The Development of Bispecific Hexavalent Antibodies as a Novel Class of DOCK-AND-LOCKTM (DNLTM) Complexes

Abstract
:1. Introduction
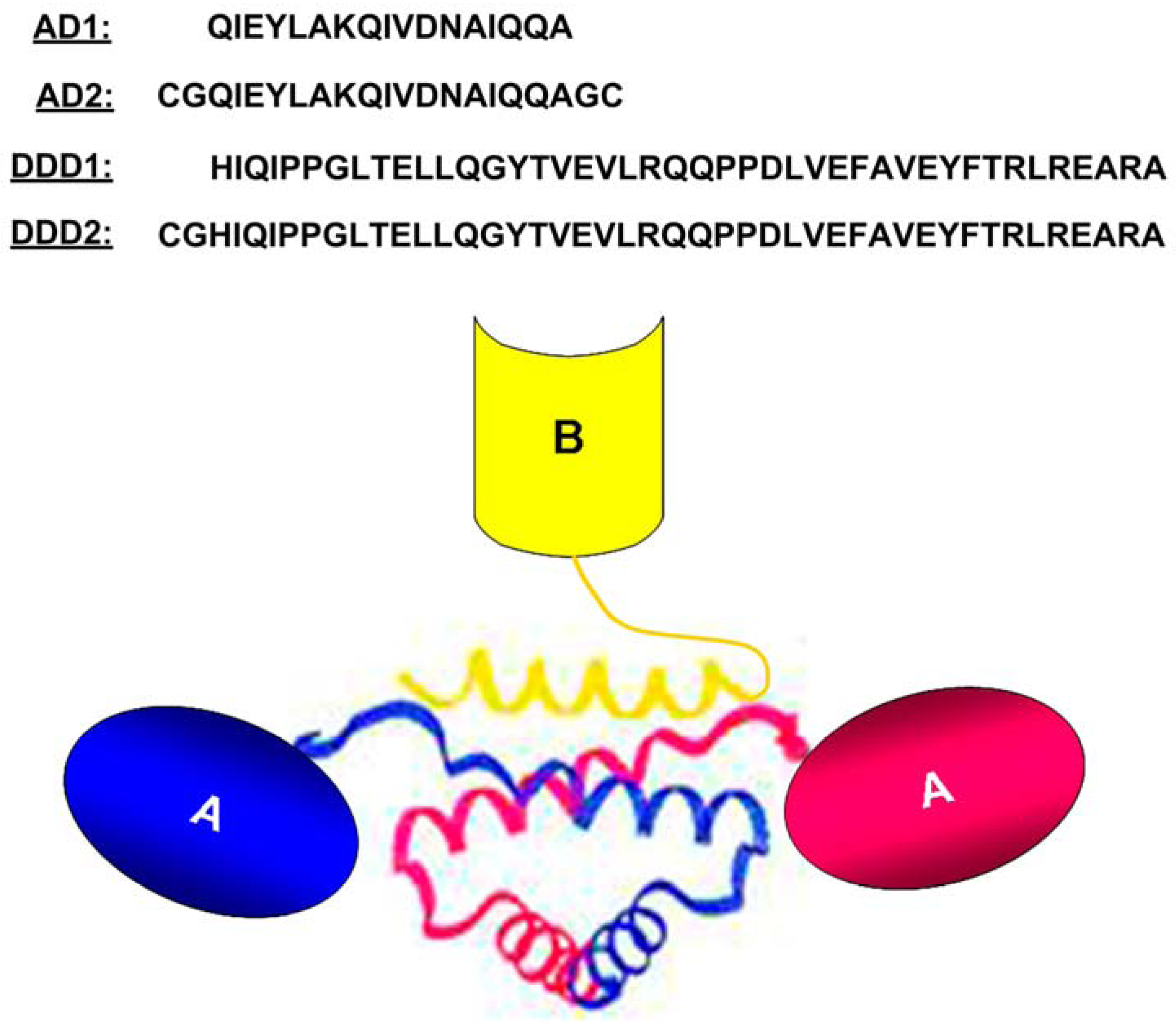
2. The DNLTM Method
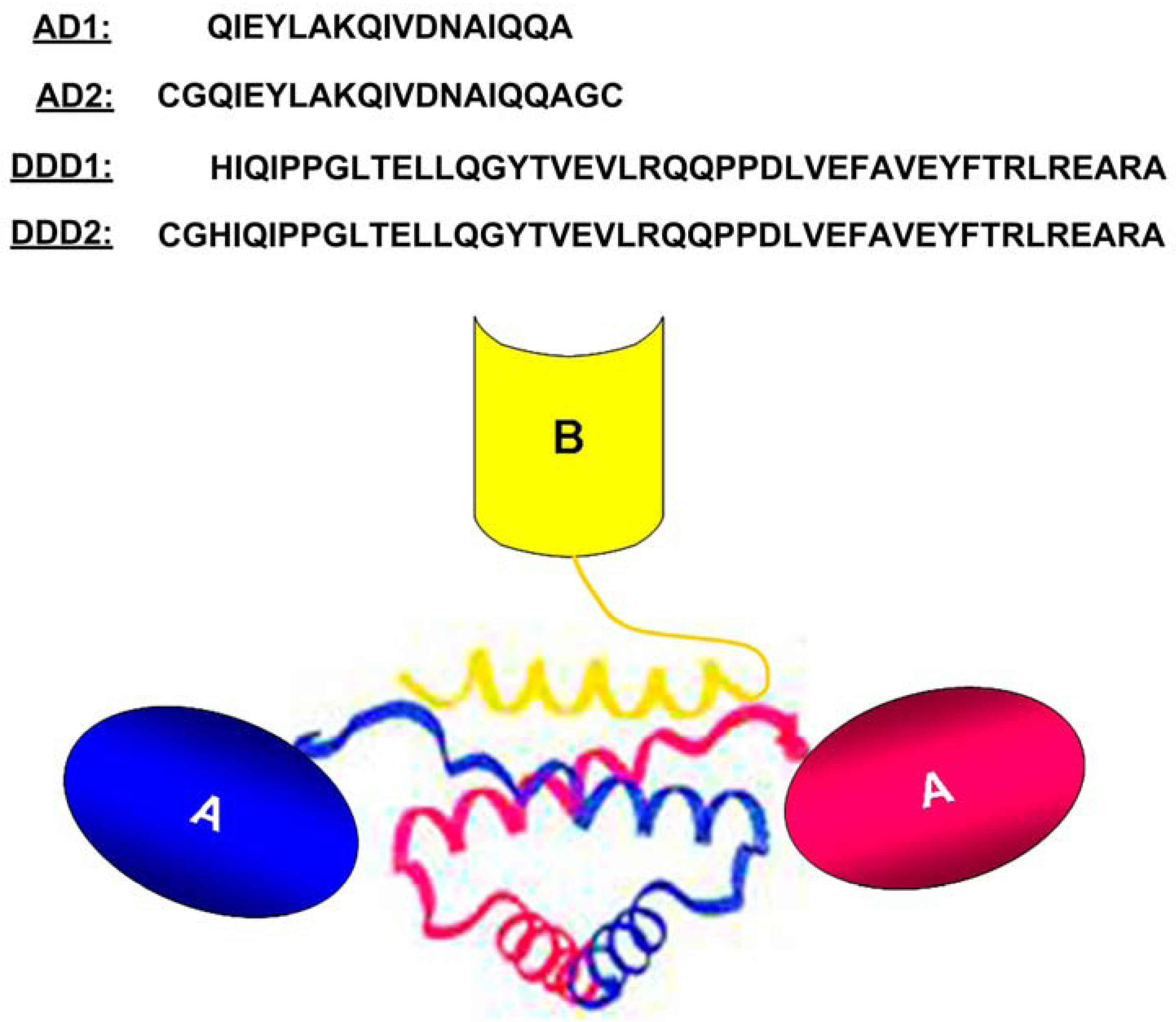
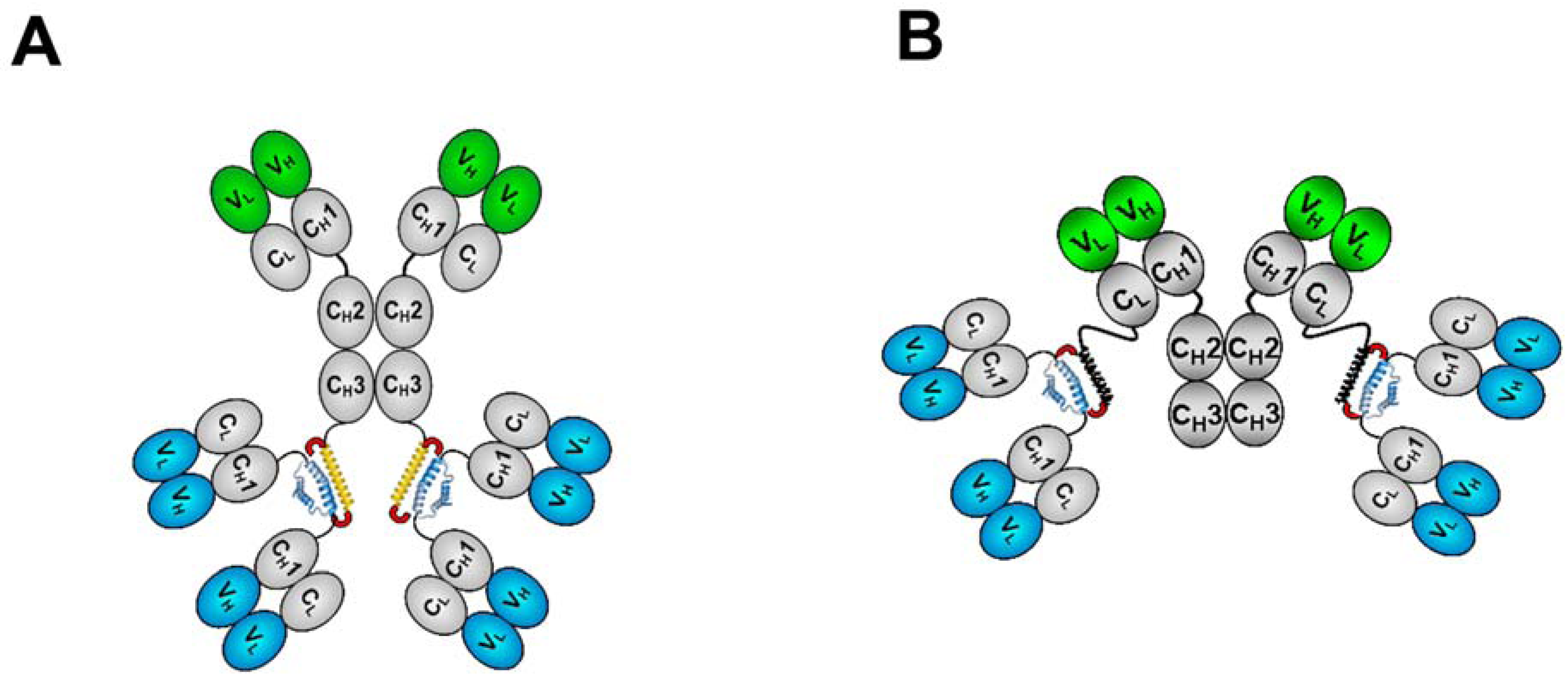
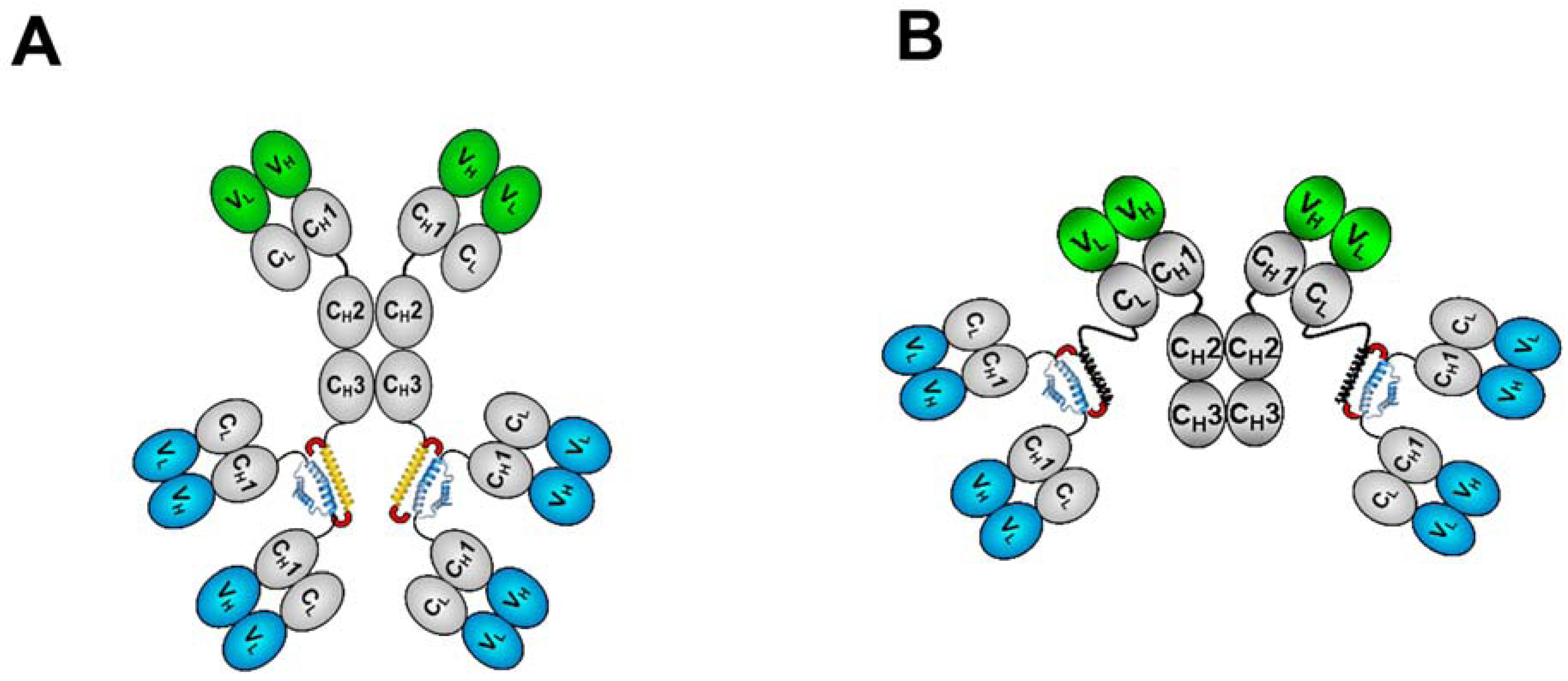
3. The CH3-format of bsHexAbs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Antibody | ||
|---|---|---|---|
| Trivial name | USAN | Designation | |
| CD19 | hA19 | - | 19 |
| CD20 | hA20 | Veltuzumab | 20 |
| CD22 | hLL2 | Epratuzumab | 22 |
| CD74 | hLL1 | Milatuzumab | 74 |
| CEACAM5 | hMN-14 | Labetuzumab | 14 |
| CEACAM6 | hMN-15 | - | 15 |
| HLA-DR | hL243 | - | C2 |
| IGF-1R | hR1 | - | 1R |
| Trop-2 | hRS7 | - | E1 |
| Indium-DTPA | h734 | - | 734 |
3.1. Generation and Biochemical Analysis
| Code | Alternative name | AD-module | DDD-module | ||
|---|---|---|---|---|---|
| Design | Target | Design | Target | ||
| 20-(22)-(22) | 20-22 | CH3-AD2-IgG-hA20 | CD20 | CH1-DDD2-Fab-hLL2 | CD22 |
| 22-(20)-(20) | 22-20 | CH3-AD2-IgG-hLL2 | CD22 | CH1-DDD2-Fab-hA20 | CD20 |
| 20-(74)-(74) | - | CH3-AD2-IgG-hA20 | CD20 | CH1-DDD2-Fab-hLL1 | CD74 |
| 74-(20)-(20) | - | CH3-AD2-IgG-hLL1 | CD74 | CH1-DDD2-Fab-hA20 | CD20 |
| 20-(14)-(14) | 20-14 | CH3-AD2-IgG-hA20 | CD20 | CH1-DDD2-Fab-hMN-14 | CEACAM5 |
| 22-(14)-(14) | 22-14 | CH3-AD2-IgG-hLL2 | CD22 | CH1-DDD2-Fab-hMN-14 | CEACAM5 |
| 734-(20)-(20) | 734-20 | CH3-AD2-IgG-h734 | Indium-DTPA | CH1-DDD2-Fab-hA20 | CD20 |
| E1-(1R)-(1R) | - | CH3-AD2-IgG-hRS7 | Trop-2 | CH1-DDD2-Fab-hR1 | IGF-1R |
| 1R-(E1)-(E1) | - | CH3-AD2-IgG-hR1 | IGF-1R | CH1-DDD2-Fab-hRS7 | Trop-2 |
| 1R-(15)-(15) | - | CH3-AD2-IgG-hR1 | IGF-1R | CH1-DDD2-Fab-hMN-15 | CEACAM6 |
| 74-(1R)-(1R) | - | CH3-AD2-IgG-hLL1 | CD74 | CH1-DDD2-Fab-hR1 | IGF-1R |
| 20-(20)-(20) | Hex-hA20 | CH3-AD2-IgG-hA20 | CD20 | CH1-DDD2-Fab-hA20 | CD20 |
| 22-(22)-(22) | Hex-hLL2 | CH3-AD2-IgG-hLL2 | CD22 | CH1-DDD2-Fab-hLL2 | CD22 |
| 1R-(1R)-(1R) | Hex-hR1 | CH3-AD2-IgG-hR1 | IGF-1R | CH1-DDD2-Fab-hR1 | IGF-1R |
3.2. Functional Characterizations Based on in Vitro Studies
3.2.1. bsHexAbs that Target CD20 and CD22
3.2.2. bsHexAbs that Target CD20 and CD74
3.3. Pharmacokinetics (PK) and in Vivo Anti-Tumor Activity
4. The CK-format of bsHexAbs
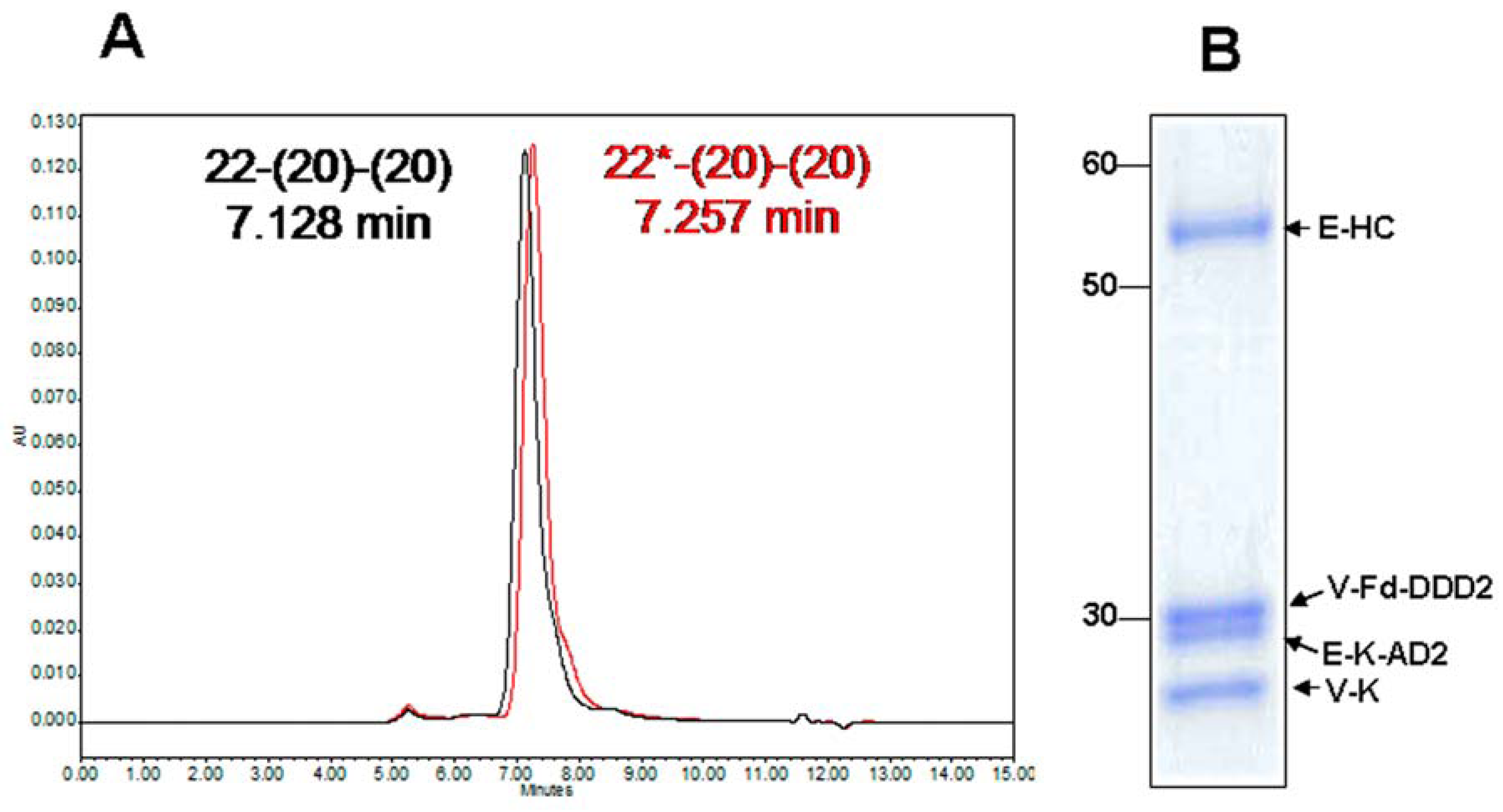
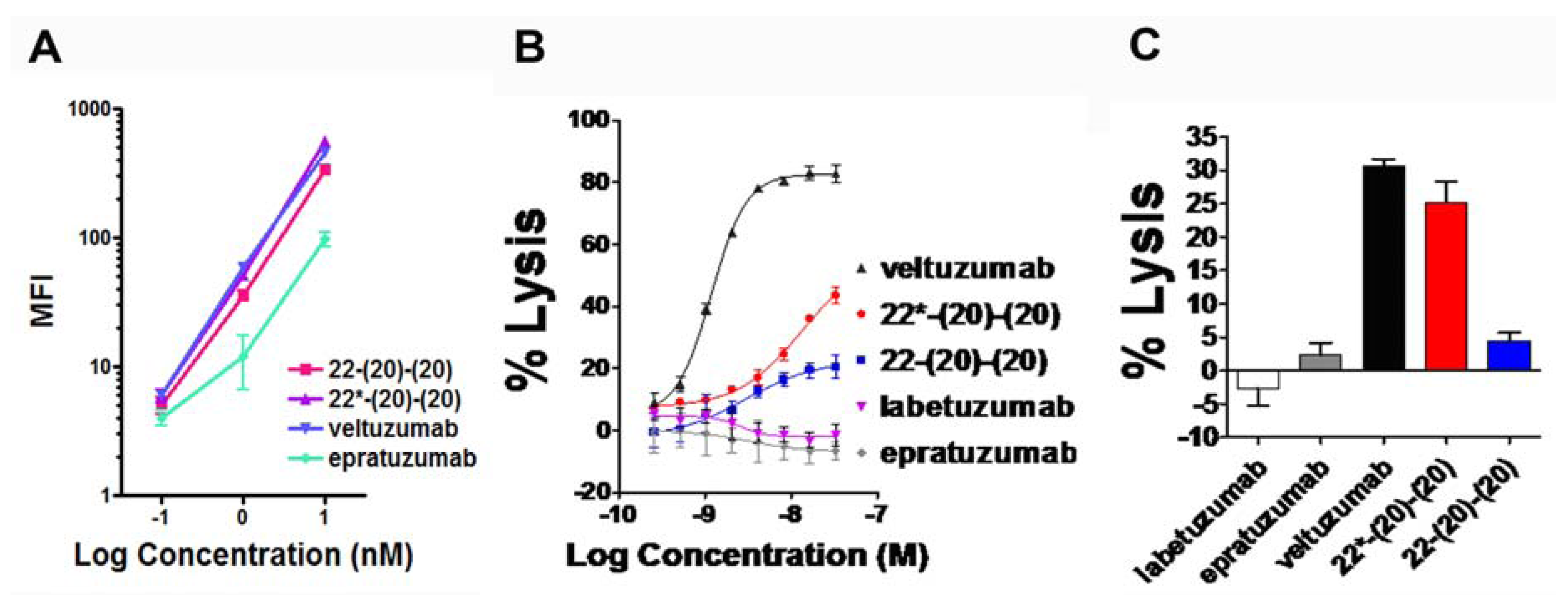
