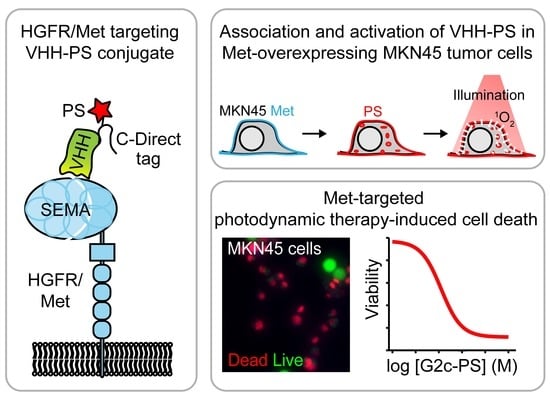
VHH-Photosensitizer Conjugates for Targeted Photodynamic Therapy of Met-Overexpressing Tumor Cells

 , , and
, , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Molecular Modeling
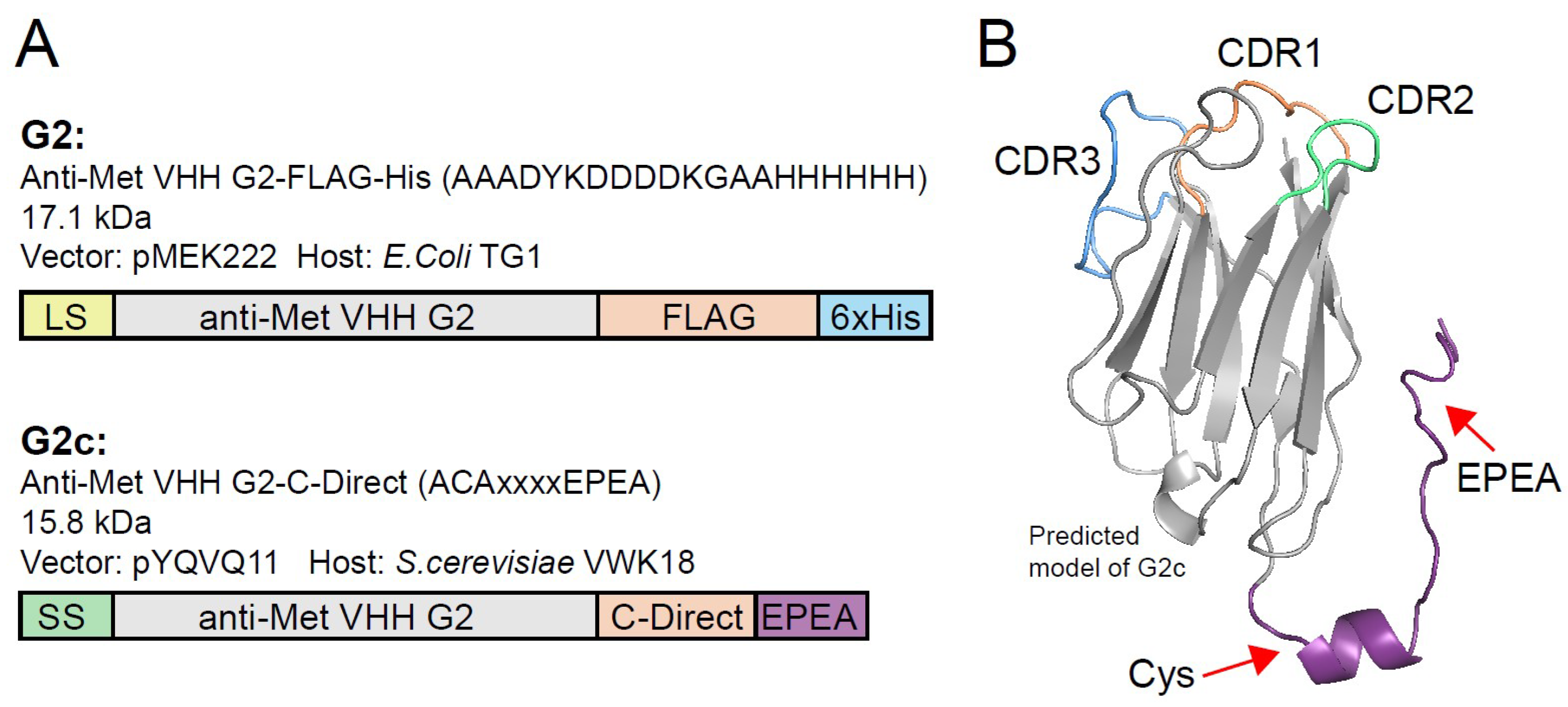
2.2. VHH Production and Purification
2.3. Cell Culture
2.4. VHH Binding ELISA
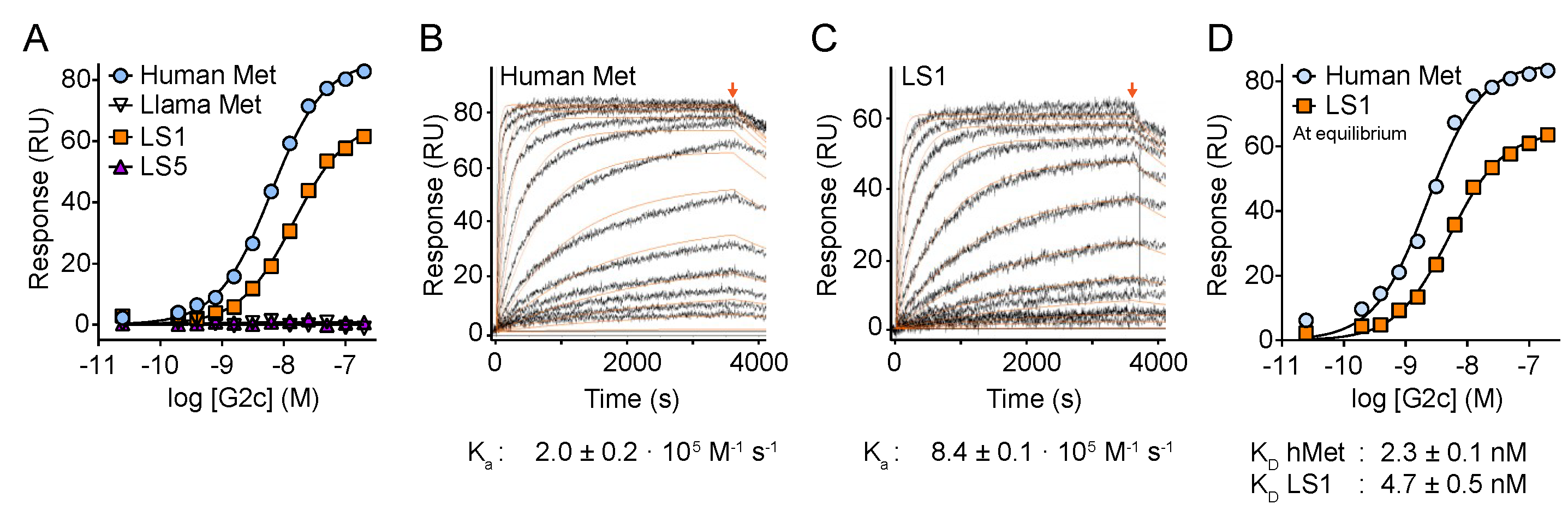
2.5. Surface Plasmon Resonance (SPR)
2.6. Conjugation to PS
2.7. Internalization Assay
2.8. Photodynamic Therapy
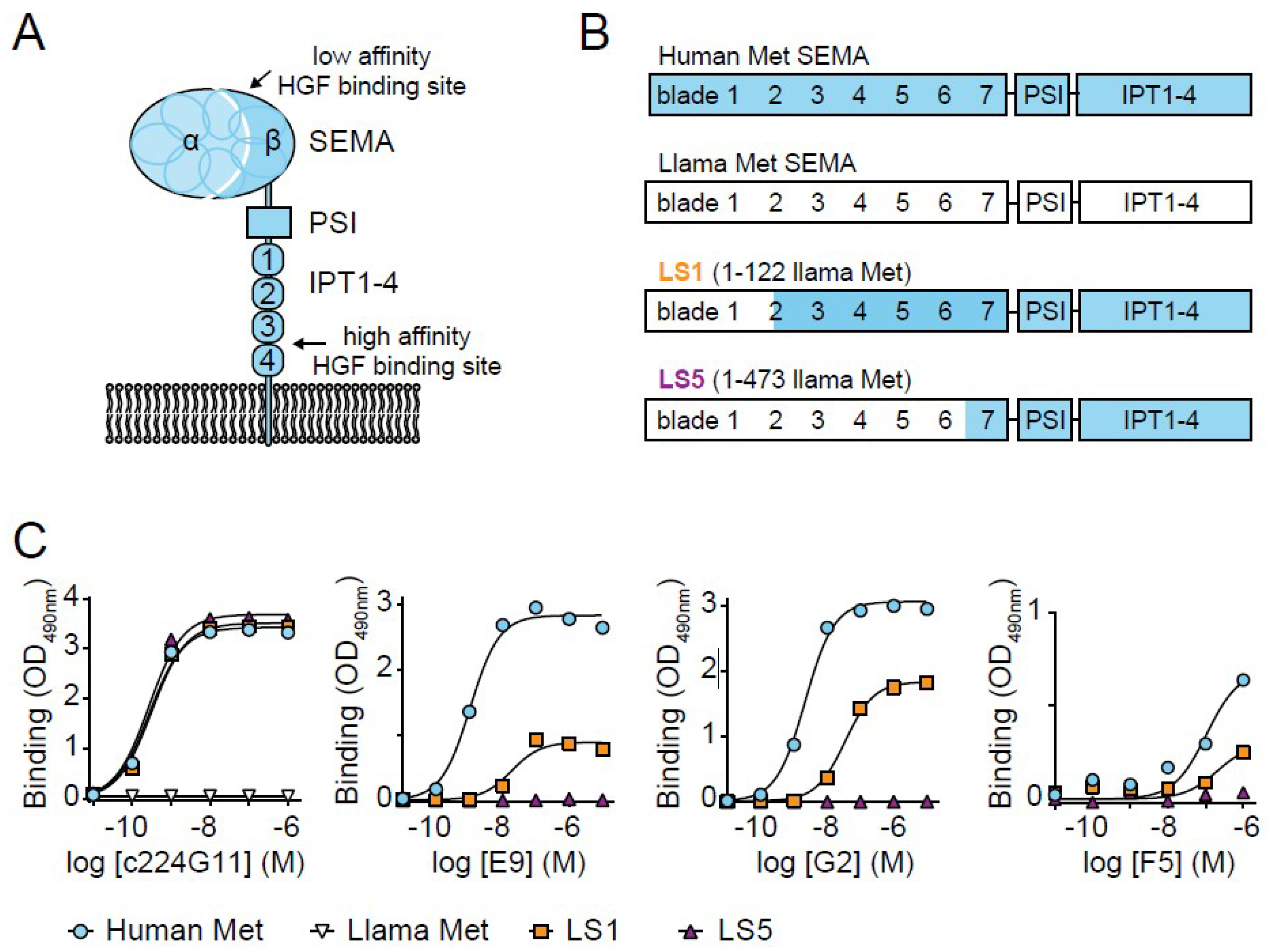
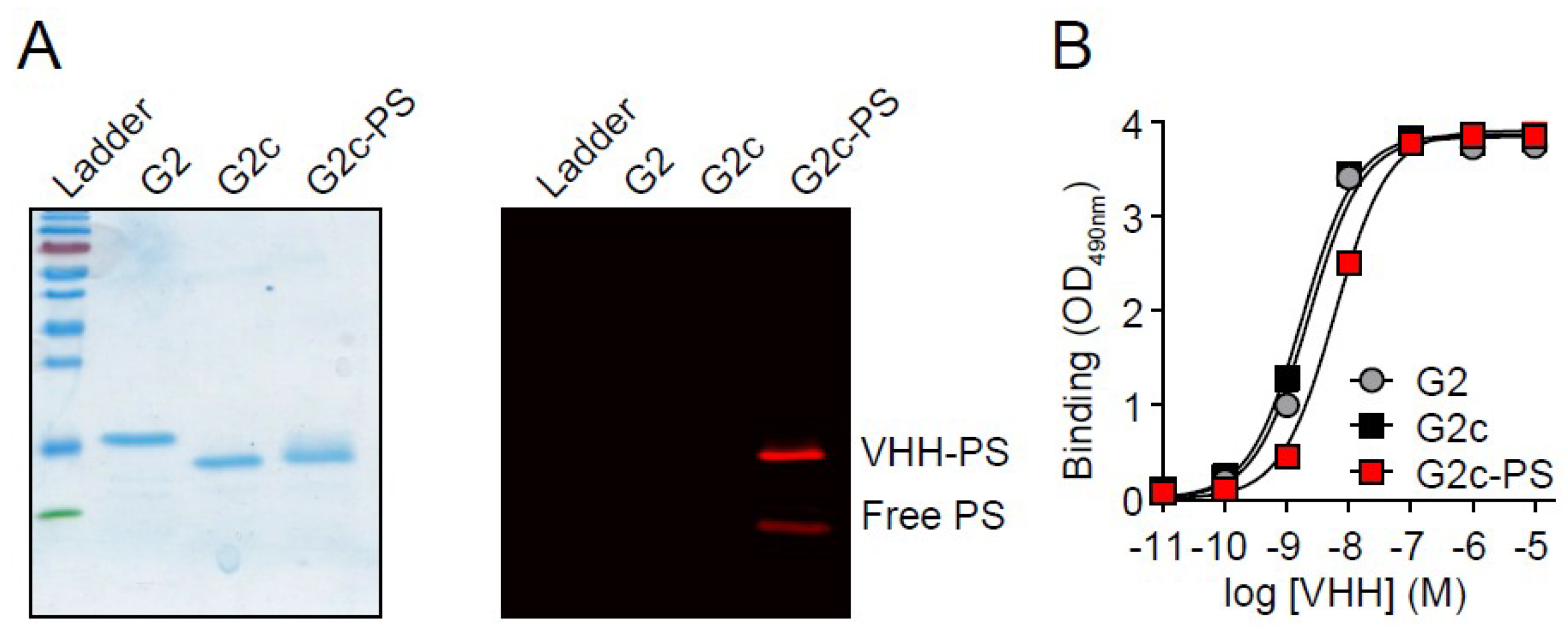
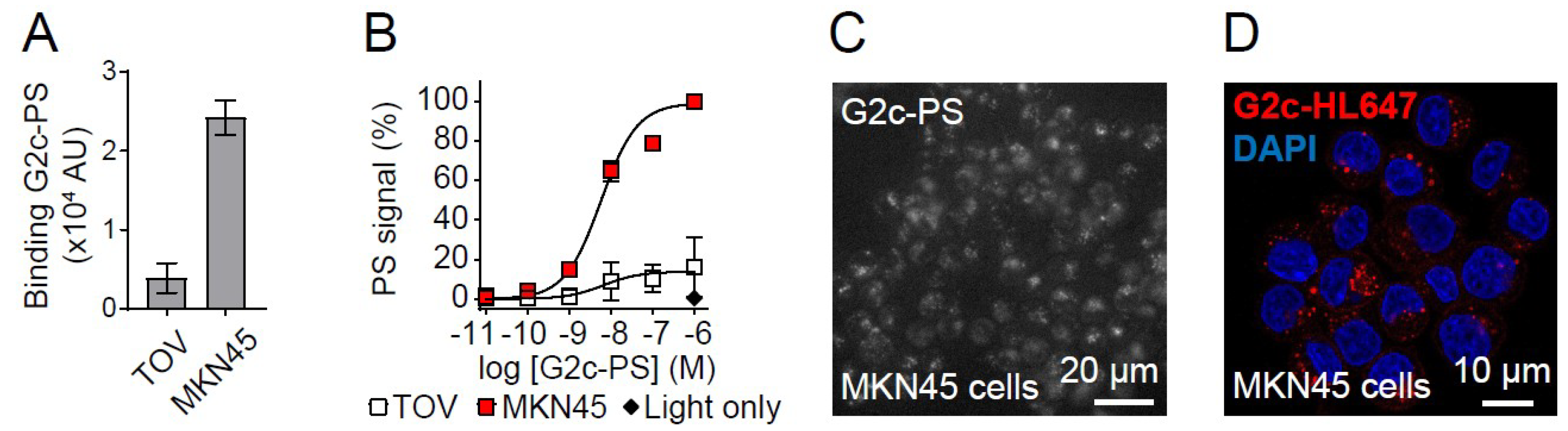
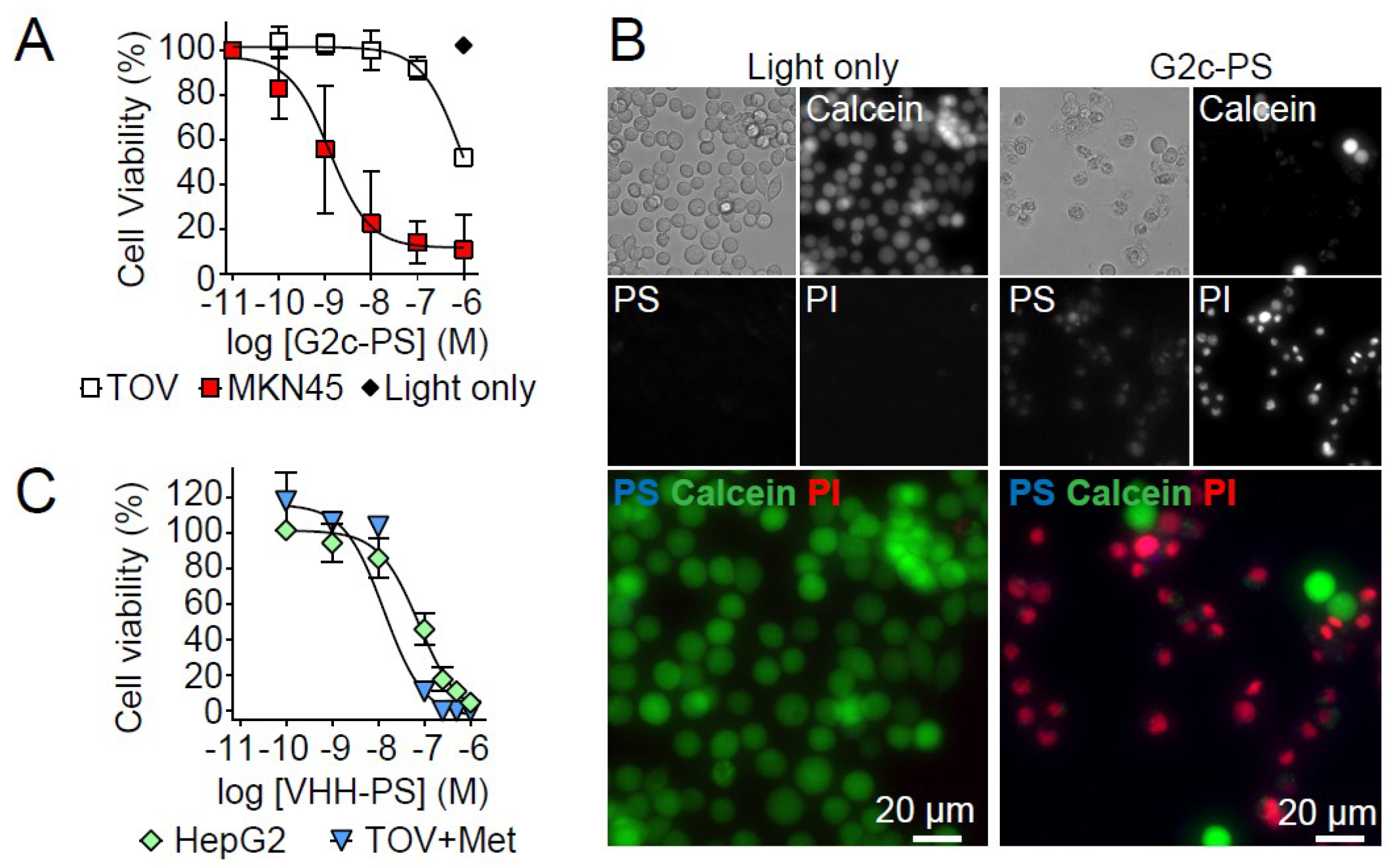
3. Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Van Straten, D.; Mashayekhi, V.; de Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic photodynamic therapy: Basic principles, current clinical status and future directions. Cancers (Basel) 2017, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kedzierska, E.; Knap-Czop, K.; Kotlinska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Gomer, C.J. Preclinical examination of first and second generation photosensitizers used in photodynamic therapy. Photochem. Photobiol. 1991, 54, 1093–1107. [Google Scholar] [CrossRef]
- Githaka, N.; Konnai, S.; Skilton, R.; Kariuki, E.; Kanduma, E.; Murata, S.; Ohashi, K. Genotypic variations in field isolates of Theileria species infecting giraffes (Giraffa camelopardalis tippelskirchi and Giraffa camelopardalis reticulata) in Kenya. Parasitol. Int. 2013, 62, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Bechet, D.; Couleaud, P.; Frochot, C.; Viriot, M.L.; Guillemin, F.; Barberi-Heyob, M. Nanoparticles as vehicles for delivery of photodynamic therapy agents. Trends Biotechnol. 2008, 26, 612–621. [Google Scholar] [CrossRef]
