Selective Induction of Cancer Cell Death by Targeted Granzyme B

Abstract
:1. Introduction
2. Granzyme B Fusion Proteins for Targeted Cancer Therapy
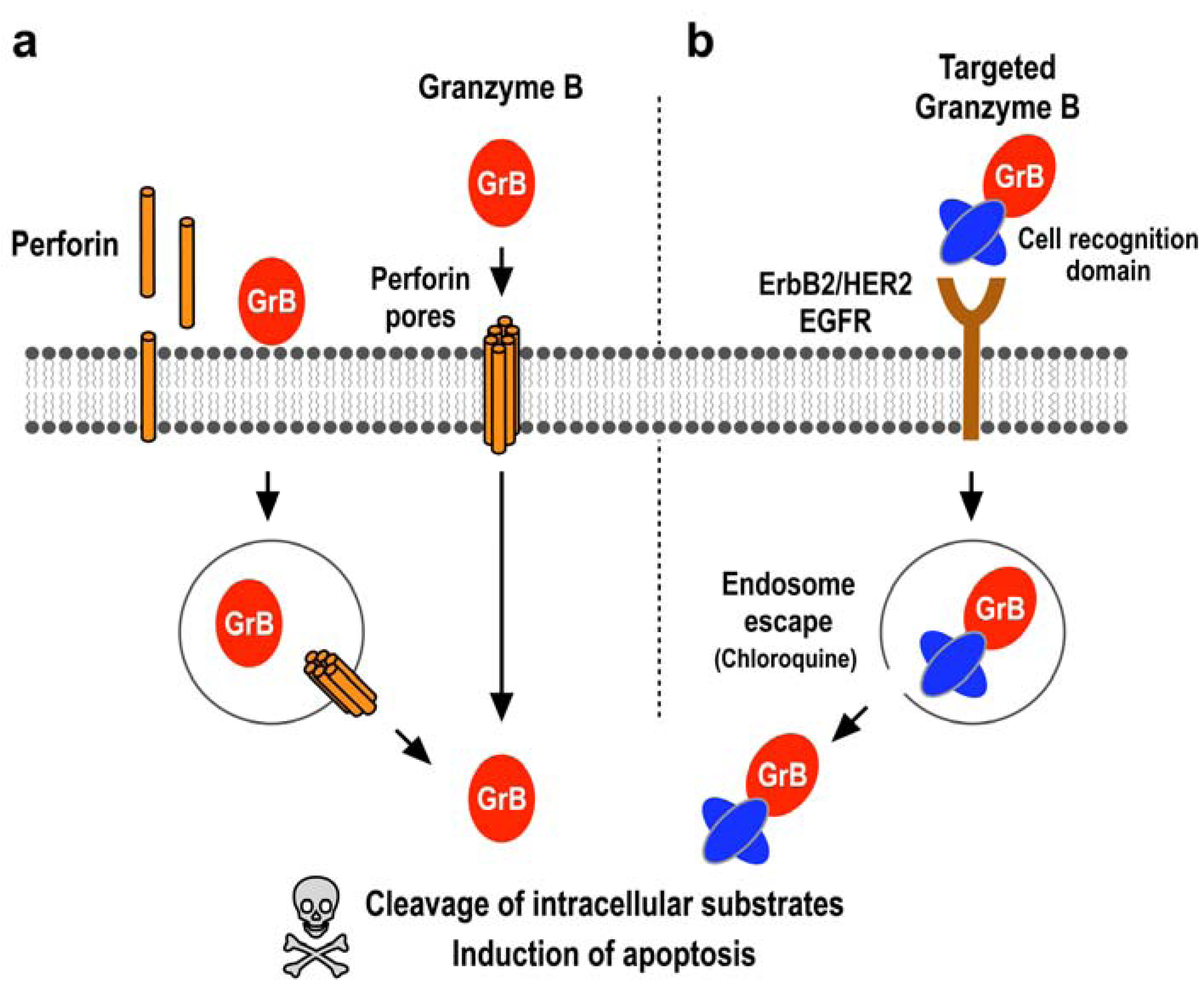
2.1. Induction of Programmed Cell Death by the Serine Protease Granzyme B