4.1. Generation and in Vitro Characterization
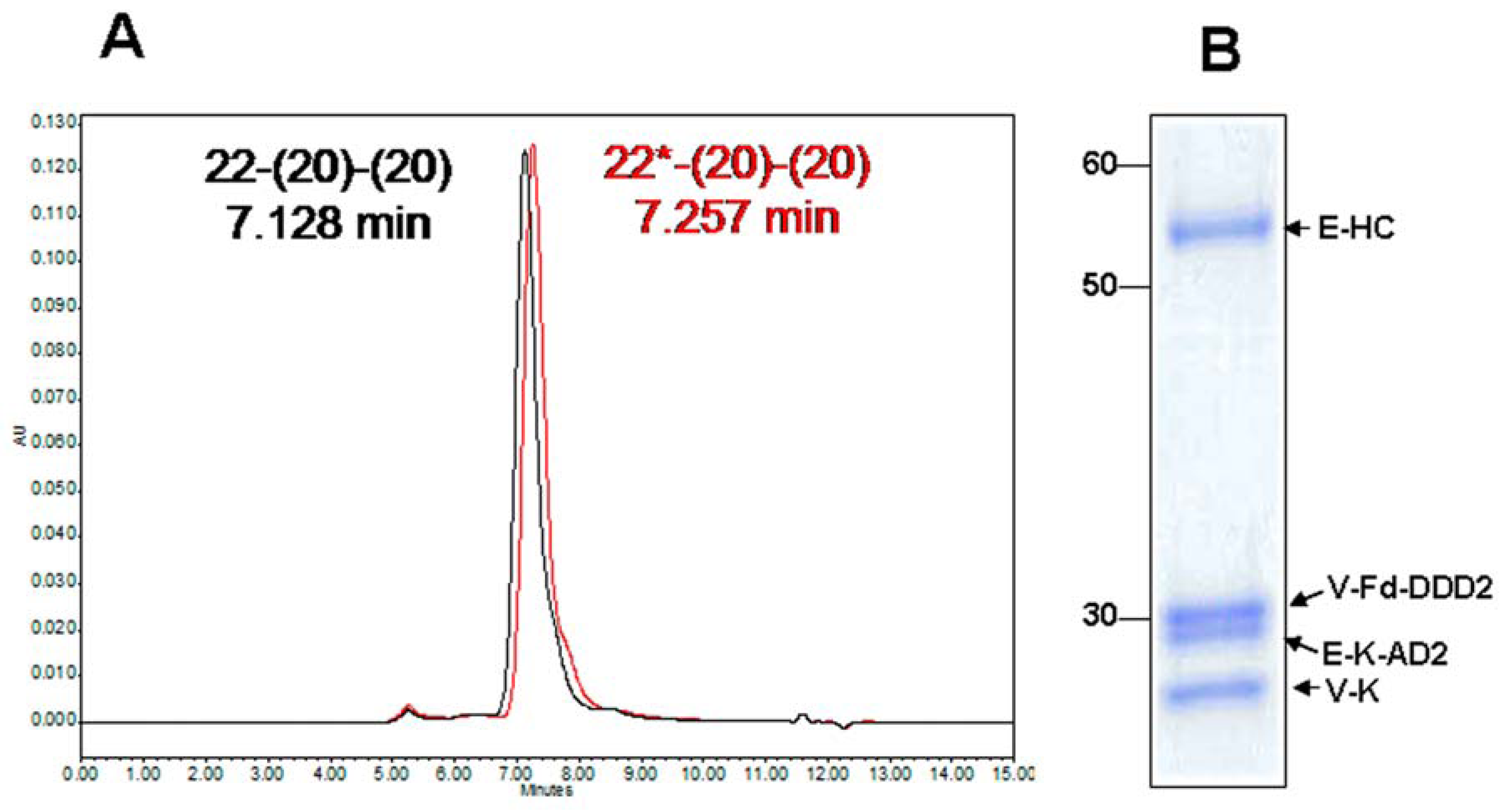
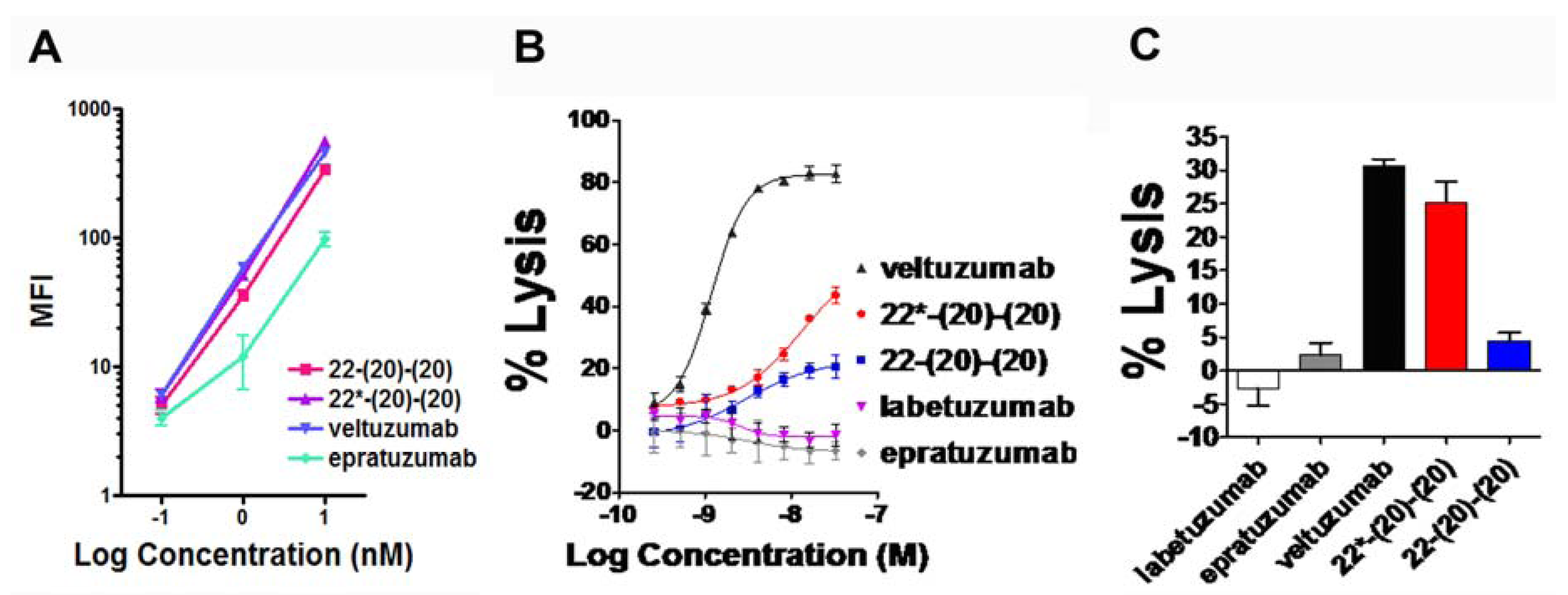
4.2. Evaluation of PK and in Vivo Anti-Tumor Activity
5. Conclusions and Future Challenges
Conflict of Interest
References
- Reichert, J.M. What are the antibodies to watch in 2013? mAbs 2013, 5, 1–4. [Google Scholar]
- Leonard, J.P.; Coleman, M.; Ketas, J.; Ashe, M.; Fiore, J.M.; Furman, R.R.; Niesvizky, R.; Shore, T.; Chadburn, A.; Horne, H.; et al. Combination antibody therapy with epratuzumab and rituximab in relapsed or refractory non-Hodgkin’s lymphoma. J. Clin. Oncol. 2005, 23, 5044–5051. [Google Scholar] [CrossRef]
- Strauss, S.J.; Morschhauser, F.; Rech, J.; Repp, R.; Solal-Celigny, P.; Zinzani, P.L.; Engert, A.; Coiffier, B.; Hoelzer, D.F.; Wegener, W.A.; et al. Multicenter phase II trial of immunotherapy with the humanized anti-CD22 antibody, epratuzumab, in combination with rituximab, in refractory or recurrent non-Hodgkin’s lymphoma. J. Clin. Oncol. 2006, 24, 3880–3886. [Google Scholar] [CrossRef]
- Leonard, J.P.; Schuster, S.J.; Emmanouilides, C.; Couture, F.; Teoh, N.; Wegener, W.A.; Coleman, M.; Goldenberg, D.M. Durable complete responses from therapy with combined epratuzumab and rituximab: Final results from an international multicenter, phase 2 study in recurrent, indolent, non-Hodgkin lymphoma. Cancer 2008, 113, 2714–2723. [Google Scholar] [CrossRef]
- Tonra, J.R.; Corcoran, E.; Deevi, D.S.; Steiner, P.; Kearney, J.; Li, H.; Ludwig, D.L.; Zhu, Z.; Witte, L.; Surguladze, D.; et al. Prioritization of EGFR/IGF-IR/VEGFR2 combination targeted therapies utilizing cancer models. Anticancer Res. 2009, 29, 1999–2008. [Google Scholar]
- Alinari, L.; Yu, B.; Christian, B.A.; Yan, F.; Shin, J.; Lapalombella, R.; Hertlein, E.; Lustberg, M.E.; Quinion, C.; Zhang, X.; et al. Combination anti-CD74 (milatuzumab) and anti-CD20 (rituximab) monoclonal antibody therapy has in vitro and in vivo activity in mantle cell lymphoma. Blood 2011, 117, 4530–4541. [Google Scholar] [CrossRef]
