot Springs (Angel Terrace, Mammoth Hot Springs), Yellowstone National Park, U.S.A. J. Sediment. Res. 2000, 70, 565–585. [Google Scholar] [CrossRef]
- Pentecost, A.; Jones, B.; Renaut, R.W. What is hot spring? Can. J. Earth Sci. 2003, 40, 1443–1446. [Google Scholar] [CrossRef]
- White, D.E.; Hutchinson, R.A.; Keith, T.E.C. The Geology and Remarkable Thermal Activity of Norris Geyser Basin, Yellowstone National Park, Wyoming; US Geological Survey Professional Paper 1456; USGS: Reston, VA, USA, 1988; pp. 1–84.
- Taran, Y.; Kalacheva, E. Acid sulphate-chloride volcanic waters; Formation and potential for monitoring of volcanic activity. J. Volcanol. Geotherm. Res. 2020, 405, 107036. [Google Scholar] [CrossRef]
- Jones, B.; Renaut, R.W.; Rosen, M.R. Stromatolites forming in acidic hot-spring waters, North Island, New Zealand. Palaios 2000, 15, 450–475. [Google Scholar] [CrossRef]
- Mountain, B.; Seward, T.M. Hydrosulfide/sulfide complexes of copper(I): Experimental confirmation of the stoichiometry and stability of Cu(HS)2− to elevated temperatures. Geochim. Cosmochim. Acta 2003, 67, 3005–3014. [Google Scholar] [CrossRef]
- Lynne, B.Y.; Campbell, K.A.; Perry, R.S.; Browne, P.R.; Moore, J.N. Acceleration of sinter diagenesis in an active fumarole, Taupo Volcanic Zone, New Zealand. Geology 2006, 34, 749–752. [Google Scholar] [CrossRef]
- Schinteie, R.; Campbell, K.A.; Browne, P.R. Microfacies of stromatolitic sinter from acid-sulphate-chloride springs at Parariki Stream, Rotokawa geothermal field, New Zealand. Palaeontol. Electron. 2007, 10, 1–33. [Google Scholar]
- Jones, B.; Renaut, R.W. Facies architecture in depositional systems resulting from the interaction of acidic springs, alkaline springs, and acidic lakes: Case study of Lake Roto-a-Tamaheke, Rotorua, New Zealand. Can. J. Earth Sci. 2012, 49, 1217–1250. [Google Scholar] [CrossRef]
- Murphy, R.J.; Van Kranendonk, M.J.; Baumgartner, R.; Ryan, C. Biogenicity of Spicular Geyserite from Te Kopia, New Zealand: Integrated Petrography, High-Resolution Hyperspectral and Elemental Analysis. Astrobiology 2021, 21, 115–135. [Google Scholar] [CrossRef]
- Zhu, Y.; An, F.; Tan, J. Geochemistry of hydrothermal gold deposits: A review. Geosci. Front. 2011, 2, 367–374. [Google Scholar] [CrossRef]
- Hamilton, A.R.; Campbell, K.A.; Guido, D.M. Atlas of Siliceous Hot Spring Deposits (Sinter) and Other Silicified Surface Manifestations in Epithermal Environments; GNS Science Report 2019/06; GNS Science: Lower Hutt, New Zealand, 2019; 56p. [Google Scholar] [CrossRef]
- Seedorff, E.; Dilles, J.H.; Proffett, J.M.; Einaudi, M.T.; Zurcher, L.; Stavast, W.J.A.; Johnson, D.A.; Barton, M.D. Porphyry deposits: Characteristics and origin of hypogene features. In Economic Geology 100th Anniversary Volume; Society of Economic Geologists, Inc.: Littleton, CO, USA, 2005; pp. 251–298. [Google Scholar]
- Jambor, J.L. Nomenclature of the alunite supergroup. Can. Mineral. 1999, 37, 1323–1341. [Google Scholar]
- Rattray, K.J.; Taylor, M.R.; Bevan, D.J.M. Compositional segregation and solid solution in the lead dominant alunite-type minerals from Broken Hill, N.S.W. Mineral. Mag. 1996, 60, 779–785. [Google Scholar] [CrossRef]
- Hanson, R.F.; Zamora, R.; Keller, W.D. Nacrite, dickite and kaolinite in one deposit in Nayarit, Mexico. Clays Clay Miner. 1981, 29, 451–453. [Google Scholar] [CrossRef]
- Hemley, J.J.; Hostetler, P.B.; Gude, A.J.; Mountjo, W.T. Some stability relations of alunite. Econ. Geol. 1969, 64, 599–612. [Google Scholar] [CrossRef]
- Aoki, M. Mineralogical features and genesis of alunite solid solution in high temperature magmatic hydrothermal systems. Geol. Surv. Jpn. Rep. 1991, 277, 35–37. [Google Scholar]
- Tsuzuki, Y. Solubility diagrams for explaining zone sequences in bauxite, kaolin and pyrophyllite-diaspore deposits. Clays Clay Miner. 1976, 24, 297–302. [Google Scholar] [CrossRef]
- Kennedy, G.C. Phase relations in the system Al2O3 H2O at higth temperatures and pressures. Am. Jour. Sci. 1959, 257, 563–573. [Google Scholar] [CrossRef]
- Tsuzuki, Y.; Mizutani, S. A study of rock alteration process based on kinetics of hydrothermal experiment. Contrib. Mineral. Petrol. 1971, 30, 15–33. [Google Scholar] [CrossRef]
- Winkler, H.G.F. Petrogenesis of Metamorphic Rocks; Springer: Berlin/Heidelberg, Germany, 1976; 348p. [Google Scholar]
- Bozkaya, O.; Yalcin, H.; Basibuyuk, Z.; Bozkaya, G. Metamorphic-hosted pyrophyllite and dickite occurrences from the hydrous Al-silicate deposits of the Malatya–Puturge region, Central Eastern Turkey. Clays Clay Miner. 2007, 55, 423–442. [Google Scholar] [CrossRef]
- Sykes, M.L.; Moody, B.J. Pyrophyllite and Metamorphism in the Carolina Slate Belt. Am. Mineral. 1978, 63, 96–108. [Google Scholar]
- Evans, B.W.; Guggenheim, S. Talc, pyrophyllite, and related minerals. In Hydrous Phyllosilicates (Exclusive of Micas); Bailey, S.W., Ed.; Reviews in Mineralogy; Mineralogical Society of America: Washington, DC, USA, 1988; Volume 19, pp. 225–294. [Google Scholar]
- Hemley, J.J.; Montoya, J.W.; Marinenko, J.W.; Luce, R.W. Equilibria in the system Al2O3-SiO2-H2O and some general implications for alteration/mineralization processes. Econ. Geol. 1980, 75, 210–228. [Google Scholar] [CrossRef]
- Berman, R.G. Internally-consistent thermodynamic data for minerals in the system Na2O-K2O-CaO-MgO-FeO-Fe2O3-Al2O3-SiO2-TiO2-H2O-CO2. J. Petrol. 1988, 29, 445–522. [Google Scholar] [CrossRef]
- Eberl, D. Synthesis of pyrophyllite polytypes and mixed layers. Am. Mineral. 1979, 64, 1091–1096. [Google Scholar]
- Nagasawa, K. Kaolin minerals. In Clays and Clay Minerals in Japan; Sudo, T., Shimoda, S., Eds.; Development in Sedimentology; Elsevier: Amsterdam, The Netherlands, 1978; Volume 26, pp. 189–219. [Google Scholar]
- Nabetani, A.; Shikazono, N. Chemical process and environment of hydrothermal alteration of acidic volcanic rocks in the Mitsuishi district, southwest Japan. Geochem. J. 2002, 36, 255–269. [Google Scholar] [CrossRef]
- Marumo, K. Genesis of kaolin minerals and pyrophyllite in Kuroko deposits of Japan. Implications for the origins of the hydrothermal fluids from mineralogical and stable isotope data. Geochim. Cosmochim. Acta 1989, 53, 2915–2924. [Google Scholar] [CrossRef]