- Heukers, R.; van Bergen en Henegouwen, P.M.P.; Oliveira, S. Nanobody-targeted photodynamic therapy for oncology. Photodiagnosis Photodyn. Ther. 2015, 12, 339. [Google Scholar] [CrossRef]
- Van Driel, P.B.A.A.; Boonstra, M.C.; Slooter, M.D.; Heukers, R.; Stammes, M.A.; Snoeks, T.J.A.; De Bruijn, H.S.; Van Diest, P.J.; Vahrmeijer, A.L.; Van Bergen En Henegouwen, P.M.P.; et al. EGFR targeted nanobody-photosensitizer conjugates for photodynamic therapy in a pre-clinical model of head and neck cancer. J. Control. Release 2016, 229, 93–105. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef]
- De Groeve, K.; Deschacht, N.; De Koninck, C.; Caveliers, V.; Lahoutte, T.; Devoogdt, N.; Muyldermans, S.; De Baetselier, P.; Raes, G. Nanobodies as tools for in vivo imaging of specific immune cell types. J. Nucl. Med. 2010, 51, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.; Vikani, N.; Betti, C.; Ballet, S.; Vanderhaegen, S.; Steyaert, J.; Descamps, B.; Vanhove, C.; Bunschoten, A.; van Leeuwen, F.W.B.; et al. Sortase A-mediated site-specific labeling of camelid single-domain antibody-fragments: A versatile strategy for multiple molecular imaging modalities. Contrast Media Mol. Imaging 2016, 11, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.; Xavier, C.; De Vos, J.; Caveliers, V.; Lahoutte, T.; Muyldermans, S.; Devoogdt, N. Site-specific labeling of cysteine-tagged camelid single-domain antibody-fragments for use in molecular imaging. Bioconjug. Chem. 2014, 25, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Van Lith, S.A.M.; Van Duijnhoven, S.M.J.; Navis, A.C.; Leenders, W.P.J.; Dolk, E.; Wennink, J.W.H.; Van Nostrum, C.F.; Van Hest, J.C.M. Legomedicine—A Versatile