- Loisel, S.; Andre, P.A.; Golay, J.; Buchegger, F.; Kadouche, J.; Cerutti, M.; Bologna, L.; Kosinski, M.; Viertl, D.; Delaloye, A.B.; et al. Antitumour effects of single or combined monoclonal antibodies directed against membrane antigens expressed by human B cells leukaemia. Mol. Cancer 2011, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Skartved, N.J.; Jacobsen, H.J.; Pedersen, M.W.; Jensen, P.F.; Sen, J.W.; Jorgensen, T.K.; Hey, A.; Kragh, M. Preclinical pharmacokinetics and safety of Sym004: A synergistic antibody mixture directed against epidermal growth factor receptor. Clin. Cancer Res. 2011, 17, 5962–5972. [Google Scholar]
- Fuentes, G.; Scaltriti, M.; Baselga, J.; Verma, C.S. Synergy between trastuzumab and pertuzumab for human epidermal growth factor 2 (Her2) from colocalization: An in silico based mechanism. Breast Cancer Res. 2011, 13, R54. [Google Scholar]
- Dong, J.; Demarest, S.J.; Sereno, A.; Tamraz, S.; Langley, E.; Doern, A.; Snipas, T.; Perron, K.; Joseph, I.; Glaser, S.M.; et al. Combination of two insulin-like growth factor-I receptor inhibitory antibodies targeting distinct epitopes leads to an enhanced antitumor response. Mol. Cancer Ther. 2010, 9, 2593–2604. [Google Scholar] [CrossRef]
- Kontermann, R.B. Dual targeting strategies with bispecific antibodies. mAbs 2012, 4, 182–197. [Google Scholar] [CrossRef]
- Schubert, I.; Stein, C.; Fey, G.H. Dual-targeting for the elimination of cancer cells with increased selectivity. Antibodies 2012, 1, 1–18. [Google Scholar]
- van der Neut Kolfschoten, M.; Schuurman, J.; Losen, M.; Bleeker, W.K.; Martinez-Martinex, P.; Vermeulen, E.; den Bleker, T.H.; Wiegman, L.; Vink, T.; Aarden, L.A.; et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007, 317, 1554–1557. [Google Scholar]
- Burton, D.R.; Wilson, I.A. Immunology. Square-dancing antibodies. Science 2007, 317, 1507–1508. [Google Scholar]
- Labrijn, A.F.; Buijsse, A.O.; van den Bremer, E.T.J.; Verwilligen, A.Y.W.; Bleeker, W.K.; Thorpe, S.J.; Killestein, J.; Polman, C.H.; Aalberse, R.C.; Schuurman, J.; et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat. Biotechnol. 2009, 27, 767–771. [Google Scholar]
- Milstein, C.; Cuello, A.C. Hybrid hybridomas and their use in immunochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef]
- Staerz, U.D.; Bevan, M.J. Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc. Nat. Acad. Sci. USA 1986, 83, 1453–1457. [Google Scholar] [CrossRef]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef]
- Brennan, M.; Davison, P.F.; Paulus, H. Preparation of bispecific antibodies by chemical recombination of monoclonal immunoglobulin G1 fragments. Science 1985, 229, 81–83. [Google Scholar]
