Human Cytolytic Fusion Proteins: Modified Versions of Human Granzyme B and Angiogenin Have the Potential to Replace Bacterial Toxins in Targeted Therapies against CD64+ Diseases

Abstract
:1. Introduction
2. CD64 in Cancer, Chronic Inflammation, and Autoimmune Diseases
2.1. Acute Myeloid Leukemia (AML)
2.2. Macrophages and Inflammation
3. Targeting CD64
3.1. Classical Immunotoxins
3.2. “Humanization” of Immunotoxins Using Granzyme B and Angiogenin
4. Comparative Efficacy of Human Effector Domains and Bacterial Toxins in Targeted Therapy

{kind=link}
{kind=link}
| Target | Cell line | Construct | IC50 | Reference |
|---|---|---|---|---|
| CD64 | HL-60 | M22(scFv)-ETA' | 0.17 nM | [77] |
| U937 | ||||
| H22(scFv)-Ang | 0.2 nM | [25] | ||
| EGFR | A431 | |||
| Ang EGF** | 70 nM | [118] | ||
| ErbB2/Her2 | A431 | scFv(FRP5)-ETA (5-ETA) | 0.5 nM | [116] |
| LeY | SK-BR3 | |||
| MCF-7 | ||||
| B3-GzmB | 140 nM | [119] | ||
| CD30 | L540cy | |||
| L540 | Ang-CD30L | 0.23 nM | [15] |