- Okamoto, A.; Saishu, H.; Hirano, N.; Tsuchiya, N. Mineralogical and textural variation of silica minerals in hydrothermal flow-through experiments: Implications for quartz vein formation. Geochim. Cosmochim. Acta 2010, 74, 3692–3706. [Google Scholar] [CrossRef]
- Morey, G.W.; Hesselgesser, J.M. The solubility of quartz and some other substances in superheated steam at high pressures. Econ. Geol. 1951, 46, 821–835. [Google Scholar] [CrossRef]
- Edmond, J.M.; Measures, C.I.; McDuff, R.E.; Chan, L.H.; Collier, R.W.; Grant, B.; Gordon, L.I.; Corliss, J.B. Ridge crest hydrothermal activity and the balances of the major and minor elements in the ocean: The Galapagos data. Earth Planet. Sci. Lett. 1979, 46, 1–18. [Google Scholar] [CrossRef]
- Michard, G.; Albarede, F.; Michard, A.; Minster, J.F.; Charlou, J.L.; Tan, N. Chemistry of solutions from the 13*N East Pacific Rise hydrothermal site. Earth Planet. Sci. Lett. 1984, 67, 297–307. [Google Scholar] [CrossRef]
- Von Damm, K.L.; Bischoff, J.L. Chemistry of hydrothermal solutions from the southern Juan de Fuca Ridge. J. Geophys. Res. 1987, 92, 334–346. [Google Scholar] [CrossRef]
- Von Damm, K.L.; Edmond, J.M.; Grant, B.; Measures, C.I.; Walden, B.; Weiss, R.F. Chemistry of submarine hydrothermal solutions at 21° N, East Pacific Rise. Geochim. Cosmochim. Acta 1985, 49, 2197–2220. [Google Scholar] [CrossRef]
- Hedenquist, J.W. Boiling and dilution in the shallow portion of the Waiotopu geothermal system, New Zealand. Geochim. Cosmochim. Acta 1991, 55, 2753–2765. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; McCleskey, R.; Ball, J.W. Sulfur geochemistry of hydrothermal waters in Yellowstone National Park: IV acid-sulfate waters. Appl. Geochem. 2009, 24, 191–207. [Google Scholar] [CrossRef]
- Inoue, A.; Utada, M. Hydrothermal alteration related to Kuroko mineralization in the Kamikita area, Northern Honshu, Japan, with special reference to the acid-sulfate alteration. Geol. Surv. Jpn. Rep. 1991, 277, 39–48. [Google Scholar]
- Inoue, A.; Utada, M. Hydrothermal alteration in Kamikita Kuroko mineralization area, Northern Honshu, Japan. Min. Geol. 1991, 41, 203–218. [Google Scholar]
- Armannsson, H. The fluid geochemistry of Icelandic high temperature geothermal areas. Appl. Geochem. 2016, 66, 14–64. [Google Scholar] [CrossRef]
- Fournier, R.O.; Rowe, J.J. Estimation of underground temperatures from the silica content of water from hot springs and wet-steam wells. Am. J. Sci. 1966, 264, 685–697. [Google Scholar] [CrossRef]
- Joseph, E.P.; Fournier, N.; Lindsay, J.M.; Fischer, T.P. Gas and water geochemistry of geothermal systems in Dominica, Lesser Antilles island arc. J. Volcanol. Geotherm. Res. 2011, 206, 1–14. [Google Scholar] [CrossRef]
- Okada, H.; Yasuda, Y.; Yagi, M.; Kai, K. Geology and fluid chemistry of the Fushime geothermal field, Kyushu, Japan. Geothermics 2000, 29, 279–311. [Google Scholar] [CrossRef]
- Saibi, H.; Ehara, S. Temperature and chemical changes in the fields of the Obama geothermal field (SW Japan) in response to field utilization. Geothermics 2010, 39, 228–241. [Google Scholar] [CrossRef]
- Taran, Y.; Fischer, T.P.; Pokrovsky, B.; Sano, Y.; Aurora Armienta, M.; Macias, J.L. Geochemistry of the volcano-hydrothermal system of El Chichón volcano, Chiapas, Mexico. Bull. Volcanol. 1998, 59, 436–449. [Google Scholar] [CrossRef]
- Takenaka, T.; Furuya, S. Geochemical model of the Takigami geothermal system, Northeast Kyushu, Japan. Geochem. J. 1991, 25, 267–281. [Google Scholar] [CrossRef]
- Yoshida, Y. Geochemistry of the Nigorikawa geothermal system, southwest Hokkaido, Japan. Geochem. J. 1991, 25, 203–222. [Google Scholar] [CrossRef]
- Hedenquist, J.W.; Arribas, A.; Reynolds, T.J. Evolution of an intrusion-centered hydrothermal system: Far Southeast-Lepanto porphyry and epithermal Cu-Au deposits, Philippines. Econ. Geol. 1998, 93, 373–404. [Google Scholar] [CrossRef]
- Drake, B.D.; Campbell Rowland, J.V.; Guido, D.M.; Browne, P.R.L.; Rae, A. Evolution of a dynamic paleo-hydrothermal systems at mangatete, Taupo volcanic zone, New Zealand. J. Volcanol. Geotherm. Res. 2014, 281, 19–35. [Google Scholar] [CrossRef]
- Cady, S.L.; Farmer, J.D. Fossilization processes in siliceous thermal springs: Trends in preservation along thermal gradients. Ciba Found. Symp. 1996, 202, 150–173. [Google Scholar]
- Lynne, B.Y.; Campbell, K.A.; Moore, J.; Browne, P.R.L. Origins and evolution of the Steamboat Springs siliceous sinter deposit, Nevada, U.S.A. Sediment. Geol. 2008, 210, 111–131. [Google Scholar] [CrossRef]
- Ercan, H.Ü.; Ece, Ö.I.; Çiftçi, E.; Aydın, A. Comparison of Epithermal Kaolin Deposits from the Etili Area (Çanakkale, Turkey): Mineralogical, Geochemical, and Isotopic Characteristics. Clays Clay Miner. 2022, 70, 753–779. [Google Scholar] [CrossRef]
- Sillitoe, R.H. Epithermal models: Genetic types, geothermal controls and shallow features. In Mineral Deposit Modeling; Kirkham, R.V., Sinclair, W.D., Thorpe, R.I., Duke, J.M., Eds.; Special Paper; Geological Association of Canada: St. John’s, NL, Canada, 1993; Volume 40, pp. 403–417. [Google Scholar]
- Guido, D.M.; Campbell, K.A. Jurassic hot spring deposits of the Deseado Massif (Patagonia, Argentina): Characteristics and controls on regional distribution. J. Volcanol. Geotherm. Res. 2011, 203, 35–47. [Google Scholar] [CrossRef]
- Africano, F.; Bernard, A. Acid alteration in the fumarolic environment of Usu volcano, Hokkaido, Japan. J. Volcanol. Geotherm. Res. 2000, 97, 475–495. [Google Scholar] [CrossRef]
- Gunnarsson, I.; Arnórsson, S. Amorphous silica solubility and the thermodynamic properties of H4SiO°4 in the range of 0 °C to 350 °C at Psat. Geochim. Cosmochim. Acta 2000, 64, 2295–2307. [Google Scholar] [CrossRef]
- Herdianita, N.R.; Browne, P.R.L.; Rodgers, K.A.; Campbell, K.A. Mineralogical and textural changes accompanying ageing of silica sinter. Miner. Depos. 2000, 35, 48–62. [Google Scholar] [CrossRef]