- Glennie, M.J.; McBride, H.M.; Worth, A.T.; Stevenson, G.T. Preparation and performance of bispecific F(ab' gamma)2 antibody containing thioether-linked Fab' gamma fragments. J. Immunol. 1987, 139, 2367–2375. [Google Scholar]
- Kriangkum, J.; Xu, B.; Nagata, L.P.; Fulton, R.E.; Suresh, M.R. Bispecific and bifunctional single chain recombinant antibodies. Biomol. Eng. 2001, 18, 31–40. [Google Scholar] [CrossRef]
- Muller, D.; Kontermann, R.E. Recombinant bispecific antibodies for cellular cancer immunotherapy. Curr. Opin. Mol. Ther. 2007, 9, 319–326. [Google Scholar]
- Hollander, N. Bispecific antibodies for cancer therapy. Immunotherapy 2009, 1, 211–222. [Google Scholar] [CrossRef]
- Marvin, J.S.; Zhu, Z. Recombinant approaches to IgG-like bispecific antibodies. Acta Pharmacol. Sin. 2005, 26, 649–658. [Google Scholar] [CrossRef]
- Coloma, M.J.; Morrison, S.L. Design and production of novel tetravalent bispecific antibodies. Nat. Biotechnol. 1997, 15, 159–163. [Google Scholar]
- Lu, D.; Zhang, H.; Ludwig, D.; Persaud, A.; Jimenez, X.; Burtrum, D.; Balderes, P.; Liu, M.; Bohlen, P.; Witte, L.; et al. Simultaneous blockade of both the epidermal growth factor receptor and the insulin-like growth factor receptor signaling pathways in cancer cells with a fully human recombinant bispecific antibody. J. Biol. Chem. 2004, 279, 2856–2865. [Google Scholar]
- Lu, D.; Zhang, H.; Koo, H.; Tonra, J.; Balderes, P.; Prewett, M.; Corcoran, E.; Mangalampalli, V.; Bassi, R.; Anselma, D.; et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J. Biol. Chem. 2005, 280, 19665–19672. [Google Scholar] [CrossRef]
- Shen, J.; Vil, M.D.; Jimenez, X.; Iacolina, M.; Zhang, H.; Zhu, Z. Single variable domain-IgG fusion. A novel recombinant approach to Fc domain-containing bispecific antibodies. J. Biol. Chem. 2006, 281, 10706–10714. [Google Scholar]
- Shen, J.; Vil, M.D.; Jimenez, X.; Zhang, H.; Lacolina, M.; Mangalampalli, V.; Balderes, P.; Ludwig, D.L.; Zhu, Z. Single variable domain antibody as a versatile building block for the construction of IgG-like bispecific antibodies. J. Immunol. Methods 2007, 318, 65–74. [Google Scholar] [CrossRef]