5. Optimization of Endosomal Release Using Multifunctional, Cleavable Adapters
6. Comparative Analysis of Anti-CD64 hCFPs and H22(scFv)-ETA'
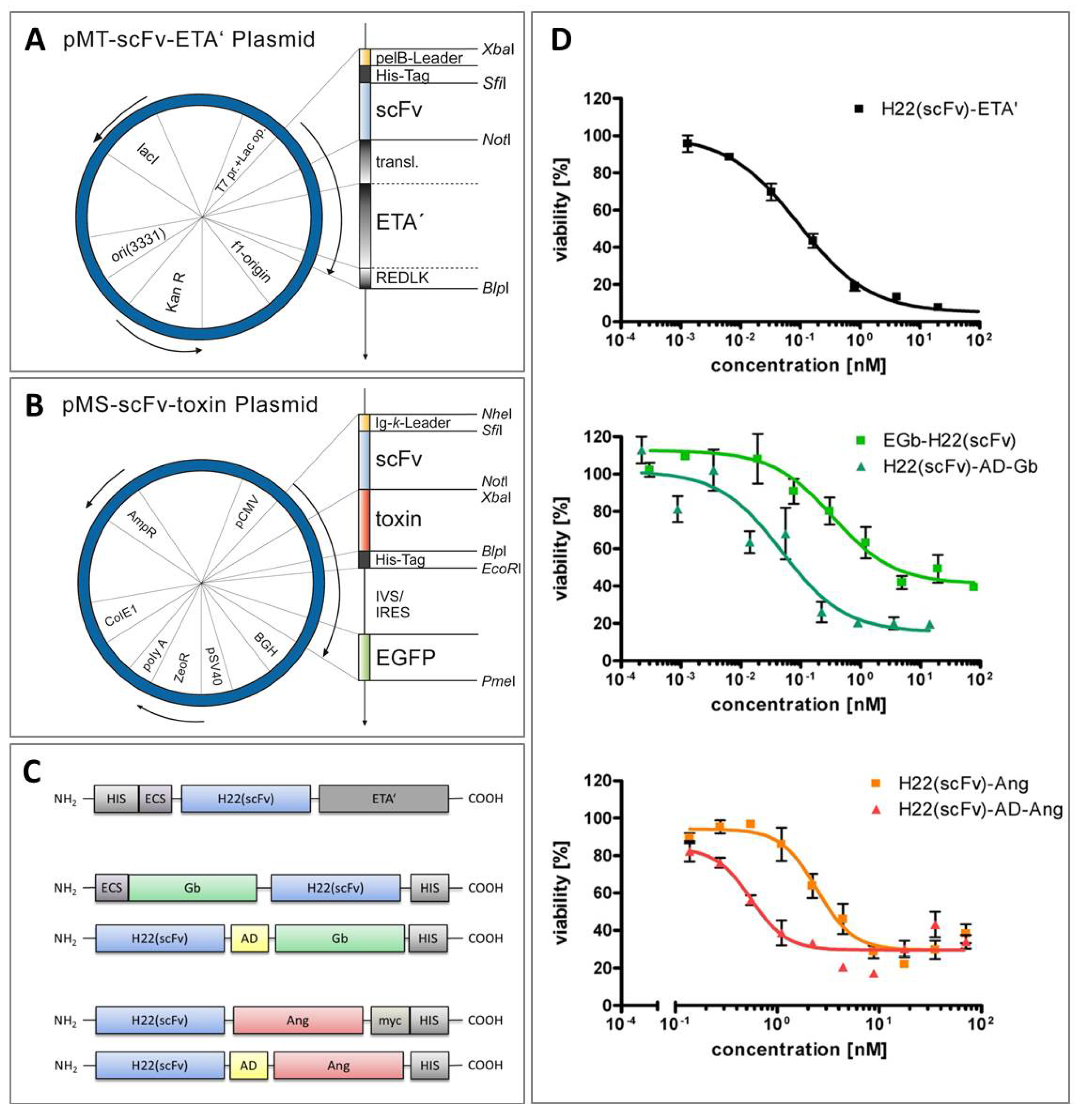
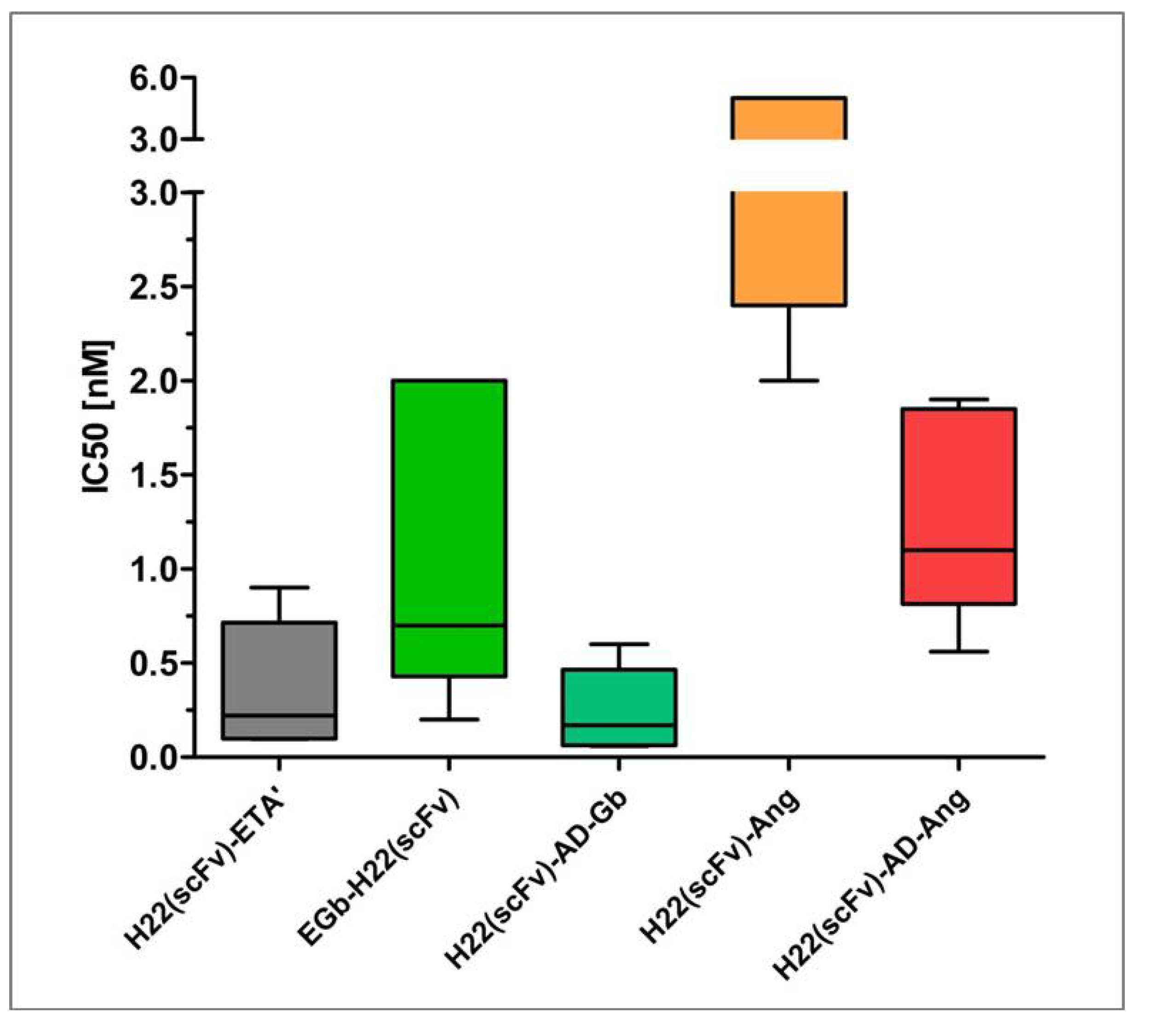
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mullard, A. Maturing antibody-drug conjugate pipeline hits 30. Nat. Rev. Drug Discov. 2013, 12, 329–332. [Google Scholar] [CrossRef]
- Kanellos, J.; MacKenzie, I.F.; Pietersz, G.A. In vivo studies of whole ricin monoclonal antibody immunoconjugates for the treatment of murine tumours. Immun. Cell Biol. 1988, 66, 403–415. [Google Scholar] [CrossRef]
- Becker, N.; Benhar, I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies 2012, 1, 39–69. [Google Scholar] [CrossRef]
- Eisai, P.D. FDA Grants Full Approval to ONTAK® (denileukin diftitox) For Use in Patients with Cutaneous T-Cell Lymphoma (CTCL). Available online: http://www.eisai.com/news/news200852.html ( accessed on 3 January 2014).