- Lynne, B.Y.; Boudreau, A.; Smith, I.J.; Smith, G.J. Silica accumulation rates for siliceous sinter at Orakei Korako geothermal field, Taupo Volcanic Zone, New Zealand. Geothermics 2019, 78, 50–61. [Google Scholar] [CrossRef]
- Rimstidt, J.D.; Cole, D.R. Geothermal mineralization I: The mechanism of formation of the Beowawe Nevada, siliceous sinter deposit. Am. J. Sci. 1983, 283, 861–875. [Google Scholar] [CrossRef]
- Berger, B.R.; Henley, R.W. Magmatic-vapor expansion and the formation of high sulfidation gold deposits: Structural controls on hydrothermal alteration and ore mineralization. Ore Geol. Rev. 2011, 39, 75–90. [Google Scholar] [CrossRef]
- Orange, F.; Lalonde, S.V.; Konhauser, K.O. Experimental simulation of evaporation-driven silica sinter formation and microbial silicification in hot spring systems. Astrobiology 2013, 13, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Stoffynegli, P.; Mackenzie, F.T. Mass balance of dissolved lithium in the oceans. Geochim. Cosmochim. Acta 1984, 48, 859–872. [Google Scholar] [CrossRef]
- Stoffregen, R.E. Genesis of acid–sulfate alteration and Au–Cu–Ag mineralization at Summitville, Colorado. Econ. Geol. 1987, 82, 1575–1591. [Google Scholar] [CrossRef]
- Jannas, R.R.; Bowers, T.S.; Petersen, U.; Beane, R.E. High-sulfidation deposit types in the El Indio district, Chile. In Geology and Ore Deposits of the Central Andes; Skinner, B.J., Ed.; Society of Economic Geologists Special Publication; Society of Economic Geologists: Littleton, CO, USA, 1999; Volume 7, pp. 219–266. [Google Scholar]
- Africano, F.; Van Rompaey, G.; Bernard, A.; Le Guern, F. Deposition of trace elements from high temperature gases of Satsuma-Iwojima volcano. Earth Planets Space 2002, 54, 275–286. [Google Scholar] [CrossRef]
- Fournier, R.O. The behavior of silica in hydrothermal solutions. In Geology and Geochemistry of Epithermal Systems; Berger, B.R., Bethke, P.M., Eds.; Reviews in Economic Geology; Society of Economic Geologists: Littleton, CO, USA, 1985; Volume 2, pp. 45–61. [Google Scholar]
- Fournier, R.O.; Potter, R.W., II. A Revised and Expanded Silica (quartz) Geothermometer. Geotherm. Resour. Counc. Bull. 1982, 11, 3–12. [Google Scholar]
- Fournier, R.O. Hydrothermal processes related to movement of fluid from plastic into brick rock in the magmatic-epithermal environment. Econ. Geol. 1999, 94, 1193–1211. [Google Scholar] [CrossRef]
- Okay, A.I.; Satır, M.; Maluski, H.; Siyako, M.; Metzger, R.; Akyüz, S. Paleo-and Neo-Tethyan events in Northwest Turkey: Geological and geochronological constrains. In The Tectonic Evolution of Asia; Yin, A., Harrison, T.M., Eds.; Cambridge University Press: Cambridge, UK, 1996; pp. 420–441. [Google Scholar]
- Okay, A.I.; Tansel, I.; Tüysüz, O. Obduction, subduction and colision as reflected in the Upper Cretaceous–Lower Eocene sedimantary record of western Turkey. Geol. Mag. 2001, 138, 117–142. [Google Scholar] [CrossRef]
- Karacık, Z.; Yılmaz, Y. Geology of the ignimbrites and the associated volcano-plutonic complex of the Ezine area, northwestern Anatolia. J. Volcanol. Geotherm. Res. 1998, 85, 251–264. [Google Scholar] [CrossRef]
- Karacık, Z.; Yılmaz, Y.; Pearce, J.A.; Ece, O.I. Petrochemistry of the south Marmara granitoids, northwest Anatolia, Turkey. Int. J. Earth Sci. 2008, 97, 1181–1200. [Google Scholar] [CrossRef]
- Menant, A.; Jolivet, L.; Tuduri, J.; Loiselet, C.; Bertrand, G.; Guillou-Frottier, L. 3D subduction dynamics: A first-order parameter of the transition from copper- to gold-rich deposits in the eastern Mediterranean region. Ore Geol. Rev. 2018, 94, 118–135. [Google Scholar] [CrossRef]
- Voudouris, P.; Mavrogonatos, C.; Spry, P.G.; Baker, T.; Melfos, V.; Klemd, R.; Melfou, M. Porphyry and epithermal deposits in Greece: An overview, new discoveries, and mineralogical constraints on their genesis. Ore Geol. Rev. 2019, 107, 654–691. [Google Scholar] [CrossRef]
- Delibaş, O.; Moritz, R.; Chiaradia, M.; Selby, D.; Ulianov, A.; Revan, M.K. Post-collisional magmatism and ore-forming systems in the Menderes massif: New constraints from the Miocene porphyry Mo-Cu PA +/- narbaAYA +/- system, Gediz-Kütahya, western Turkey. Miner. Depos. 2017, 52, 1157–1178. [Google Scholar] [CrossRef]
- Boucher, K.S. The Structural and Fluid Evolution of the Efemçukuru Epithermal Gold Deposit, Western Turkey. Master’s Thesis, University of British Columbia, Vancouver, BC, Canada, 2016; 452p. unpublished. [Google Scholar]
- Yigit, Ö. Mineral deposits of Turkey in relation to Tethyan Metallogeny: Implications for future mineral exploration. Econ. Geol. 2009, 104, 19–51. [Google Scholar] [CrossRef]
- Hanilci, N.; Öztürk, H.; Banks, D. Geological, geochemical and microthermometric characteristics of the Hakkari region Zn-Pb deposits, SE Turkey. Ore Geol. Rev. 2020, 125, 103667. [Google Scholar] [CrossRef]
- Kuşçu, I.; Tosdal, R.M.; Gençalioğlu-Kuşçu, G. Episodic porphyry Cu (-Mo-Au) formation and associated magmatic evolution in Turkish Tethyan collage. Ore Geol. Rev. 2019, 107, 119–154. [Google Scholar] [CrossRef]
- Baker, T.; Bickford, D.; Juras, S.; Lewis, P.; Oztas, Y.; Ross, K.; Spikings, R. The geology of the Kisladag porphyry gold deposit, Turkey. In Tectonics and Metallogeny of the Tethyan Orogenic Belt; Society of Economic Geologists Special Publications; Society of Economic Geologists: Littleton, CO, USA, 2016; Volume 19, pp. 57–83. [Google Scholar]
- Juras, S.; Millers, R.; Skayman, P. Technical Report for the Kisladag Gold Mine, Turkey; Internal Report of the Eldorado Gold Corporation; Eldorado Gold Corporation: Vancouver, BC, Canada, 2010; 120p. [Google Scholar]
- Bozkaya, Ö.; Bozkaya, G.; Hanilçi, N.; Laçin, D.; Banks, D.A.; Uysal, I.T. Mineralogical and geochemical evidence of late epithermal alteration in the Kisladag porphyry gold deposit, Usak, Western Turkey. In Life with Ore Deposits on Earth, Proceedings of the 15th Biennial SGA Conference, Glasgow, UK, 27–30 August 2019; Society for Geology Applied to Mineral Deposits, University of Glasgow: Glasgow, UK, 2019; Volume 3, pp. 1031–1034. [Google Scholar]