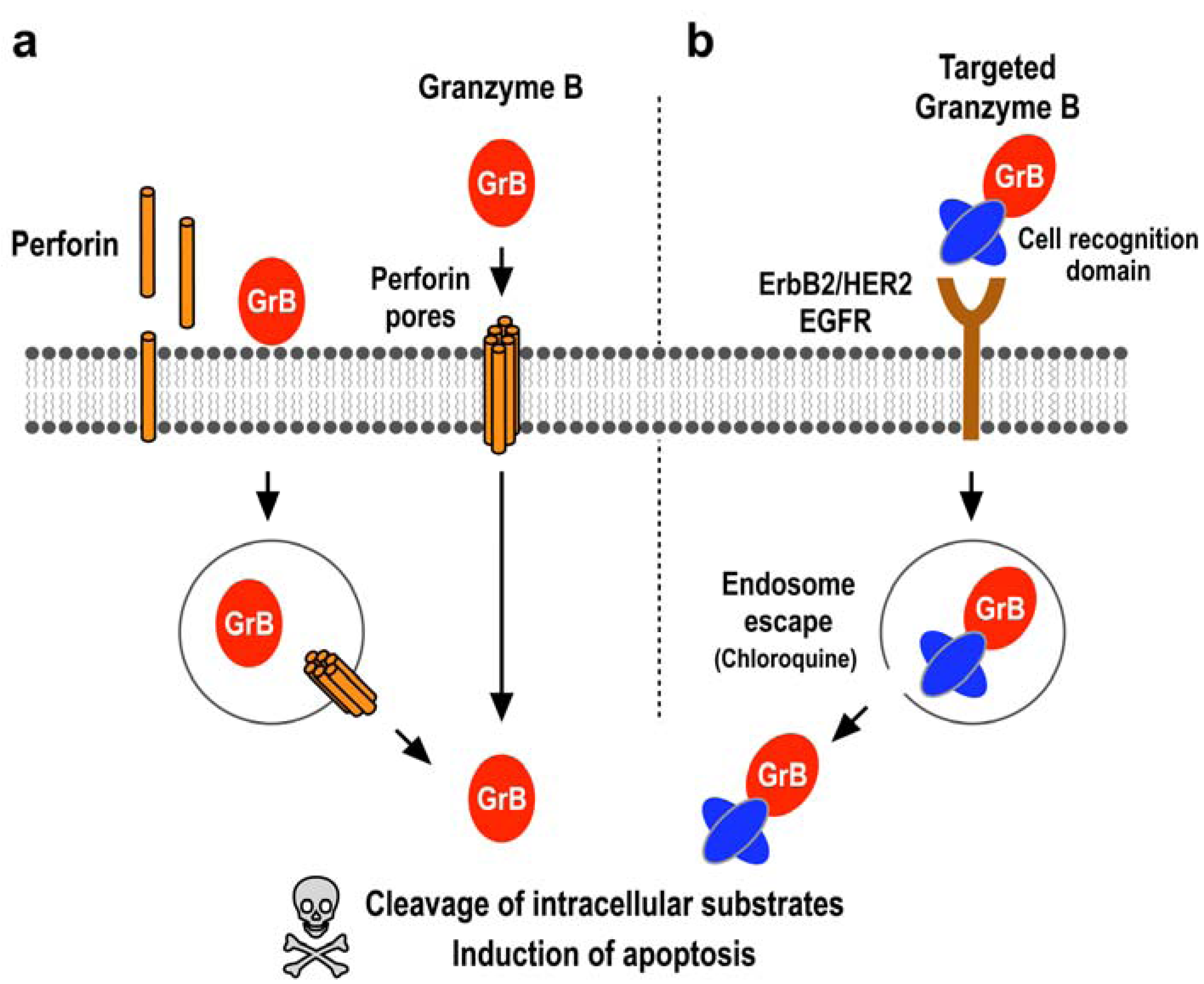
2.2. Expression Systems for Production of Granzyme B in Recombinant Form
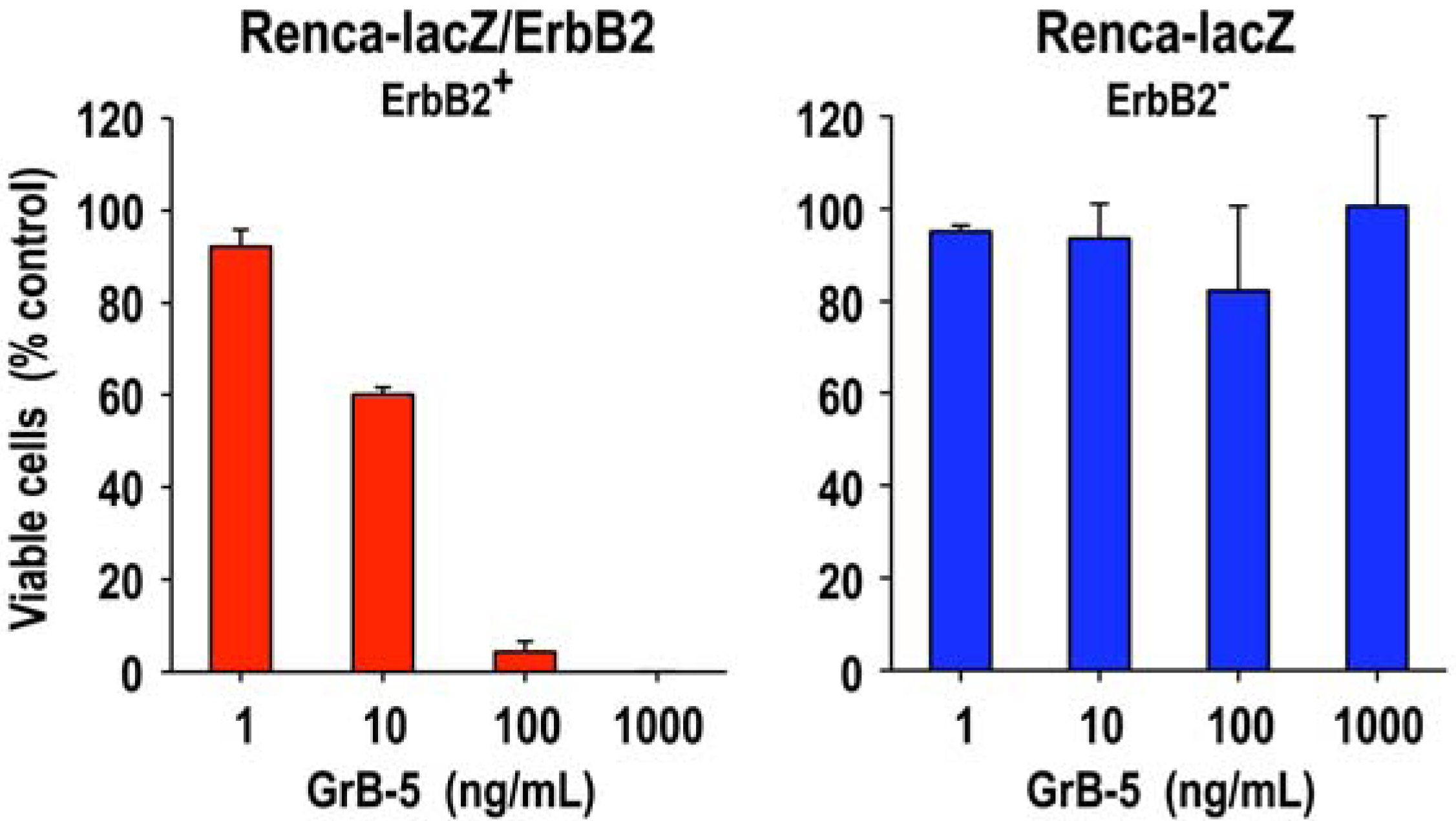
2.3. Tumor Cell-Specific Granzyme B Fusion Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor cell line | |||||
|---|---|---|---|---|---|
| Reagent a | Specificity | MDA-MB468
EGFR+, ErbB2− | A431 b EGFR+, ErbB2+ | Renca-lacZ/ErbB2
EGFR−, ErbB2+ | |
| GrB-5 | ErbB2/HER2 | no killing at 14.5 nM | 5.8 nM | 0.29 nM | |
| 5-ETA | ErbB2/HER2 | no killing at 15 nM | 0.5 nM | 0.09 nM | |
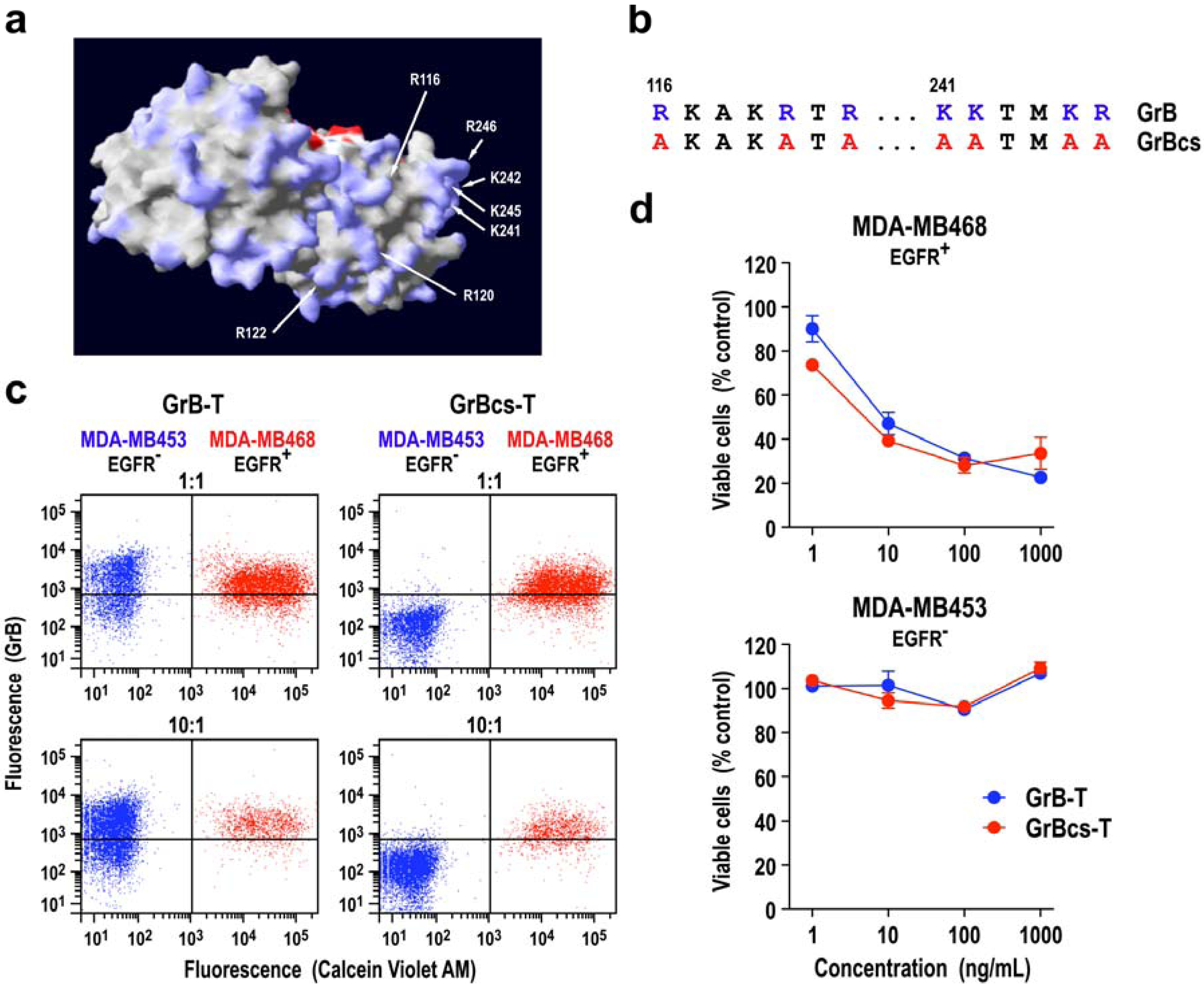
| GrB-T | EGFR | 0.25 nM | 3.5 nM | no killing at 25 nM | |
| T-ETA | EGFR | 0.06 nM | 0.02 nM | n.d.c | |