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 15, 300–309. [Google Scholar] [CrossRef]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: a new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef]
- Oratz, R.; Speyer, J.L.; Wernz, J.C.; Hochster, H.; Meyers, M.; Mischak, R.; Spitler, L.E. Antimelanoma monoclonal antibody-ricin A chain immunoconjugate (XMMME-001-RTA) plus cyclophosphamide in the treatment of metastatic malignant melanoma: Results of a phase II trial. J. Biol. Resp. Modif. 1990, 9, 345–354. [Google Scholar]
- Siegall, C.B.; Haggerty, H.G.; Warner, G.L.; Chace, D.; Mixan, B.; Linsley, P.S.; Davidson, T. Prevention of immunotoxin-induced immunogenicity by coadministration with CTLA4Ig enhances antitumor efficacy. J. Immunol. 1997, 159, 5168–5173. [Google Scholar]
- Tsutsumi, Y.; Onda, M.; Nagata, S.; Lee, B.; Kreitman, R.J.; Pastan, I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 8548–8553. [Google Scholar] [CrossRef]
- Hansen, J.K.; Weldon, J.E.; Xiang, L.; Beers, R.; Onda, M.; Pastan, I. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J. Immunother. 2010, 33, 297–304. [Google Scholar] [CrossRef]
- Liu, W.; Onda, M.; Lee, B.; Kreitman, R.J.; Hassan, R.; Xiang, L.; Pastan, I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. USA 2012, 109, 11782–11787. [Google Scholar] [CrossRef]
- Boulianne, G.L.; Hozumi, N.; Shulman, M.J. Production of functional chimaeric mouse/human antibody. Nature 1984, 312, 643–646. [Google Scholar] [CrossRef]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Lonberg, N. Fully human antibodies from transgenic mouse and phage display platforms. Curr. Opin. Immunol. 2008, 20, 450–459. [Google Scholar] [CrossRef]
- Huhn, M.; Sasse, S.; Tur, M.K.; Matthey, B.; Schinkothe, T.; Rybak, S.M.; Barth, S.; Engert, A. Human angiogenin fused to human CD30 ligand (Ang-CD30L) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res. 2001, 61, 8737–8742. [Google Scholar]
- Tur, M.K.; Neef, I.; Jost, E.; Galm, O.; Jager, G.; Stocker, M.; Ribbert, M.; Osieka, R.; Klinge, U.; Barth, S. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J. Immunother. 2009, 32, 431–441. [Google Scholar] [CrossRef]
- Dalken, B.; Giesubel, U.; Knauer, S.K.; Wels, W.S. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006, 13, 576–585. [Google Scholar] [CrossRef]
- Lorberboum-Galski, H. Human toxin-based recombinant immunotoxins/chimeric proteins as a drug delivery system for targeted treatment of human diseases. Expert Opin. Drug Del. 2011, 8, 605–621. [Google Scholar] [CrossRef]
- Weidle, U.H.; Georges, G.; Brinkmann, U. Fully human targeted cytotoxic fusion proteins: new anticancer agents on the horizon. Cancer Genomics Proteomics 2012, 9, 119–133. [Google Scholar]
- Wu, M. Enhancement of immunotoxin activity using chemical and biological reagents. Br. J. Cancer 1997, 75, 1347–1355. [Google Scholar] [CrossRef]
- Bachran, C.; Weng, A.; Bachran, D.; Riese, S.B.; Schellmann, N.; Melzig, M.F.; Fuchs, H. The distribution of saponins in vivo affects their synergy with chimeric toxins against tumours expressing human epidermal growth factor receptors in mice. Br. J. Pharmacol. 2010, 159, 345–352. [Google Scholar] [CrossRef]
- Heisler, I.; Sutherland, M.; Bachran, C.; Hebestreit, P.; Schnitger, A.; Melzig, M.F.; Fuchs, H. Combined application of saponin and chimeric toxins drastically enhances the targeted cytotoxicity on tumor cells. J. Control. Release 2005, 106, 123–137. [Google Scholar] [CrossRef]
- Wu, Y.N.; Gadina, M.; Tao-Cheng, J.H.; Youle, R.J. Retinoic acid disrupts the Golgi apparatus and increases the cytosolic routing of specific protein toxins. J. Cell Biol. 1994, 125, 743–753. [Google Scholar] [CrossRef]
- Keller, J.; Heisler, I.; Tauber, R.; Fuchs, H. Development of a novel molecular adapter for the optimization of immunotoxins. J. Control. Release 2001, 74, 259–261. [Google Scholar] [CrossRef]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stocker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daeron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Ravetch, J.V.; Kinet, J.P. Fc receptors. Annu. Rev. Immunol. 1991, 9, 457–492. [Google Scholar] [CrossRef]
- Daeron, M. Structural bases of Fc gamma R functions. Int. Rev. Immunol. 1997, 16, 1–27. [Google Scholar] [CrossRef]
- Daeron, M. Fc receptor biology. Annu. Rev. Immunol. 1997, 15, 203–234. [Google Scholar] [CrossRef]