- Kadir, S.; Erman, H.; Erkoyun, H. Mineralogical and geochemical characteristics and genesis of hydrothermal kaolinite deposits within Neogene volcanites, Kütahya (western Anatolia), Turkey. Clays Clay Miner. 2011, 59, 250–276. [Google Scholar] [CrossRef]
- Cortecci, G.; Dinelli, E.; Bolognesi, L.; Boschetti, T.; Ferrara, G. Chemical and isotopic compositions of water and dissolved sulfate from shallow wells on Vulcano Island, Aeolian Archipelago, Italy. Geothermics 2001, 30, 69–91. [Google Scholar] [CrossRef]
- Shelley, D. Igneous and Metamorphic Rocks under the Microscope; Chapman and Hall: London, UK, 1993; 445p. [Google Scholar]
- Ece, Ö.I.; Nakagawa, Z.; Schroeder, P.A. Alteration of Volcanic Rocks and Genesis of Kaolin Deposits in Şile Region, Northern Istanbul, Turkey. Part I. Clay Mineralogy. Clays Clay Miner. 2003, 51, 675–688. [Google Scholar] [CrossRef]
- Maksimovic, Z.; Panto, G.Y. Mineralogy of yttrium and lanthanide elements in karstic bauxite deposits. Trav. ICSOBA 1983, 18, 191–200. [Google Scholar]
- Uygun, A. Geology and occurrences of some halloysite deposits in limestones, NW Anatolia. MTA Derg. 1999, 121, 141–151. [Google Scholar]
- Keeling, J.L. The mineralogy, geology and occurrences of halloysite. In Natural Mineral Nanotubes: Properties and Applications; Pasbakhsh, P., Churchman, G.J., Eds.; Apple Academic Press Inc.: Oakville, ON, Canada, 2015; pp. 96–115. [Google Scholar]
- Laçin, D.; Yeniyol, M. An example to the halloysite deposits formed associated with the andesitic pyroclastics: Soğucak halloysite deposit, Yenice–Çanakkale. İstanb. Üniv. Mühendis. Fak. Yerbilim. Derg. 2006, 19, 27–41. [Google Scholar]
- Murray, H.H.; Harvey, C.; Smith, J.M. Mineralogy and geology of the Maungaparerua halloysite deposit in New Zealand. Clay Miner. 1977, 25, 1–5. [Google Scholar] [CrossRef]
- Brathwaite, R.L.; Christie, A.B.; Faure, K.; Townsend, M.G.; Terlesk, S. Geology, mineralogy and geochemistry of the rhyolite-hosted Maungaparerua clay deposit, Northland. N. Z. J. Geol. Geophys. 2014, 57, 357–368. [Google Scholar] [CrossRef]
- Harvey, C.C. Rock Alteration in the South-East, Whitiangia Area. Master’s Thesis, University of Auckland, Auckland, New Zealand, 1967, unpublished. [Google Scholar]
- Brathwaite, R.L.; Christie, A.B.; Faure, K.; Townsend, M.G.; Terlesk, S. Origin of the Matauri Bay halloysite deposit, Northland, New Zealand. Miner. Depos. 2012, 47, 897–910. [Google Scholar] [CrossRef]
- Wilson, I.; Keeling, J.L. Global occurrence, geology and characteristics of tubular halloysite deposits. Clay Miner. 2016, 51, 309–324. [Google Scholar] [CrossRef]
- Kyne, R.; Hollings, P.; Jansen, N.H.; Cooke, D.R. Supergene and hypogene in a porphyry-epithermal environment at Cerro la Mina, Chiapas, Mexico. Econ. Geol. 2013, 108, 1147–1161. [Google Scholar] [CrossRef]
- Jansen, N.H.; Gemmell, J.B.; Chang, Z.; Cooke, D.R.; Jourdan, F.; Creaser, R.A.; Hollings, P. Geology and genesis of the Cerro La Mina Porphyry-high sulfidation Au (Cu-Mo) Prospect, Mexico. Econ. Geol. 2017, 112, 799–827. [Google Scholar] [CrossRef]
- Oggiano, G.; Mameli, P. Tectonic and litho-stratigraphic controls on kaolin deposits within volcanic successions: Insights from the kaoliniferous district of north-western Sardinia (Italy). Ore Geol. Rev. 2012, 48, 151–164. [Google Scholar] [CrossRef]
- Kitagawa, R.; Köster, R. Genesis of the Tirschenreuth kaolin deposit in Germany compared with the deposit in Japan. Clay Miner. 1991, 26, 61–79. [Google Scholar] [CrossRef]
- Marumo, K.; Nagasawa, K.; Kuroda, Y. Mineralogy and hydrogen isotope geochemistry of clay minerals in the Ohnuma geothermal area, Northeastern Japan. Earth Planet. Sci. Lett. 1980, 47, 255–262. [Google Scholar] [CrossRef]
- Shikazono, N. Mineralization and chemical environment of the Toyoha lead-zinc vein-type deposits, Hokkaido, Japan. Econ. Geol. 1975, 70, 694–705. [Google Scholar] [CrossRef]
- Wilson, I.R. Kaolin and halloysite deposits of China. Clay Miner. 2004, 39, 1–15. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, D.; Feng, M.; Feng, B.; Jin, T. Kaolin deposits of China. In Proceedings of the International Clay Conference, Bologna and Pavia, Italy, 6–12 September 1981; Sedimentology 35. Elsevier: Amsterdam, The Netherlands, 1982; pp. 719–731. [Google Scholar]
- Yuan, Y.; Shi, G.; Yang, M.; Wu, Y.; Zhang, Z.; Huang, A.; Zhang, J. Formation of a hydrothermal kaolinite deposit from rhyolitic tuff in Jiangxi, China. J. Earth Sci. 2014, 25, 495–505. [Google Scholar] [CrossRef]
- Hanson, R.F.; Keller, W.D. Genesis of refractory clay near Guanajuato, Mexico. In Clays and Clay Minerals, Proceedings of the 14th National Conference, Berkeley, CA, USA, 1 January 1966; Bailey, S.W., Ed.; Pergamon Press: Oxford, UK, 1966; pp. 259–267. [Google Scholar]
- Keller, W.D.; Hanson, R.F. Hydrothermal alteration of a rhyolite flow breccia near San Luis Potosi, Mexico, to refractory kaolin. Clays Clay Miner. 1968, 16, 223–229. [Google Scholar] [CrossRef]
Figure 1.
FE-SEM morphology of kaolin- and alunite-group minerals. (a) Vermiform (book-shaped) kaolinite crystals; (b) hexagonal equidimensional kaolinite; (c) euhedral alunite rhombohedral crystals; (d,e) tubular-shaped halloysite showing a hollow-ended structure; and (f) spherical halloysite growth crystal on the edge of kaolinite flakes [31].
Figure 1.
FE-SEM morphology of kaolin- and alunite-group minerals. (a) Vermiform (book-shaped) kaolinite crystals; (b) hexagonal equidimensional kaolinite; (c) euhedral alunite rhombohedral crystals; (d,e) tubular-shaped halloysite showing a hollow-ended structure; and (f) spherical halloysite growth crystal on the edge of kaolinite flakes [31].

Figure 2.
TEM of replica of freeze-fractured cross-sectional views of halloysite tubes. (a,b) are dehydrated (spiral shape, 7 Å), and (c,d) are hydrated (original—donut shape, 10 Å) halloysites [31]. Scale bars = 100 nm.
Figure 2.
TEM of replica of freeze-fractured cross-sectional views of halloysite tubes. (a,b) are dehydrated (spiral shape, 7 Å), and (c,d) are hydrated (original—donut shape, 10 Å) halloysites [31]. Scale bars = 100 nm.
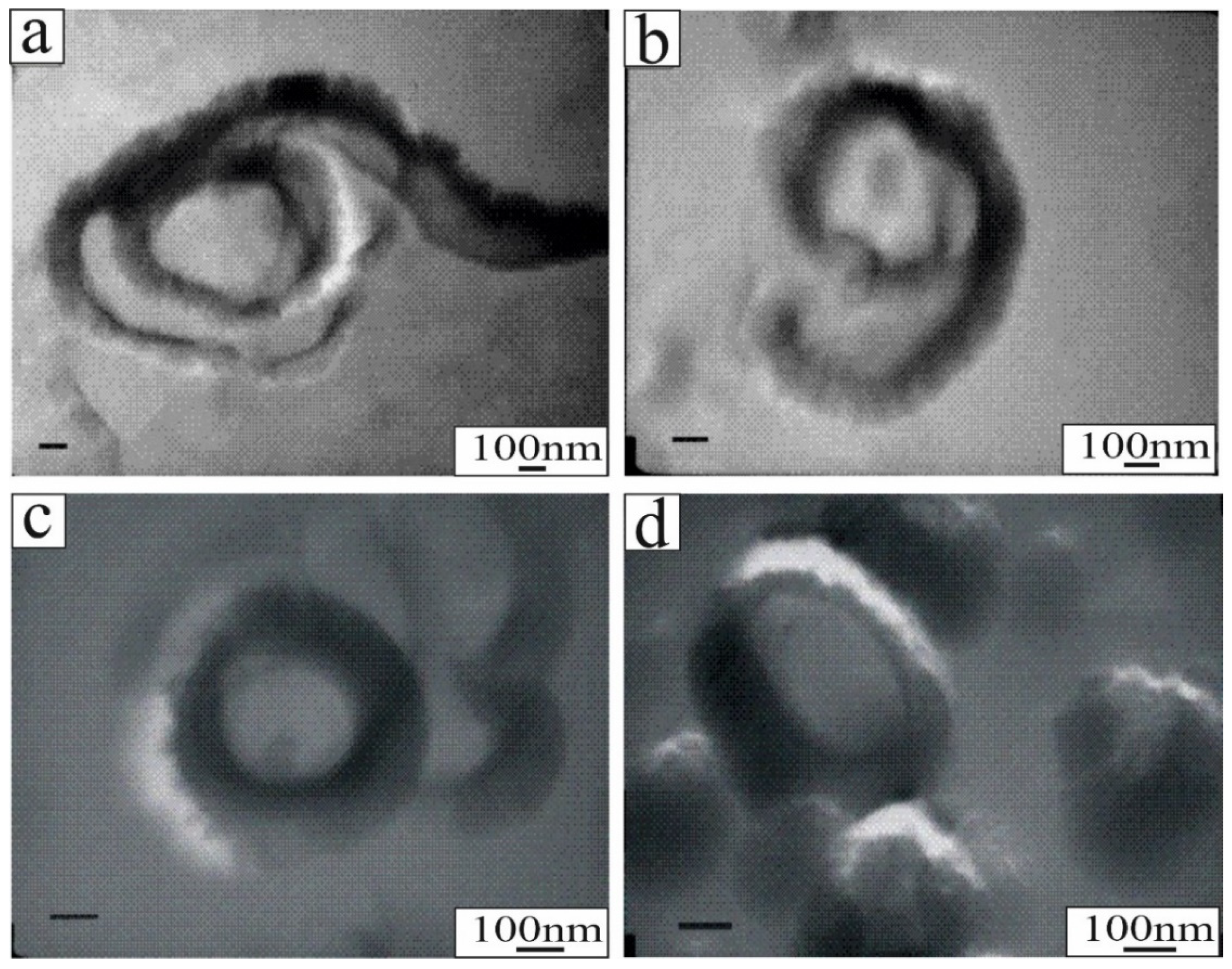
Figure 3.