Chemo-Enzymatic Approach for the Preparation of Targeted Dual-Labeled Llama Antibody-Nanoparticle Conjugates. Bioconjug. Chem. 2017, 28, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Kijanka, M.; Warnders, F.J.; El Khattabi, M.; Lub-De Hooge, M.; Van Dam, G.M.; Ntziachristos, V.; De Vries, L.; Oliveira, S.; Van Bergen En Henegouwen, P.M.P. Rapid optical imaging of human breast tumour xenografts using anti-HER2 VHHs site-directly conjugated to IRDye 800CW for image-guided surgery. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Köstler, W.J.; Yarden, Y. The epidermal growth factor receptor family. In Handbook of Cell Signaling, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 2, pp. 435–441. ISBN 9780123741455. [Google Scholar]
- Bublil, E.M.; Yarden, Y. The EGF receptor family: Spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 2007, 19, 124–134. [Google Scholar] [CrossRef]
- Peruzzi, B.; Bottaro, D.P. Targeting the c-Met signaling pathway in cancer. Clin. Cancer Res. 2006, 12, 3657–3660. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF β-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004, 23, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Youles, M.E.; Miguel, R.N.; Blundell, T.L.; Iamele, L.; Gough, J.; Bandyopadhyay, A.; Hartmann, G.; Butler, P.J.G. Functional map and domain structure of MET, the product of the c-met protooncogene and receptor for hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci. USA 2003, 100, 12039–12044. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A.; Salto-Tellez, M.; Laurent-Puig, P.; Bardelli, A.; Rolfo, C.; Tabernero, J.; Khawaja, H.A.; Lawler, M.; Johnston, P.G.; Van Schaeybroeck, S. Targeting c-MET in gastrointestinal tumours: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2017, 14, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Heukers, R.; Altintas, I.; Raghoenath, S.; De Zan, E.; Pepermans, R.; Roovers, R.C.; Haselberg, R.; Hennink, W.E.; Schiffelers, R.M.; Kok, R.J.; et al. Targeting hepatocyte growth factor receptor (Met) positive tumor cells using internalizing nanobody-decorated albumin nanoparticles. Biomaterials 2014, 35, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Budiarto, T.; Wu, M.; Pardon, E.; Steyaert, J.; Hol, W.G.J. The structure of the C-terminal domain of the largest editosome interaction protein and its role in promoting RNA binding by RNA-editing ligase L2. Nucleic Acids Res. 2012, 40, 6966–6977. [Google Scholar] [CrossRef] [PubMed]
- Gorlani, A.; Lutje Hulsik, D.; Adams, H.; Vriend, G.; Hermans, P.; Verrips, T. Antibody engineering reveals the important role of J segments in the production efficiency of llama single-domain antibodies in Saccharomyces cerevisiae. Protein Eng. Des. Sel. 2012, 25, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, Y.E.; Meijer, W.; Sierkstra, L.; Verrips, C.T. Large-scale production of VHH antibody fragments by Saccharomyces cerevisiae. Enzyme Microb. Technol. 2002, 30, 273–278. [Google Scholar] [CrossRef]
- Van De Laar, T.; Visser, C.; Holster, M.; López, C.G.; Kreuning, D.; Sierkstra, L.; Lindner, N.; Verrips, T. Increased heterologous protein production by Saccharomyces cerevisiae growing on ethanol as sole carbon source. Biotechnol. Bioeng. 2007, 96, 483–494. [Google Scholar] [CrossRef]
- Gorlani, A.; De Haard, H.; Verrips, T. Expression of VHHs in saccharomyces cerevisiae. Methods Mol. Biol. 2012, 911, 277–286. [Google Scholar] [CrossRef]