- van de Winkel, J.G.; Anderson, C.L. Biology of human immunoglobulin G Fc receptors. J. Leukoc Biol. 1991, 49, 511–524. [Google Scholar]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Krasinskas, A.M.; Wasik, M.A.; Kamoun, M.; Schretzenmair, R.; Moore, J.; Salhany, K.E. The usefulness of CD64, other monocyte-associated antigens, and CD45 gating in the subclassification of acute myeloid leukemias with monocytic differentiation. Am. J. Clin. Pathol. 1998, 110, 797–805. [Google Scholar]
- Ball, E.D.; McDermott, J.; Griffin, J.D.; Davey, F.R.; Davis, R.; Bloomfield, C.D. Expression of the three myeloid cell-associated immunoglobulin G Fc receptors defined by murine monoclonal antibodies on normal bone marrow and acute leukemia cells. Blood 1989, 73, 1951–1956. [Google Scholar]
- Frasnelli, M.; So, A. Toll-like receptor 2 and toll-like receptor 4 expression on CD64+ monocytes in rheumatoid arthritis: Comment on the article by Iwahashi et al. Arthritis Rheum. 2005, 52, 2227–2228. [Google Scholar] [CrossRef]
- Van Roon, J.A.; Bijlsma, J.W.; van de Winkel, J.G.; Lafeber, F.P. Depletion of synovial macrophages in rheumatoid arthritis by an anti-FcgammaRI-calicheamicin immunoconjugate. Ann. Rheum. Dis. 2005, 64, 865–870. [Google Scholar] [CrossRef]
- Van Vuuren, A.J.; van Roon, J.A.; Walraven, V.; Stuij, I.; Harmsen, M.C.; McLaughlin, P.M.; van de Winkel, J.G.; Thepen, T. CD64-directed immunotoxin inhibits arthritis in a novel CD64 transgenic rat model. J. Immunol. 2006, 176, 5833–5838. [Google Scholar]
- Dialynas, D.P.; Rodgers, V.D. Anomalous leukopoiesis in two patients with Crohn’s disease. J. Clin. Gastroenterol. 2002, 34, 64–71. [Google Scholar] [CrossRef]
- Wojtal, K.A.; Rogler, G.; Scharl, M.; Biedermann, L.; Frei, P.; Fried, M.; Weber, A.; Eloranta, J.J.; Kullak-Ublick, G.A.; Vavricka, S.R. Fc gamma receptor CD64 modulates the inhibitory activity of infliximab. PLoS One 2012, 7, e43361. [Google Scholar] [CrossRef] [Green Version]
- Tillinger, W.; Jilch, R.; Jilma, B.; Brunner, H.; Koeller, U.; Lichtenberger, C.; Waldhor, T.; Reinisch, W. Expression of the high-affinity IgG receptor FcRI (CD64) in patients with inflammatory bowel disease: a new biomarker for gastroenterologic diagnostics. Am. J. Gastroenterol. 2009, 104, 102–109. [Google Scholar] [CrossRef]
- Li, Y.; Lee, P.Y.; Sobel, E.S.; Narain, S.; Satoh, M.; Segal, M.S.; Reeves, W.H.; Richards, H.B. Increased expression of FcgammaRI/CD64 on circulating monocytes parallels ongoing inflammation and nephritis in lupus. Arthritis Res. Ther. 2009, 11, R6. [Google Scholar]
- Li, Y.; Lee, P.Y.; Kellner, E.S.; Paulus, M.; Switanek, J.; Xu, Y.; Zhuang, H.; Sobel, E.S.; Segal, M.S.; Satoh, M.; et al. Monocyte surface expression of Fcgamma receptor RI (CD64), a biomarker reflecting type-I interferon levels in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, R90. [Google Scholar] [CrossRef]
- Tan Sardjono, C.; Mottram, P.L.; van de Velde, N.C.; Powell, M.S.; Power, D.; Slocombe, R.F.; Wicks, I.P.; Campbell, I.K.; McKenzie, S.E.; Brooks, M.; et al. Development of spontaneous multisystem autoimmune disease and hypersensitivity to antibody-induced inflammation in Fcgamma receptor IIa-transgenic mice. Arthritis Rheum. 2005, 52, 3220–3229. [Google Scholar] [CrossRef]
- van Royen-Kerkhof, A.; Sanders, E.A.; Walraven, V.; Voorhorst-Ogink, M.; Saeland, E.; Teeling, J.L.; Gerritsen, A.; van Dijk, M.A.; Kuis, W.; Rijkers, G.T.; et al. A novel human CD32 mAb blocks experimental immune haemolytic anaemia in FcgammaRIIA transgenic mice. Br. J. Haematol. 2005, 130, 130–137. [Google Scholar] [CrossRef]
- Stone, R.M.; O’Donnell, M.R.; Sekeres, M.A. Acute myeloid leukemia. Hematology Am. Soc. Hematol. Educ. Program 2004, 1, 98–117. [Google Scholar]
- Behre, G.; Zhang, P.; Zhang, D.E.; Tenen, D.G. Analysis of the modulation of transcriptional activity in myelopoiesis and leukemogenesis. Methods 1999, 17, 231–237. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Vardiman, J.W. The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: An overview with emphasis on the myeloid neoplasms. Chem-Biol. Inter. 2010, 184, 16–20. [Google Scholar] [CrossRef]
- Robak, T.; Wierzbowska, A. Current and emerging therapies for acute myeloid leukemia. Clin. Ther. 2009, 31, 2349–2370. [Google Scholar] [CrossRef]
- Smith, M.; Barnett, M.; Bassan, R.; Gatta, G.; Tondini, C.; Kern, W. Adult acute myeloid leukaemia. Crit. Rev. Oncol. Hematol. 2004, 50, 197–222. [Google Scholar] [CrossRef]