Idealized morphological change in halloysite particles due to dehydration. Cross-sectional views of hydrated and dehydrated halloysite tubes [40].
Figure 3.
Idealized morphological change in halloysite particles due to dehydration. Cross-sectional views of hydrated and dehydrated halloysite tubes [40].

Figure 4.
A summary of hydrothermal alteration and dissolution processes of andesitic tuffs to kaolinite, halloysite and alunite—summarized according to FE̶ SEM studies [33].
Figure 4.
A summary of hydrothermal alteration and dissolution processes of andesitic tuffs to kaolinite, halloysite and alunite—summarized according to FE̶ SEM studies [33].
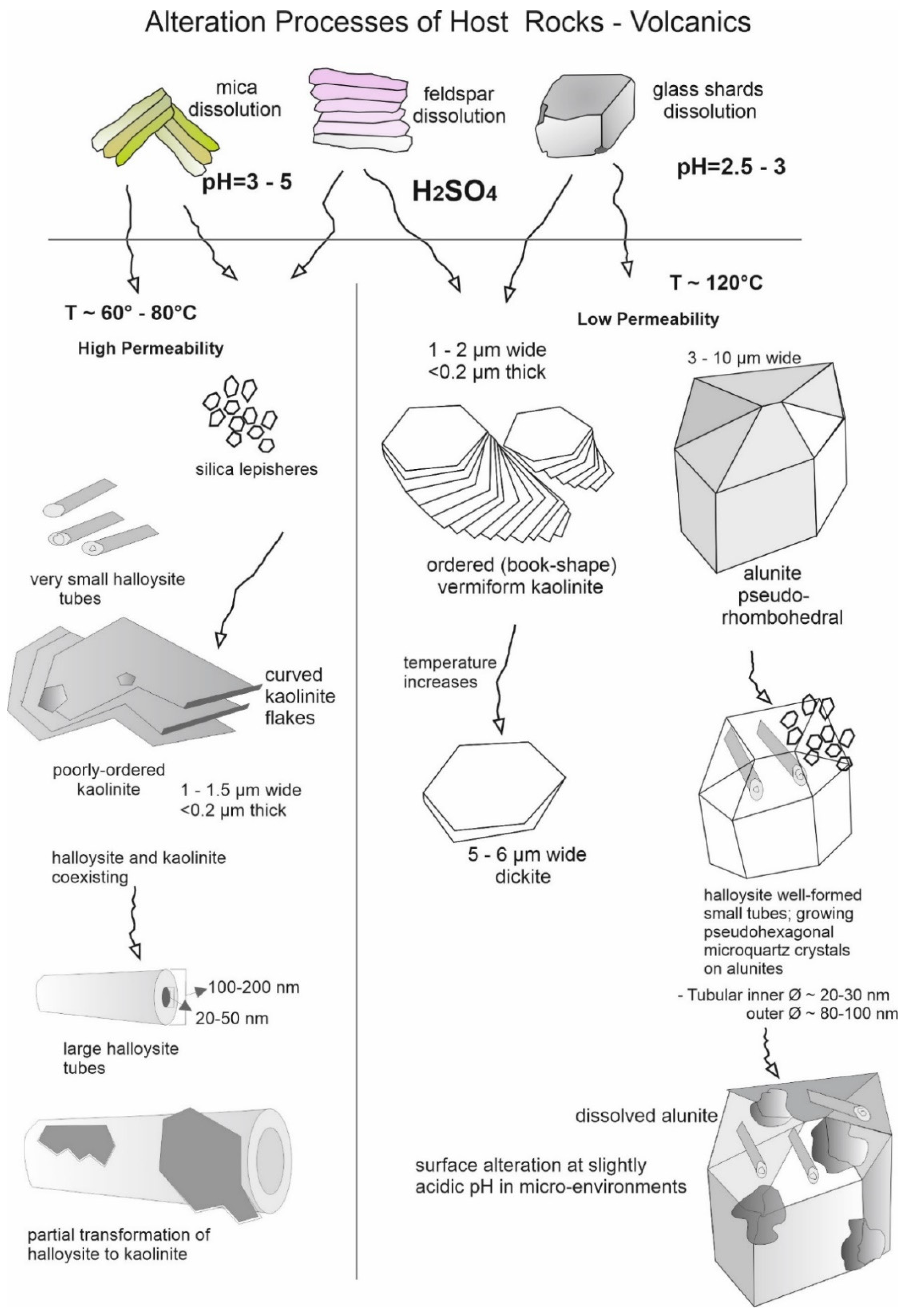
Figure 5.
The δD and δ18O values of halloysites from the Turplu mine, and hydrothermal kaolinites from active geothermal fields and thermal waters near kaolinite localities in Japan [61]. The meteoric water line is δD = 6.8 δ18O + 10.5 [59]. The light dashed line shows the δD-δ18O relation of kaolinite in equilibrium with meteoric water at 100 °C. Supergene vs. hypogene lines are taken from [52]. The area to the left of the S/H line indicates a hypogene origin, with the right-hand side indicating supergene origin due to weathering effects on isotope exchange after clay mineralization (modified from [60]).
Figure 5.
The δD and δ18O values of halloysites from the Turplu mine, and hydrothermal kaolinites from active geothermal fields and thermal waters near kaolinite localities in Japan [61]. The meteoric water line is δD = 6.8 δ18O + 10.5 [59]. The light dashed line shows the δD-δ18O relation of kaolinite in equilibrium with meteoric water at 100 °C. Supergene vs. hypogene lines are taken from [52]. The area to the left of the S/H line indicates a hypogene origin, with the right-hand side indicating supergene origin due to weathering effects on isotope exchange after clay mineralization (modified from [60]).