- Basilico, C.; Hultberg, A.; Blanchetot, C.; De Jonge, N.; Festjens, E.; Hanssens, V.; Osepa, S.I.; De Boeck, G.; Mira, A.; Cazzanti, M.; et al. Four individually druggable MET hotspots mediate HGF-driven tumor progression. J. Clin. Investig. 2014, 124, 3172–3186. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Broussas, M.; Beau-Larvor, C.; Haeuw, J.F.; Boute, N.; Robert, A.; Champion, T.; Beck, A.; Bailly, C.; Corvaïa, N.; et al. A novel antagonist anti-cMet antibody with antitumor activities targeting both ligand-dependent and ligand-independent c-Met receptors. Int. J. Cancer 2016, 139, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Prat, M.; Oltolina, F.; Basilico, C. Monoclonal Antibodies against the MET/HGF Receptor and Its Ligand: Multitask Tools with Applications from Basic Research to Therapy. Biomedicines 2014, 2, 359–383. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.; van Dongen, G.A.M.S.; Stigter-van Walsum, M.; Roovers, R.C.; Stam, J.C.; Mali, W.; van Diest, P.J.; van Bergen en Henegouwen, P.M.P. Rapid visualization of human tumor xenografts through optical imaging with a near-infrared fluorescent anti-epidermal growth factor receptor nanobody. Mol. Imaging 2012, 11, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef]
- Allison, R.R.; Sibata, C.H. Oncologic photodynamic therapy photosensitizers: A clinical review. Photodiagnosis Photodyn. Ther. 2010, 7, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Shirasu, N.; Nam, S.O.; Kuroki, M. Tumor-targeted photodynamic therapy. Anticancer Res. 2013, 33, 2823–2832. [Google Scholar]
- Bugaj, A.M. Targeted photodynamic therapy—A promising strategy of tumor treatment. Photochem. Photobiol. Sci. 2011, 10, 1097–1109. [Google Scholar] [CrossRef]