- Fritsch, S.; Buske, C.; Wormann, B.; Wedding, U.; Hiddemann, W.; Spiekermann, K. Therapy of acute myeloid leukemia (AML) for medically non-fit patients. Med. Klin. 2007, 102, 324–329. [Google Scholar] [CrossRef]
- Yin, J.A.; Grimwade, D. Minimal residual disease evaluation in acute myeloid leukaemia. Lancet 2002, 360, 160–162. [Google Scholar] [CrossRef]
- Carter, P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 2001, 1, 118–129. [Google Scholar] [CrossRef]
- Morris, J.C.; Waldmann, T.A. Antibody-based therapy of leukaemia. Expert Rev. Mol. Med. 2009, 11, e29. [Google Scholar] [CrossRef]
- Gianni, L.; Herman, E.H.; Lipshultz, S.E.; Minotti, G.; Sarvazyan, N.; Sawyer, D.B. Anthracycline cardiotoxicity: From bench to bedside. J. Clin. Oncol. 2008, 26, 3777–3784. [Google Scholar] [CrossRef]
- Dunphy, C.H.; Tang, W. The value of CD64 expression in distinguishing acute myeloid leukemia with monocytic differentiation from other subtypes of acute myeloid leukemia: A flow cytometric analysis of 64 cases. Arch. Pathol. Lab. Med. 2007, 131, 748–754. [Google Scholar]
- Menendez, P.; del Canizo, M.C.; Orfao, A. Immunophenotypic characteristics of PB-mobilised CD34+ hematopoietic progenitor cells. J. Biol. Regul. Homeost. Agents 2001, 15, 53–61. [Google Scholar]
- Gorczyca, W.; Sun, Z.Y.; Cronin, W.; Li, X.; Mau, S.; Tugulea, S. Immunophenotypic pattern of myeloid populations by flow cytometry analysis. Methods Cell Biol. 2011, 103, 221–266. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Hamilton, T.A.; Ohmori, Y.; Tebo, J. Regulation of chemokine expression by antiinflammatory cytokines. Immunol. Res. 2002, 25, 229–245. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Verreck, F.A.; de Boer, T.; Langenberg, D.M.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef]
- Mackaness, G.B. Cellular immunity and the parasite. Adv. Exp. Med. Biol. 1977, 93, 65–73. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Edwards, J.P.; Zhang, X.; Frauwirth, K.A.; Mosser, D.M. Biochemical and functional characterization of three activated macrophage populations. J. Leukoc. Biol. 2006, 80, 1298–1307. [Google Scholar] [CrossRef]
- Loots, M.A.; Lamme, E.N.; Zeegelaar, J.; Mekkes, J.R.; Bos, J.D.; Middelkoop, E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J. Invest. Dermatol. 1998, 111, 850–857. [Google Scholar] [CrossRef]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: molecular and cellular mechanisms. J. Invest. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef]
- Ambarus, C.A.; Noordenbos, T.; de Hair, M.J.; Tak, P.P.; Baeten, D.L. Intimal lining layer macrophages but not synovial sublining macrophages display an IL-10 polarized-like phenotype in chronic synovitis. Arthritis Res. Ther. 2012, 14, R74. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Lammers, T.; Morgan, M.E.; Chaabane, L.; Carboni, S.; Greco, B.; Zaratin, P.; Kraneveld, A.D.; Storm, G. Macrophages and liposomes in inflammatory disease: Friends or foes? Int. J. Pharm. 2011, 416, 499–506. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Orme, J.; Mohan, C. Macrophage subpopulations in systemic lupus erythematosus. Discov. Med. 2012, 13, 151–158. [Google Scholar]
- Kiekens, R.C.; Thepen, T.; Bihari, I.C.; Knol, E.F.; van de Winkel, J.G.; Bruijnzeel-Koomen, C.A. Expression of Fc receptors for IgG during acute and chronic cutaneous inflammation in atopic dermatitis. Br. J. Dermatol. 2000, 142, 1106–1113. [Google Scholar] [CrossRef]
- Graziano, R.F.; Tempest, P.R.; White, P.; Keler, T.; Deo, Y.; Ghebremariam, H.; Coleman, K.; Pfefferkorn, L.C.; Fanger, M.W.; Guyre, P.M. Construction and characterization of a humanized anti-gamma-Ig receptor type I (Fc gamma RI) monoclonal antibody. J. Immunol. 1995, 155, 4996–5002. [Google Scholar]
- Somasundaram, C.; Sundarapandiyan, K.; Keler, T.; Deo, Y.M.; Graziano, R.F. Development of a trispecific antibody conjugate that directs two distinct tumor-associated antigens to CD64 on myeloid effector cells. Hum. Antibodies 1999, 9, 47–54. [Google Scholar]
- Tur, M.K.; Huhn, M.; Thepen, T.; Stocker, M.; Krohn, R.; Vogel, S.; Jost, E.; Osieka, R.; van de Winkel, J.G.; Fischer, R.; et al. Recombinant CD64-specific single chain immunotoxin exhibits specific cytotoxicity against acute myeloid leukemia cells. Cancer Res. 2003, 63, 8414–8419. [Google Scholar]