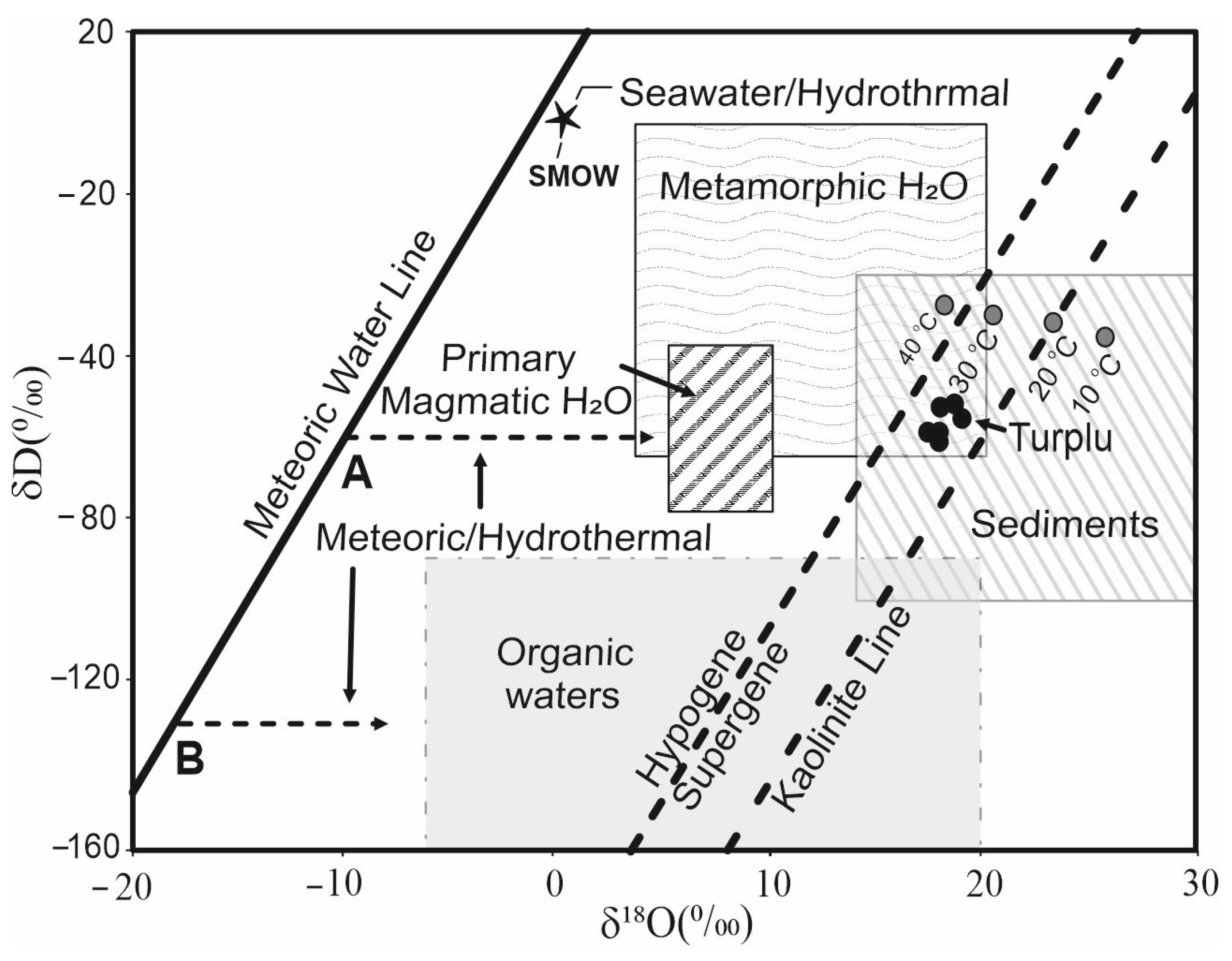
Figure 6.
A schematic diagram showing the alteration spaces in the porphyry Cu-Au system at high temperatures and in the epithermal Au-As-Sb system at low temperatures. The phase boundaries are schematically constrained (from [125,127]). The question mark (?) represents uncertainty.
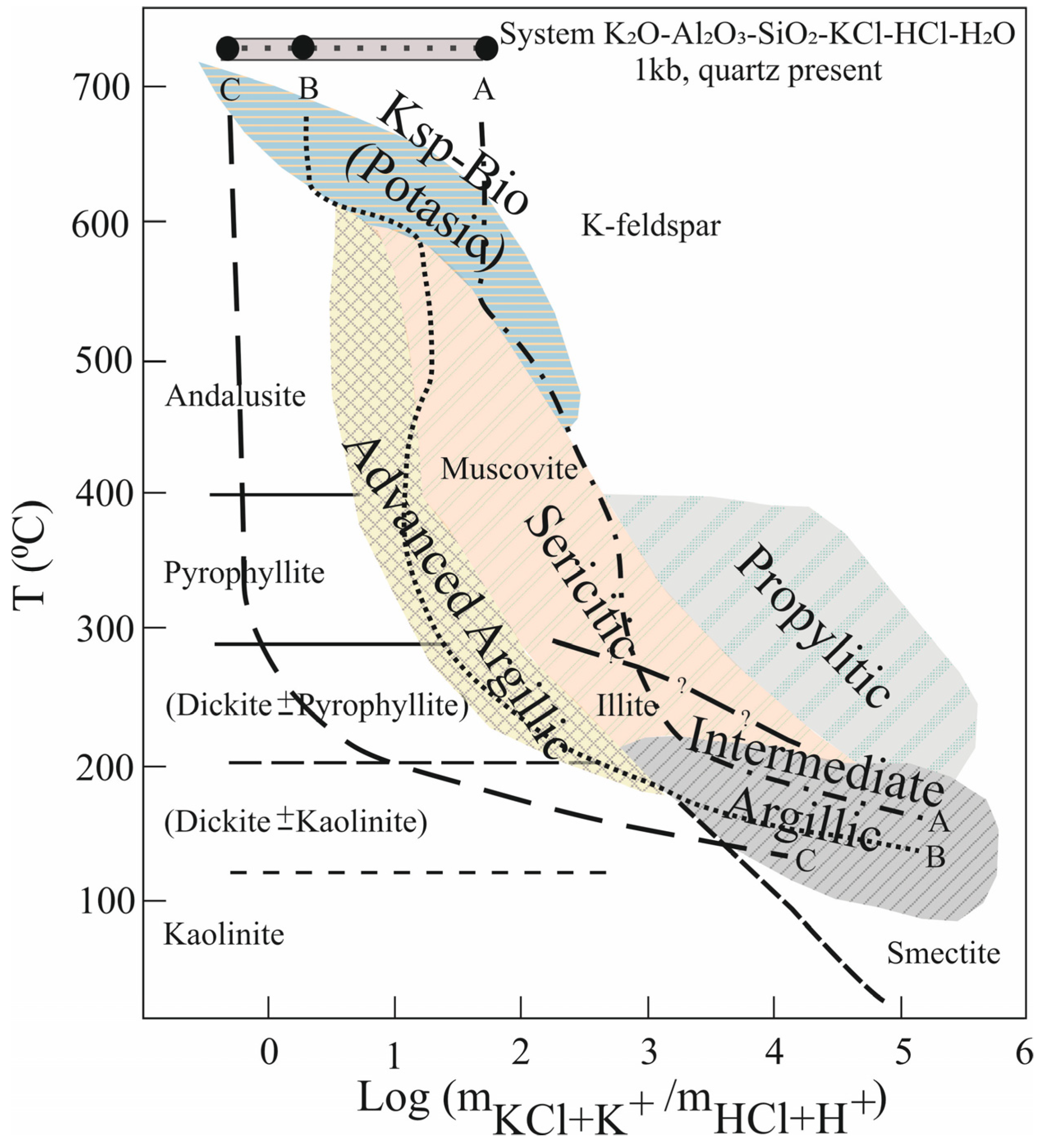
Figure 7.
Stability relations at PH2O = 250 bars with the addition of silica activity–temperature relations for the common polymorphs of quartz at temperatures >300 °C [146]. The point MH shows, for illustration purposes, the concentration of silica when a liquid condensate fraction of 10% separates from the vapor at 550 °C based on the vapor phase solubility data of [147]. The plotted rectangle represents hydrothermal endmembers (data from [148,149,150,151]. The arrows generalize the mixing line of seawater and the “hydrothermal endmembers” (after [145]). The composition of steam-heated acid–sulfate solutions presents at ≤100 °C above the water table [152,153] (modified from [82]).
Figure 7.
Stability relations at PH2O = 250 bars with the addition of silica activity–temperature relations for the common polymorphs of quartz at temperatures >300 °C [146]. The point MH shows, for illustration purposes, the concentration of silica when a liquid condensate fraction of 10% separates from the vapor at 550 °C based on the vapor phase solubility data of [147]. The plotted rectangle represents hydrothermal endmembers (data from [148,149,150,151]. The arrows generalize the mixing line of seawater and the “hydrothermal endmembers” (after [145]). The composition of steam-heated acid–sulfate solutions presents at ≤100 °C above the water table [152,153] (modified from [82]).

Figure 8.