- Lucky, S.S.; Soo, K.C.; Zhang, Y. Nanoparticles in photodynamic therapy. Chem. Rev. 2015, 115, 1990–2042. [Google Scholar] [CrossRef]
- Savellano, M.D.; Hasan, T. Photochemical targeting of epidermal growth factor receptor: A mechanistic study. Clin. Cancer Res. 2005, 11, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Sano, K.; Choyke, P.L.; Kobayashi, H. Improving the efficacy of photoimmunotherapy (PIT) using a cocktail of antibody conjugates in a multiple antigen tumor model. Theranostics 2013, 3, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Savellano, M.D.; Pogue, B.W.; Hoopes, P.J.; Vitetta, E.S.; Paulsen, K.D. Multiepitope HER2 targeting enhances photoimmunotherapy of HER2-overexpressing cancer cells with pyropheophorbide-a immunoconjugates. Cancer Res. 2005, 65, 6371–6379. [Google Scholar] [CrossRef] [PubMed]
- Shirasu, N.; Yamada, H.; Shibaguchi, H.; Kuroki, M.; Kuroki, M. Potent and specific antitumor effect of CEA-targeted photoimmunotherapy. Int. J. Cancer 2014, 135, 2697–2710. [Google Scholar] [CrossRef] [PubMed]
- Maawy, A.A.; Hiroshima, Y.; Zhang, Y.; Heim, R.; Makings, L.; Garcia-Guzman, M.; Luiken, G.A.; Kobayashi, H.; Hoffman, R.M.; Bouvet, M. Near infra-red photoimmunotherapy with anti-CEA-IR700 results in extensive tumor lysis and a significant decrease in tumor burden in orthotopic mouse models of pancreatic cancer. PLoS ONE 2015, 10, e0121989. [Google Scholar] [CrossRef] [PubMed]
- Carcenac, M.; Larroque, C.; Langlois, R.; Van Lier, J.E.; Artus, J.C.; Pèlegrin, A. Preparation, phototoxicity and biodistribution studies of anti-carcinoembryonic antigen monoclonal antibody-phthalocyanine conjugates. Photochem. Photobiol. 1999, 70, 930–936. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to Anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Yan, F.; Liu, D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front. Med. 2016, 10, 383–388. [Google Scholar] [CrossRef]