- Asano, R.; Watanabe, Y.; Kawaguchi, H.; Fukuzawa, H.; Nakanishi, T.; Umetzu, M.; Hayashi, H.; Katayose, Y.; Unno, M.; Kudo, T.; et al. Highly effective recombinant format of a humanized IgG-like bispecific antibody for cancer immunotherapy with retargeting of lymphocytes to tumor cells. J. Biol. Chem. 2007, 282, 27659–27665. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R. R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; McBride, W.J.; Sharkey, R.M.; Chang, C.H. Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting. Proc. Natl. Acad. Sci. USA 2006, 103, 6841–6846. [Google Scholar]
- Chang, C.H.; Rossi, E.A.; Goldenberg, D.M. The dock and lock method: A novel platform technology for building multivalent, multifunctional structures of defined composition with retained bioactivity. Clin. Cancer Res. 2007, 13, 5586s–5591s. [Google Scholar] [CrossRef]
- Rossi, E.A.; Goldenberg, D.M.; Chang, C.H. Complex and defined biostructures with the dock-and-lock method. Trends Pharmacol. Sci. 2012, 33, 474–481. [Google Scholar] [CrossRef]
- Rossi, E.A.; Goldenberg, D.M.; Chang, C.H. The Dock-and-Lock method combines recombinant engineering with site-specific covalent conjugation to generate multifunctional structures. Bioconjug. Chem. 2012, 23, 309–323. [Google Scholar]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; Stein, R.; Chang, C.H. Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: 1. Properties of anti-CD20/CD22 antibodies in lymphoma. Blood 2009, 113, 6161–6171. [Google Scholar] [CrossRef]
- Gupta, P.; Goldenberg, D.M.; Rossi, E.A.; Chang, C.H. Multiple signaling pathways induced by hexavalent, monospecific, anti-CD20 and hexavalent, bispeciifc, anti-CD20/CD22 humanized antibodies correlate with enhanced cytotoxicity to B-cell lymphomas and leukemia. Blood 2010, 116, 3258–3267. [Google Scholar] [CrossRef]
- Gupta, P.; Goldenberg, D.M.; Rossi, E.A.; Cardillo, T.M.; Byrd, J.C.; Muthusamy, N.; Furman, R.R.; Chang, C.H. Dual-targeting immunotherapy of lymphoma: Potent cytotoxicity of anti-CD20/CD74 bispecific antibodies in mantle cell and other lymphomas. Blood 2012, 119, 3767–3778. [Google Scholar] [CrossRef]
- Chang, C.H.; Rossi, E.A.; Sharkey, R.M.; Goldenberg, D.M. The Dock-and-Lock (DNL) approach to novel bispecific antibodies. In Bispecific Antibodies; Kontermann, R.E., Ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2011; Chapter 12; pp. 199–216. [Google Scholar]