The distribution of alteration zones in the Kamikita area, Japan, shown in the top image, and a cross-section showing alteration zones along the A-B line in the bottom image (the figure’s numerical labels represent drilling locations and their corresponding well names) (from [154,155]).

Figure 9.
Genetic model for the occurrence of kaolin and alunite and silica sinter deposits in the Düvertepe area, the Simav Graben, Aegean Region of Turkey. Red arrows show early-stage H2SO4-, HF- and HCl-rich solutions, green arrows show late-stage neutral–alkaline Si-rich solutions and blue arrows show supergene overprint of the circulation of meteoric waters [9]; modified from [61,107,200].
Figure 9.
Genetic model for the occurrence of kaolin and alunite and silica sinter deposits in the Düvertepe area, the Simav Graben, Aegean Region of Turkey. Red arrows show early-stage H2SO4-, HF- and HCl-rich solutions, green arrows show late-stage neutral–alkaline Si-rich solutions and blue arrows show supergene overprint of the circulation of meteoric waters [9]; modified from [61,107,200].
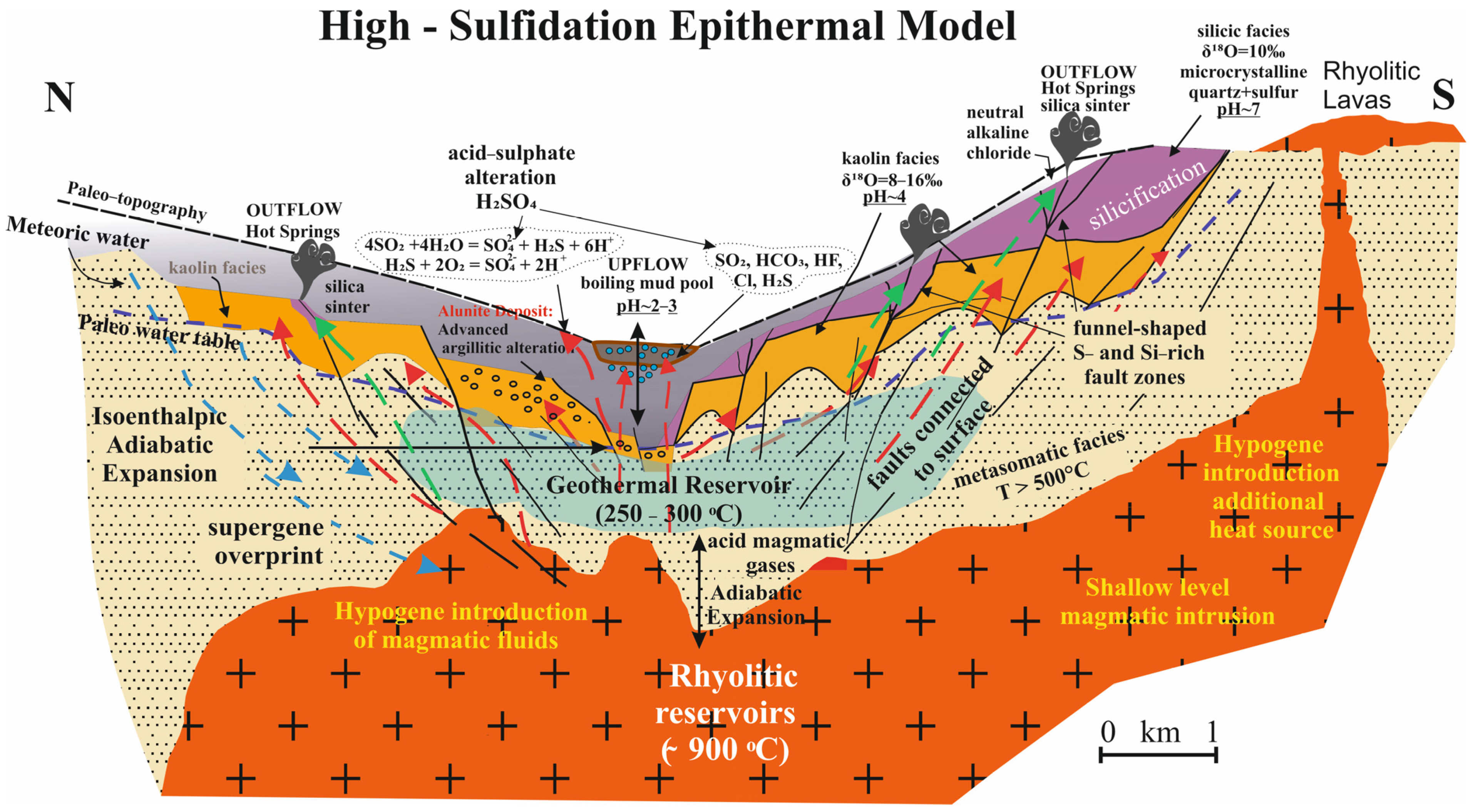
Figure 10.
Geologic cross-section of the upper quarry of the Taban halloysite deposit, Turkey [204].
Figure 10.
Geologic cross-section of the upper quarry of the Taban halloysite deposit, Turkey [204].
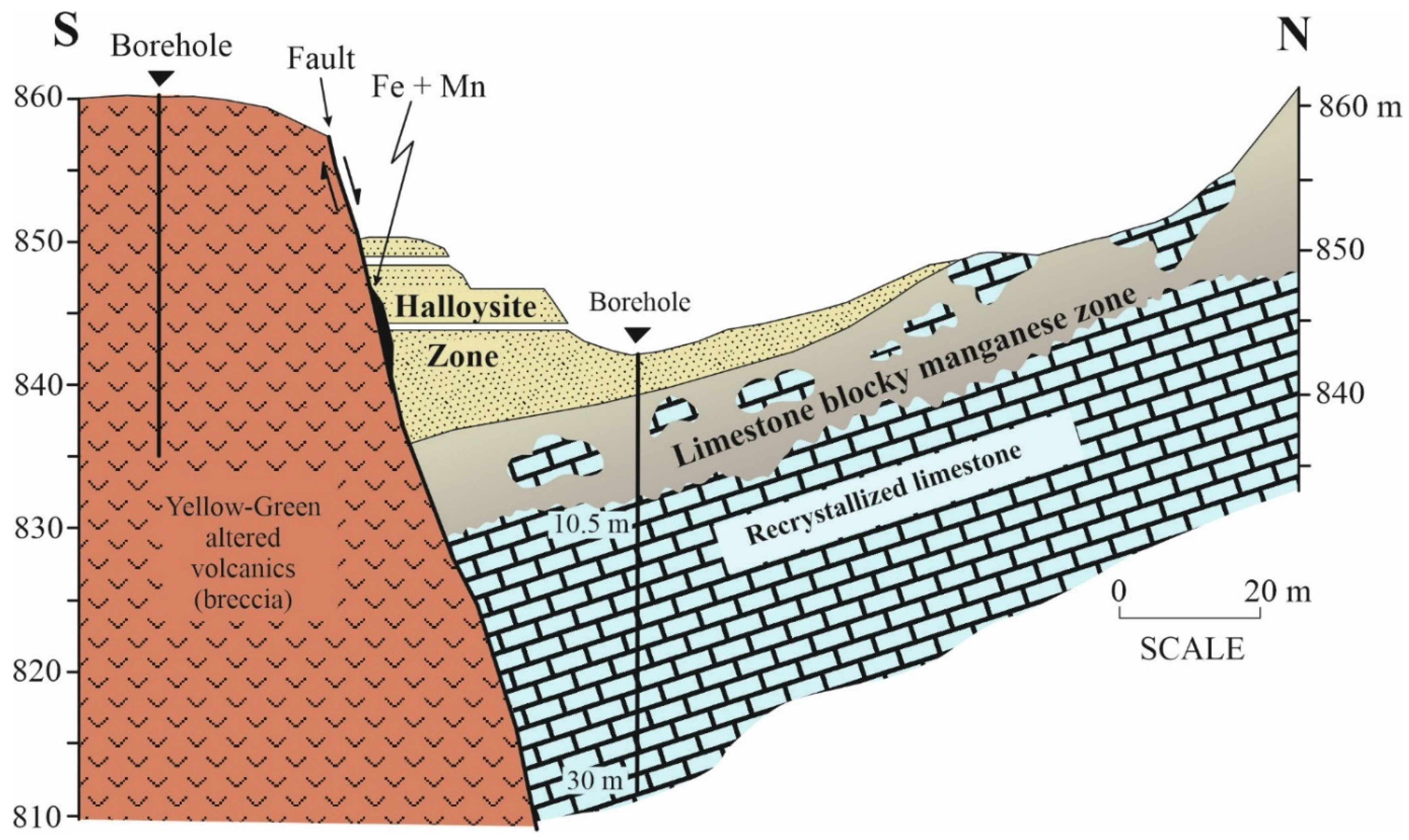
Figure 11.