- Matsubara, D.; Ishikawa, S.; Sachiko, O.; Aburatani, H.; Fukayama, M.; Niki, T. Co-activation of epidermal growth factor receptor and c-MET defines a distinct subset of lung adenocarcinomas. Am. J. Pathol. 2010, 177, 2191–2204. [Google Scholar] [CrossRef] [PubMed]
- Hultberg, A.; Morello, V.; Huyghe, L.; De Jonge, N.; Blanchetot, C.; Hanssens, V.; De Boeck, G.; Silence, K.; Festjens, E.; Heukers, R.; et al. Depleting MET-expressing tumor cells by ADCC provides a therapeutic advantage over inhibiting HGF/MET signaling. Cancer Res. 2015, 75, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, G.A.M.S.; Visser, G.W.M.; Vrouenraets, M.B. Photosensitizer-antibody conjugates for detection and therapy of cancer. Adv. Drug Deliv. Rev. 2004, 56, 31–52. [Google Scholar] [CrossRef]
- Wu, A.M.; Senter, P.D. Arming antibodies: Prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Redmond, R.W.; Kochevar, I.E. Spatially Resolved Cellular Responses to Singlet Oxygen. Photochem. Photobiol. 2006, 82, 1178. [Google Scholar] [CrossRef] [PubMed]
- Louryan, S. La formation de la tête des vertébrés: Faits et hypothèses. Rev. Med. Brux. 2005, 26, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- van Brussel, A.S.A.; Adams, A.; Oliveira, S.; Dorresteijn, B.; El Khattabi, M.; Vermeulen, J.F.; van der Wall, E.; Mali, W.P.T.M.; Derksen, P.W.B.; van Diest, P.J.; et al. Hypoxia-Targeting Fluorescent Nanobodies for Optical Molecular Imaging of Pre-Invasive Breast Cancer. Mol. Imaging Biol. 2016, 18, 535–544. [Google Scholar] [CrossRef]