- Rossi, E.A.; Chang, C.H.; Cardillo, T.M.; Goldenberg, D.M. Optimization of multivalent bispecific antibodies and immunocytokines with improved in vivo properties. Bioconjug. Chem. 2013, 24, 63–71. [Google Scholar]
- Baillie, G.S.; Scott, J.D.; Houslay, M.D. Compartmentalisation of phosphodiesterases and protein kinase A: opposites attract. FEBS Lett. 2005, 579, 3264–3270. [Google Scholar]
- Wong, W.; Scott, J.D. AKAP signaling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Newlon, M.G.; Roy, M.; Morikis, D.; Hausken, Z.E.; Coghlan, V.; Scott, J.D.; Jennings, P.A. The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat. Struct. Biol. 1999, 6, 222–227. [Google Scholar] [CrossRef]
- Carr, D.W.; Stofko-Hahn, R.E.; Fraser, I.D.; Bishop, S.M.; Acott, T.S.; Brennan, R.G.; Scott, J.D. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 1991, 266, 14188–14192. [Google Scholar]
- Colledge, M.; Scott, J.D. AKAPs: From structure to function. Trends Cell Biol. 1999, 9, 216–222. [Google Scholar] [CrossRef]
- Newlon, M.G.; Roy, M.; Morikis, D.; Carr, D.W.; Westphal, R.R.; Scott, J.D.; Jennings, P.A. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001, 20, 1651–1662. [Google Scholar] [CrossRef]
- Rossi, E.A.; Sharkey, R.M.; McBride, W.; Karacay, H.; Zeng, L.; Hansen, H.J.; Goldenberg, D.M.; Chang, C.H. Development of new multivalent-bispecific agents for pretargeting tumor localization and therapy. Clin. Cancer Res. 2003, 9, 3886s–3896s. [Google Scholar]
- Rossi, E.A.; Chang, C.H.; Losman, M.J.; Sharkey, R.M.; Karacay, H.; McBride, W.; Cardillo, T.M.; Hansen, H.J; Qu, Z.; Horak, I.D.; et al. Pretargeting of carcinoembryonic antigen-expressing cancers with a trivalent bispecific fusion protein produced in myeloma cells. Clin. Cancer Res. 2005, 11, 7122s–7129s. [Google Scholar]
- Sharkey, R.M.; Cardillo, T.M.; Rossi, E.A.; Chang, C.H.; Karacay, H.; McBride, W.J.; Hansen, H.J.; Horak, I.D.; Goldenberg, D.M. Signal amplification in molecular imaging by pretargeting a multivalent, bispecific antibody. Nat. Med. 2005, 11, 1250–1255. [Google Scholar] [CrossRef]
- Alto, N.M.; Soderling, S.H.; Hoshi, N.; Langeberg, L.K.; Fayos, R.; Jennings, P.A.; Scott, J.D. Bioinformatic design of A-kinase anchoring protein-in silico: A potent and selective peptide antagonist of type II protein kinase A anchoring. Proc. Natl. Acad. Sci. USA 2003, 100, 4445–4450. [Google Scholar]