A genetic model for the occurrence of halloysite and alunite deposits in the Biga Peninsula. A four-stage genetic model proposed for the origin of the magmatic hydrothermal alunite deposit. (a) The initial setting of andesitic tuffs as the parent rock to the base karstic limestone; geochemical leaching processes of andesitic tuffs and differential mobility of certain elements in different Eh/pH conditions. Both sides of the fault zone were affected by SO4-rich hydrothermal solutions. (b) Altered rocks involve ~50 wt.% mass reduction in the initial parent rocks to form gibbsite via progressive vertically downward Si-leaching up to depressions in the karstic limestone, hence upgrading the gibbsite through Si-leaching at pH < 2–3. (c) The alunitization process from the intermediate parent rock of gibbsite, with high-temperature kaolin-group minerals nacrite and dickite being detected. Some alunite samples are rich in phosphate. Gibbsite is found randomly between limestone blocks and halloysite. (d) Mainly pyrite mineralization is observed along the fault zone and the bottom zone to the consolidated limestone. Gypsum zones on the surface of limestone blocks are a common feature as supergene oxidation causes Fe migration during the latest diagenetic stage. No scale is used (modified from [60]).
Figure 11.
A genetic model for the occurrence of halloysite and alunite deposits in the Biga Peninsula. A four-stage genetic model proposed for the origin of the magmatic hydrothermal alunite deposit. (a) The initial setting of andesitic tuffs as the parent rock to the base karstic limestone; geochemical leaching processes of andesitic tuffs and differential mobility of certain elements in different Eh/pH conditions. Both sides of the fault zone were affected by SO4-rich hydrothermal solutions. (b) Altered rocks involve ~50 wt.% mass reduction in the initial parent rocks to form gibbsite via progressive vertically downward Si-leaching up to depressions in the karstic limestone, hence upgrading the gibbsite through Si-leaching at pH < 2–3. (c) The alunitization process from the intermediate parent rock of gibbsite, with high-temperature kaolin-group minerals nacrite and dickite being detected. Some alunite samples are rich in phosphate. Gibbsite is found randomly between limestone blocks and halloysite. (d) Mainly pyrite mineralization is observed along the fault zone and the bottom zone to the consolidated limestone. Gypsum zones on the surface of limestone blocks are a common feature as supergene oxidation causes Fe migration during the latest diagenetic stage. No scale is used (modified from [60]).

Table 1.
Zonal arrangement of paragenesis in the three types of hydrothermal systems defined by the value of the cations/H+ ratio in solutions [48].
Table 1.
Zonal arrangement of paragenesis in the three types of hydrothermal systems defined by the value of the cations/H+ ratio in solutions [48].
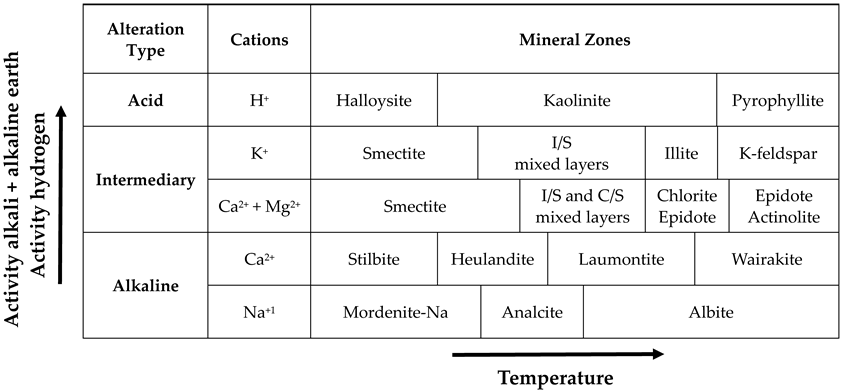
Table 4.
Temperatures at depth and dissolved silica after cooling by steam formation from natural springs. Dissolved SiO2 in natural meteoric waters is about 2.57 ppm. (*) These temperatures were measured at the surface [118,156,157,158,159,160,161,162,163].
| Locations | Dissolved SiO2 (ppm) | Estimated Temperature (°C) | pH (20 °C) |
|---|---|---|---|
| Well, Seltun, Iceland | 425 | 215–220 | – |
| Well, Kolbeinsey, Iceland | 601 | 70–110 | – |
| Well, Steinaholl, Iceland | 120 | ~220 | – |
| Well, Grimsey, Iceland | 663 | ~250 | 5.9–6.8 |
| Well, Kairaki, New Zeland | 660 | 246–252 | – |
| Well, Steamboat Spring, USA | 245 | 178–180 | – |
| Spring No.24, Steamboat Spring, USA | 345 | 201–205 | – |
| Boiling Lake, Dominica | 361 | * 84 | 4.1 |
| Fred’s pool, Penville, Dominica | 63.7 | * 27.2 | 1.6 |
| Nico’s spring, Sulphur Spring, Dominica | 166.7 | * 62.5 | 1.4 |
| Ellie’s pool, Watten Waven, Dominica | 42.5 | * 39 | 5.1 |
| Jan’s pool, Watten Waven, Dominica | 46.7 | * 90.1 | 2.9 |
| El Chichon volcano, Mexico | 439 | * 99 | 3.3 |
| El Chichon volcano, Mexico | 306 | * 99 | 3.09 |
| El Chichon volcano, Mexico | 238 | * 29 | 2.36 |
| El Chichon volcano, Mexico | 257 | * 55 | 0.56 |
| El Chichon volcano, Mexico | 264 | * 32 | 2.63 |
| Fushime, Kuyushu, Japan | 922 | >300 | 4.15 |
| Fushime, Kuyushu, Japan | 571 | 250 | 7.23 |
| Fushime, Kuyushu, Japan | 1274 | >300 | 3.99 |
| Fushime, Kuyushu, Japan | 922 | 330–340 | 4.88 |
| Well S–2, Sumikawa, Japan | 592 | 240–289 | 2.61 |
| Well S–3, Sumikawa, Japan | 547 | 223–239 | 8.48 |
| Well S–A–2, Sumikawa, Japan | 926 | 279–302 | 5.8 |
| Spring, Obama, Japan | 116 | 100 | 8 |
| Spring, Obama, Japan | 117 | * 79 | 7.5 |
| Well TT–1, Takigami, Kyushu, Japan | 437 | 216 | 9.4 |
| Well TT–2, Takigami, Kyushu, Japan | 365 | 203 | 9 |
| Well TT–14, Takigami, Kyushu, Japan | 593 | 238 | 9.1 |
| Well KT–1, Uenotai, Japan | 775 | 259 | 9.8 |
| Well T–41, Uenotai, Japan | 894 | 271 | 9.9 |
| Well T–45, Uenotai, Japan | 943 | 275 | 9.5 |
| Well ND–6, Nigorikawa, Japan | 768 | 259 | 7.97 |
| Well ND–1, Nigorikawa, Japan | 677 | 249 | 8.21 |
| Well NF–1, Nigorikawa, Japan | 559 | 235 | 8.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ece, Ö.I.; Ercan, H.Ü. Global Occurrence, Geology and Characteristics of Hydrothermal-Origin Kaolin Deposits. Minerals 2024, 14, 353. https://doi.org/10.3390/min14040353
AMA Style
Ece ÖI, Ercan HÜ. Global Occurrence, Geology and Characteristics of Hydrothermal-Origin Kaolin Deposits. Minerals. 2024; 14(4):353. https://doi.org/10.3390/min14040353
Chicago/Turabian StyleEce, Ömer Işık, and Hatice Ünal Ercan. 2024. "Global Occurrence, Geology and Characteristics of Hydrothermal-Origin Kaolin Deposits" Minerals 14, no. 4: 353. https://doi.org/10.3390/min14040353
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.