- Wallace, P.K.; Keler, T.; Coleman, K.; Fisher, J.; Vitale, L.; Graziano, R.F.; Guyre, P.M.; Fanger, M.W. Humanized mAb H22 binds the human high affinity Fc receptor for IgG (FcgammaRI), blocks phagocytosis, and modulates receptor expression. J. Leukoc. Biol. 1997, 62, 469–479. [Google Scholar]
- Tur, M.K.; Huhn, M.; Jost, E.; Thepen, T.; Brummendorf, T.H.; Barth, S. In vivo efficacy of the recombinant anti-CD64 immunotoxin H22(scFv)-ETA' in a human acute myeloid leukemia xenograft tumor model. Int. J. Cancer 2011, 129, 1277–1282. [Google Scholar] [CrossRef]
- Thepen, T.; van Vuuren, A.J.; Kiekens, R.C.; Damen, C.A.; Vooijs, W.C.; van de Winkel, J.G. Resolution of cutaneous inflammation after local elimination of macrophages. Nat. Biotechnol. 2000, 18, 48–51. [Google Scholar] [CrossRef]
- Ribbert, T.; Thepen, T.; Tur, M.K.; Fischer, R.; Huhn, M.; Barth, S. Recombinant, ETA'-based CD64 immunotoxins: Improved efficacy by increased valency, both in vitro and in vivo in a chronic cutaneous inflammation model in human CD64 transgenic mice. Br. J. Dermatol. 2010, 163, 279–286. [Google Scholar] [CrossRef]
- Fet, N.G.; Fiebeler, A.; Klinge, U.; Park, J.K.; Barth, S.; Thepen, T.; Tolba, R.H. Reduction of activated macrophages after ischaemia-reperfusion injury diminishes oxidative stress and ameliorates renal damage. Nephrol. Dial. Transplant. 2012.
- van de Winkel, J.G.; Capel, P.J. Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications. Immunol. Today 1993, 14, 215–221. [Google Scholar] [CrossRef]
- Leemans, J.C.; Thepen, T.; Weijer, S.; Florquin, S.; van Rooijen, N.; van de Winkel, J.G.; van der Poll, T. Macrophages play a dual role during pulmonary tuberculosis in mice. J. Infect. Dis. 2005, 191, 65–74. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef]
- Allured, V.S.; Collier, R.J.; Carroll, S.F.; McKay, D.B. Structure of exotoxin A of pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. USA 1986, 83, 1320–1324. [Google Scholar] [CrossRef]
- Frankel, A.E.; Kreitman, R.J.; Sausville, E.A. Targeted toxins. Clin. Cancer Res. 2000, 6, 326–334. [Google Scholar]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Squires, D.R.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.; Wilson, W.H.; Pastan, I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J. Clin. Oncol. 2005, 23, 6719–6729. [Google Scholar] [CrossRef]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin. Cancer Res. 2002, 8, 3092–3099. [Google Scholar]
- Wei, H.; Bera, T.K.; Wayne, A.S.; Xiang, L.; Colantonio, S.; Chertov, O.; Pastan, I. A modified form of diphthamide causes immunotoxin resistance in a lymphoma cell line with a deletion of the WDR85 gene. J. Biol. Chem. 2013, 288, 12305–12312. [Google Scholar]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef]
- Schiffer, S.; Hristodorov, D.; Mladenov, R.; Aslanian, E.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Species-Dependent Functionality of the Human Cytolytic Fusion Proteins Granzyme B-H22(scFv) and H22(scFv)-Angiogenin in Macrophages. Antibodies 2013, 2, 9–18. [Google Scholar] [CrossRef]
- Susanto, O.; Trapani, J.A.; Brasacchio, D. Controversies in granzyme biology. Tissue Antigens 2012, 80, 477–487. [Google Scholar] [CrossRef]
- Andrade, F.; Casciola-Rosen, L.A.; Rosen, A. Granzyme B-induced cell death. Acta Haematol. 2004, 111, 28–41. [Google Scholar] [CrossRef]
- Chowdhury, D.; Lieberman, J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef]
- Bots, M.; Medema, J.P. Granzymes at a glance. J. Cell Sci. 2006, 119, 5011–5014. [Google Scholar] [CrossRef]
- Trapani, J.A.; Sutton, V.R. Granzyme B: Pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 2003, 15, 533–543. [Google Scholar] [CrossRef]
- Hehmann-Titt, G.; Schiffer, S.; Berges, N.; Melmer, G.; Barth, S. Improving the Therapeutic Potential of Human Granzyme B for Targeted Cancer Therapy. Antibodies 2013, 2, 19–49. [Google Scholar] [CrossRef]
- Cao, Y.; Mohamedali, K.A.; Marks, J.W.; Cheung, L.H.; Hittelman, W.N.; Rosenblum, M.G. Construction and characterization of novel, completely human serine protease therapeutics targeting Her2/neu. Mol. Cancer Ther. 2013, 12, 979–991. [Google Scholar] [CrossRef]
- Schiffer, S.; Hansen, H.P.; Hehmann-Titt, G.; Huhn, M.; Fischer, R.; Barth, S.; Thepen, T. Efficacy of an adapted granzyme B-based anti-CD30 cytolytic fusion protein against PI-9-positive classical Hodgkin lymphoma cells in a murine model. Blood Cancer J. 2013, 3, e106. [Google Scholar] [CrossRef]