- Rege-Cambrin, G.; Scaravaglio, P.; Carozzi, F.; Giordano, S.; Ponzetto, C.; Comoglio, P.M.; Saglio, G. Karyotypic analysis of gastric carcinoma cell lines carrying an amplified c-met oncogene. Cancer Genet. Cytogenet. 1992, 64, 170–173. [Google Scholar] [CrossRef]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. USA 2006, 103, 2316–2321. [Google Scholar] [CrossRef]
- Heukers, R.; van Bergen en Henegouwen, P.M.P.; Oliveira, S. Nanobody-photosensitizer conjugates for targeted photodynamic therapy. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1441–1451. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heukers, R.; Mashayekhi, V.; Ramirez-Escudero, M.; de Haard, H.; Verrips, T.C.; van Bergen en Henegouwen, P.M.P.; Oliveira, S. VHH-Photosensitizer Conjugates for Targeted Photodynamic Therapy of Met-Overexpressing Tumor Cells. Antibodies 2019, 8, 26. https://doi.org/10.3390/antib8020026
Heukers R, Mashayekhi V, Ramirez-Escudero M, de Haard H, Verrips TC, van Bergen en Henegouwen PMP, Oliveira S. VHH-Photosensitizer Conjugates for Targeted Photodynamic Therapy of Met-Overexpressing Tumor Cells. Antibodies. 2019; 8(2):26. https://doi.org/10.3390/antib8020026
Chicago/Turabian StyleHeukers, Raimond, Vida Mashayekhi, Mercedes Ramirez-Escudero, Hans de Haard, Theo C. Verrips, Paul. M.P. van Bergen en Henegouwen, and Sabrina Oliveira. 2019. "VHH-Photosensitizer Conjugates for Targeted Photodynamic Therapy of Met-Overexpressing Tumor Cells" Antibodies 8, no. 2: 26. https://doi.org/10.3390/antib8020026
APA StyleHeukers, R., Mashayekhi, V., Ramirez-Escudero, M., de Haard, H., Verrips, T. C., van Bergen en Henegouwen, P. M. P., & Oliveira, S. (2019). VHH-Photosensitizer Conjugates for Targeted Photodynamic Therapy of Met-Overexpressing Tumor Cells. Antibodies, 8(2), 26. https://doi.org/10.3390/antib8020026