- Burns-Hamuro, L.L.; Ma, Y.; Kammerer, S.; Reineke, U.; Self, C.; Cook, C.; Olson, G.L.; Cantor, C.R.; Braun, A.; Taylor, S.S. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc. Natl. Acad. Sci. USA 2003, 100, 4072–4077. [Google Scholar] [CrossRef]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; Stein, R.; Chang, C.H. CD20-targeted tetrameric interferon-α, a novel and potent immunocytokine for the therapy of B-cell lymphomas. Blood 2009, 114, 3864–3871. [Google Scholar] [CrossRef]
- Binz, H.K.; Pluckthun, A. Engineered proteins as specific binding reagents. Curr. Opin. Biotechnol. 2005, 16, 459–469. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Pluckthun, A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005, 23, 1257–1268. [Google Scholar]
- Hey, T.; Fiedler, E.; Rudolph, R.; Fiedler, M. Artificial, non-antibody binding proteins for pharmaceutical and industrial applications. Trends Biotechnol. 2005, 23, 514–522. [Google Scholar] [CrossRef]
- Hosse, R.J.; Rothe, A.; Power, B.E. A new generation of protein display scaffold for molecular recognition. Protein Sci. 2006, 15, 14–27. [Google Scholar] [CrossRef]
- Chang, C.H.; Rossi, E.A.; Cardillo, T.M.; Nordstrom, D.L.; McBride, W.J.; Goldenberg, D.M. A new method to produce monoPEGylated dimeric cytokines shown with human interferon-α2b. Bioconjug. Chem. 2009, 20, 1899–1907. [Google Scholar]
- Rossi, E.A.; Goldenberg, D.M.; Cardillo, T.M.; Stein, R.; Wang, Y.; Chang, C.H. Novel designs of multivalent anti-CD20 humanized antibodies as improved lymphoma therapeutics. Cancer Res. 2008, 68, 8384–8392. [Google Scholar] [CrossRef]
- Chang, C.H.; Wang, Y.; Trisal, P.; Li, R.; Rossi, D.L.; Nair, A.; Gupta, P.; Losman, M.; Cardillo, T.M.; Rossi, E.A.; et al. Evaluation of a novel hexavalent humanized anti-IGF-1R antibody and its bivalent parental IgG in diverse cancer cell lines. PLoS One 2012, 7, e44235. [Google Scholar]
- Stein, R.; Qu, Z.; Chen, S.; Rosario, A.; Shi, V.; Hayes, M.; Horak, I.D.; Hansen, H.J.; Goldenberg, D.M. Characterization of a new humanized anti-CD20 monoclonal antibody, IMMU-106, and Its use in combination with the humanized anti-CD22 antibody, epratuzumab, for the therapy of non-Hodgkin's lymphoma. Clin. Cancer Res. 2004, 10, 2868–2878. [Google Scholar] [CrossRef]