- Tello-Montoliu, A.; Patel, J.V.; Lip, G.Y. Angiogenin: A review of the pathophysiology and potential clinical applications. J. Thromb. Haemost. 2006, 4, 1864–1874. [Google Scholar] [CrossRef]
- Saxena, S.K.; Rybak, S.M.; Davey, R.T., Jr.; Youle, R.J.; Ackerman, E.J. Angiogenin is a cytotoxic, tRNA-specific ribonuclease in the RNase A superfamily. J. Biol. Chem. 1992, 267, 21982–21986. [Google Scholar]
- Stocker, M.; Tur, M.K.; Sasse, S.; Krussmann, A.; Barth, S.; Engert, A. Secretion of functional anti-CD30-angiogenin immunotoxins into the supernatant of transfected 293T-cells. Protein Express. Purif. 2003, 28, 211–219. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Method. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Roehm, N.W.; Rodgers, G.H.; Hatfield, S.M.; Glasebrook, A.L. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Method. 1991, 142, 257–265. [Google Scholar] [CrossRef]
- Ulukaya, E.; Ozdikicioglu, F.; Oral, A.Y.; Demirci, M. The MTT assay yields a relatively lower result of growth inhibition than the ATP assay depending on the chemotherapeutic drugs tested. Toxicol. Vitro. 2008, 22, 232–239. [Google Scholar] [CrossRef]
- Marshall, N.J.; Goodwin, C.J.; Holt, S.J. A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regulat. 1995, 5, 69–84. [Google Scholar]
- Vistica, D.T.; Skehan, P.; Scudiero, D.; Monks, A.; Pittman, A.; Boyd, M.R. Tetrazolium-based assays for cellular viability: a critical examination of selected parameters affecting formazan production. Cancer Res. 1991, 51, 2515–2520. [Google Scholar]
- Bernhard, D.; Schwaiger, W.; Crazzolara, R.; Tinhofer, I.; Kofler, R.; Csordas, A. Enhanced MTT-reducing activity under growth inhibition by resveratrol in CEM-C7H2 lymphocytic leukemia cells. Cancer Lett. 2003, 195, 193–199. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Kundu, S.; Kumar, S.; Chakrabarti, R. Vitamin A as an enzyme that catalyzes the reduction of MTT to formazan by vitamin C. J. Cell. Biochem. 2001, 80, 133–138. [Google Scholar] [CrossRef]
- Funk, D.; Schrenk, H.H.; Frei, E. Serum albumin leads to false-positive results in the XTT and the MTT assay. BioTechniques 2007, 43, 178–186. [Google Scholar] [CrossRef]
- Natarajan, M.; Mohan, S.; Martinez, B.R.; Meltz, M.L.; Herman, T.S. Antioxidant compounds interfere with the 3. Cancer Detection Prev. 2000, 24, 405–414. [Google Scholar]
- Rubinstein, L.V.; Shoemaker, R.H.; Paull, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Scudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990, 82, 1113–1118. [Google Scholar] [CrossRef]
- Oberoi, P.; Jabulowsky, R.A.; Wels, W.S. Selective Induction of Cancer Cell Death by Targeted Granzyme B. Antibodies 2013, 2, 130–151. [Google Scholar] [CrossRef]
- Yoon, J.M.; Han, S.H.; Kown, O.B.; Kim, S.H.; Park, M.H.; Kim, B.K. Cloning and cytotoxicity of fusion proteins of EGF and angiogenin. Life Sci. 1999, 64, 1435–1445. [Google Scholar] [CrossRef]
- Jinno, H.; Ueda, M.; Ikeda, T.; Seno, M.; Kitajima, M. Selective cytotoxicity of human angiogenin conjugated to human EGF against EGFR-overexpressing cancer cells. Proc. Am. Assoc. Cancer Res. 2004, 2004, 1235. [Google Scholar]
- Kurschus, F.C.; Kleinschmidt, M.; Fellows, E.; Dornmair, K.; Rudolph, R.; Lilie, H.; Jenne, D.E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004, 562, 87–92. [Google Scholar] [CrossRef]
- Klimka, A.; Barth, S.; Matthey, B.; Roovers, R.C.; Lemke, H.; Hansen, H.; Arends, J.W.; Diehl, V.; Hoogenboom, H.R.; Engert, A. An anti-CD30 single-chain Fv selected by phage display and fused to Pseudomonas exotoxin A (Ki-4(scFv)-ETA') is a potent immunotoxin against a Hodgkin-derived cell line. Br. J. Cancer 1999, 80, 1214–1222. [Google Scholar] [CrossRef]
- Barth, S.; Matthey, B.; Huhn, M.; Diehl, V.; Engert, A. CD30L-ETA': A new recombinant immunotoxin based on the CD30 ligand for possible use against human lymphoma. Cytokines Cell. Mol. Ther. 1999, 5, 69–78. [Google Scholar]
- Fuchs, H.; Bachran, C.; Flavell, D.J. Diving through membranes: Molecular cunning to enforce the endosomal escape of antibody-targeted anti-tumor toxins. Antibodies 2013, 2, 209–235. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wangemail, T.Y.; Pellois, J.P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef]
- Rawat, A.; Vaidya, B.; Khatri, K.; Goyal, A.K.; Gupta, P.N.; Mahor, S.; Paliwal, R.; Rai, S.; Vyas, S.P. Targeted intracellular delivery of therapeutics: An overview. Die Pharmazie 2007, 62, 643–658. [Google Scholar]
- Torchilin, V.P. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 2006, 8, 343–375. [Google Scholar] [CrossRef]
- Enninga, J.; Rosenshine, I. Imaging the assembly, structure and activity of type III secretion systems. Cell. Microbiol. 2009, 11, 1462–1470. [Google Scholar] [CrossRef]
- Juhas, M.; Crook, D.W.; Hood, D.W. Type IV secretion systems: Tools of bacterial horizontal gene transfer and virulence. Cell. Microbiol. 2008, 10, 2377–2386. [Google Scholar] [CrossRef]
- Leopold, P.L.; Ferris, B.; Grinberg, I.; Worgall, S.; Hackett, N.R.; Crystal, R.G. Fluorescent virions: Dynamic tracking of the pathway of adenoviral gene transfer vectors in living cells. Hum. Gene Ther. 1998, 9, 367–378. [Google Scholar] [CrossRef]
- Lord, J.M.; Smith, D.C.; Roberts, L.M. Toxin entry: How bacterial proteins get into mammalian cells. Cell. Microbiol. 1999, 1, 85–91. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Plank, C.; Oberhauser, B.; Mechtler, K.; Koch, C.; Wagner, E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J. Biol. Chem. 1994, 269, 12918–12924. [Google Scholar]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef]
- Ogata, M.; Fryling, C.M.; Pastan, I.; FitzGerald, D.J. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J. Biol. Chem. 1992, 267, 25396–25401. [Google Scholar]
- Zhan, H.; Choe, S.; Huynh, P.D.; Finkelstein, A.; Eisenberg, D.; Collier, R.J. Dynamic transitions of the transmembrane domain of diphtheria toxin: disulfide trapping and fluorescence proximity studies. Biochemistry 1994, 33, 11254–11263. [Google Scholar] [CrossRef]
- van den Berg, A.; Dowdy, S.F. Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 2011, 22, 888–893. [Google Scholar] [CrossRef]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–468. [Google Scholar] [CrossRef]
- Snyder, E.L.; Meade, B.R.; Dowdy, S.F. Anti-cancer protein transduction strategies: reconstitution of p27 tumor suppressor function. J. Control. Release 2003, 91, 45–51. [Google Scholar] [CrossRef]
- Hong, F.D.; Clayman, G.L. Isolation of a peptide for targeted drug delivery into human head and neck solid tumors. Cancer Res. 2000, 60, 6551–6556. [Google Scholar]
- Hetzel, C.; Bachran, C.; Tur, M.K.; Fuchs, H.; Stocker, M. Improved immunotoxins with novel functional elements. Curr. Pharm. Design 2009, 15, 2700–2711. [Google Scholar] [CrossRef]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A cleavable adapter to reduce nonspecific cytotoxicity of recombinant immunotoxins. Int. J. Cancer 2003, 103, 277–282. [Google Scholar] [CrossRef]
- McGrath, M.S.; Rosenblum, M.G.; Philips, M.R.; Scheinberg, D.A. Immunotoxin resistance in multidrug resistant cells. Cancer Res. 2003, 63, 72–79. [Google Scholar]
- Wang, T.; Zhao, J.; Ren, J.L.; Zhang, L.; Wen, W.H.; Zhang, R.; Qin, W.W.; Jia, L.T.; Yao, L.B.; Zhang, Y.Q.; et al. Recombinant immunoproapoptotic proteins with furin site can translocate and kill HER2-positive cancer cells. Cancer Res. 2007, 67, 11830–11839. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Wang, T.; Yu, C.J.; Jia, L.T.; Duan, Y.Y.; Yao, L.B.; Chen, S.Y.; Yang, A.G. HER2-targeting recombinant protein with truncated pseudomonas exotoxin A translocation domain efficiently kills breast cancer cells. Cancer Biol. Ther. 2008, 7, 1226–1231. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Berges, N.; Hehmann-Titt, G.; Hristodorov, D.; Melmer, G.; Thepen, T.; Barth, S. Human Cytolytic Fusion Proteins: Modified Versions of Human Granzyme B and Angiogenin Have the Potential to Replace Bacterial Toxins in Targeted Therapies against CD64+ Diseases. Antibodies 2014, 3, 92-115. https://doi.org/10.3390/antib3010092
Berges N, Hehmann-Titt G, Hristodorov D, Melmer G, Thepen T, Barth S. Human Cytolytic Fusion Proteins: Modified Versions of Human Granzyme B and Angiogenin Have the Potential to Replace Bacterial Toxins in Targeted Therapies against CD64+ Diseases. Antibodies. 2014; 3(1):92-115. https://doi.org/10.3390/antib3010092
Chicago/Turabian StyleBerges, Nina, Grit Hehmann-Titt, Dmitrij Hristodorov, Georg Melmer, Theo Thepen, and Stefan Barth. 2014. "Human Cytolytic Fusion Proteins: Modified Versions of Human Granzyme B and Angiogenin Have the Potential to Replace Bacterial Toxins in Targeted Therapies against CD64+ Diseases" Antibodies 3, no. 1: 92-115. https://doi.org/10.3390/antib3010092