- Gold, D.V.; Goldenberg, D.M.; Karacay, H.; Rossi, E.A.; Chang, C.H.; Cardillo, T.M.; McBride, W.J.; Sharkey, R.M. A novel bispecific, trivalent antibody construct for targeting pancreatic carcinoma. Cancer Res. 2008, 68, 4819–4826. [Google Scholar] [CrossRef]
- Sharkey, R.M.; van Rij, C.M.; Karacay, H.; Rossi, E.A.; Frielink, C.; Regino, C.; Cardillo, T.M.; McBride, W.J.; Chang, C.H.; et al. A new tri-Fab bispecific antibody for pretargeting Trop-2-expressing epithelial cancers. J. Nucl. Med. 2012, 53, 1625–1632. [Google Scholar] [CrossRef]
- Rossi, E.A.; Rossi, D.L.; Cardillo, T.M.; Stein, R.; Goldenberg, D.M.; Chang, C.H. Preclinical studies on targeted delivery of multiple interferon-alpha-2b to HLA-DR in diverse hematological cancers. Blood 2011, 118, 1877–1884. [Google Scholar] [CrossRef]
- Rossi, E.A.; Rossi, D.L.; Stein, R.; Goldenberg, D.M.; Chang, C.H. A bispecific antibody-IFNα2b immunocytokine targeting CD20 and HLA-DR is highly toxic to human lymphoma and multiple myeloma cells. Cancer Res. 2010, 70, 7600–7609. [Google Scholar] [CrossRef]
- Chang, C.H.; Hinkula, J.; Loo, M.; Falkeborn, T.; Li, R.; Cardillo, T.M.; Rossi, E.A.; Goldenberg, D.M.; Wahren, B. A novel class of anti-HIV agents with multiple copies of enfuvirtide enhances inhibition of viral replication and cellular transmission in vitro. PLoS One 2012, 7, e41235. [Google Scholar] [CrossRef]
- Blanco-Toribio, A.; Sainz-Pastor, N.; Álvarez-Cienfuegos, A.; Merino, N.; Cuesta, A.M.; Sánchez-Martín, D.; Bonet, J.; Santos-Valle, P.; Sanz, L.; Oliva, B.; et al. Generation and characterization of monospecific and bispecific hexavalanet trimerbodies. mAbs 2013, 5, 70–79. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chang, C.-H.; Rossi, E.A.; Wang, Y.; Cardillo, T.M.; Goldenberg, D.M. The Development of Bispecific Hexavalent Antibodies as a Novel Class of DOCK-AND-LOCKTM (DNLTM) Complexes. Antibodies 2013, 2, 353-370. https://doi.org/10.3390/antib2020353
Chang C-H, Rossi EA, Wang Y, Cardillo TM, Goldenberg DM. The Development of Bispecific Hexavalent Antibodies as a Novel Class of DOCK-AND-LOCKTM (DNLTM) Complexes. Antibodies. 2013; 2(2):353-370. https://doi.org/10.3390/antib2020353
Chicago/Turabian StyleChang, Chien-Hsing, Edmund A. Rossi, Yang Wang, Thomas M. Cardillo, and David M. Goldenberg. 2013. "The Development of Bispecific Hexavalent Antibodies as a Novel Class of DOCK-AND-LOCKTM (DNLTM) Complexes" Antibodies 2, no. 2: 353-370. https://doi.org/10.3390/antib2020353