3. Opportunities and Challenges for Further Development of Targeted Granzyme B
3.1. Target Cell Specificity of Granzyme B Fusion Proteins
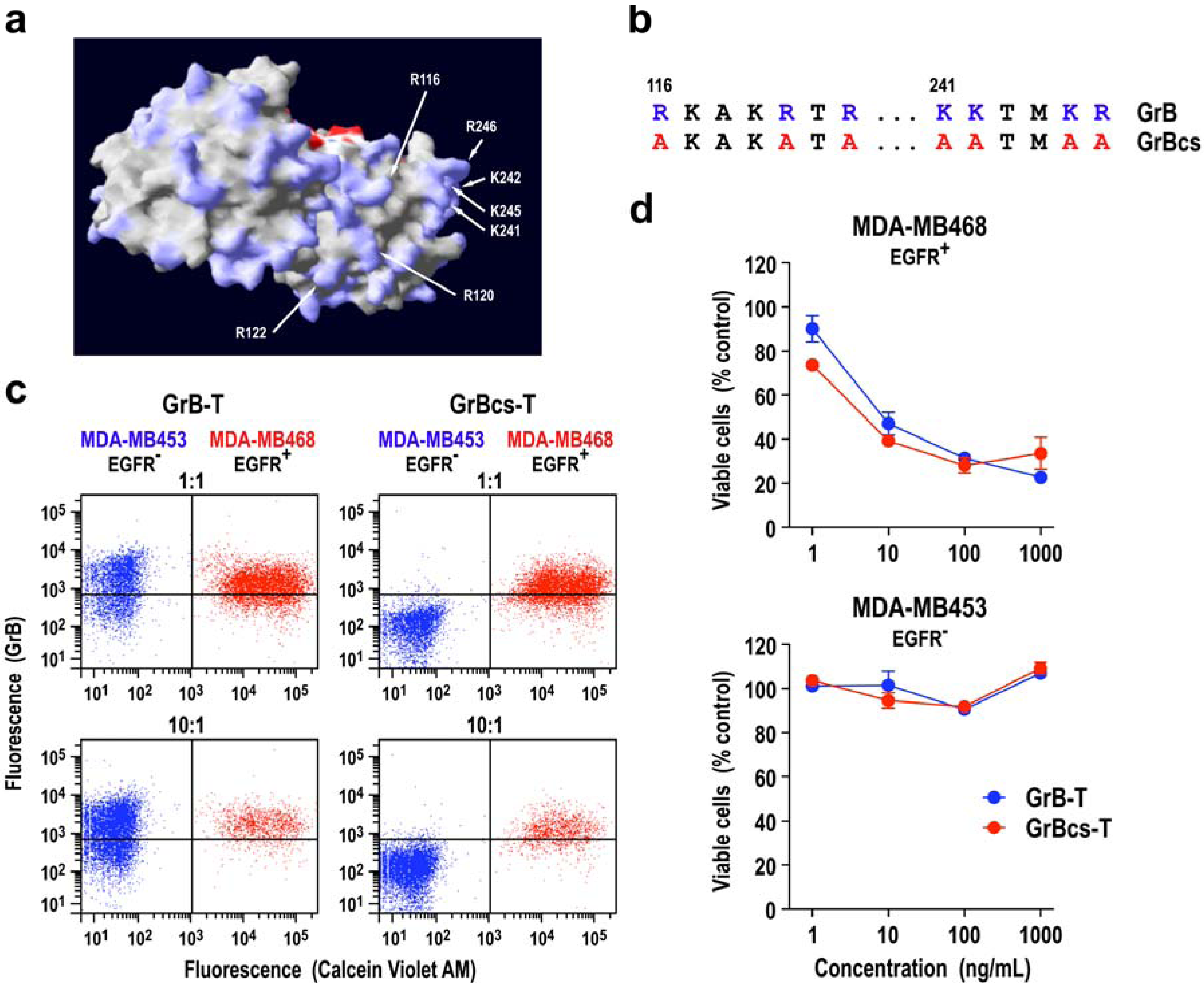
3.2. Extracellular Activity of Granzyme B
3.3. Granzyme B Resistance of Tumor Cells
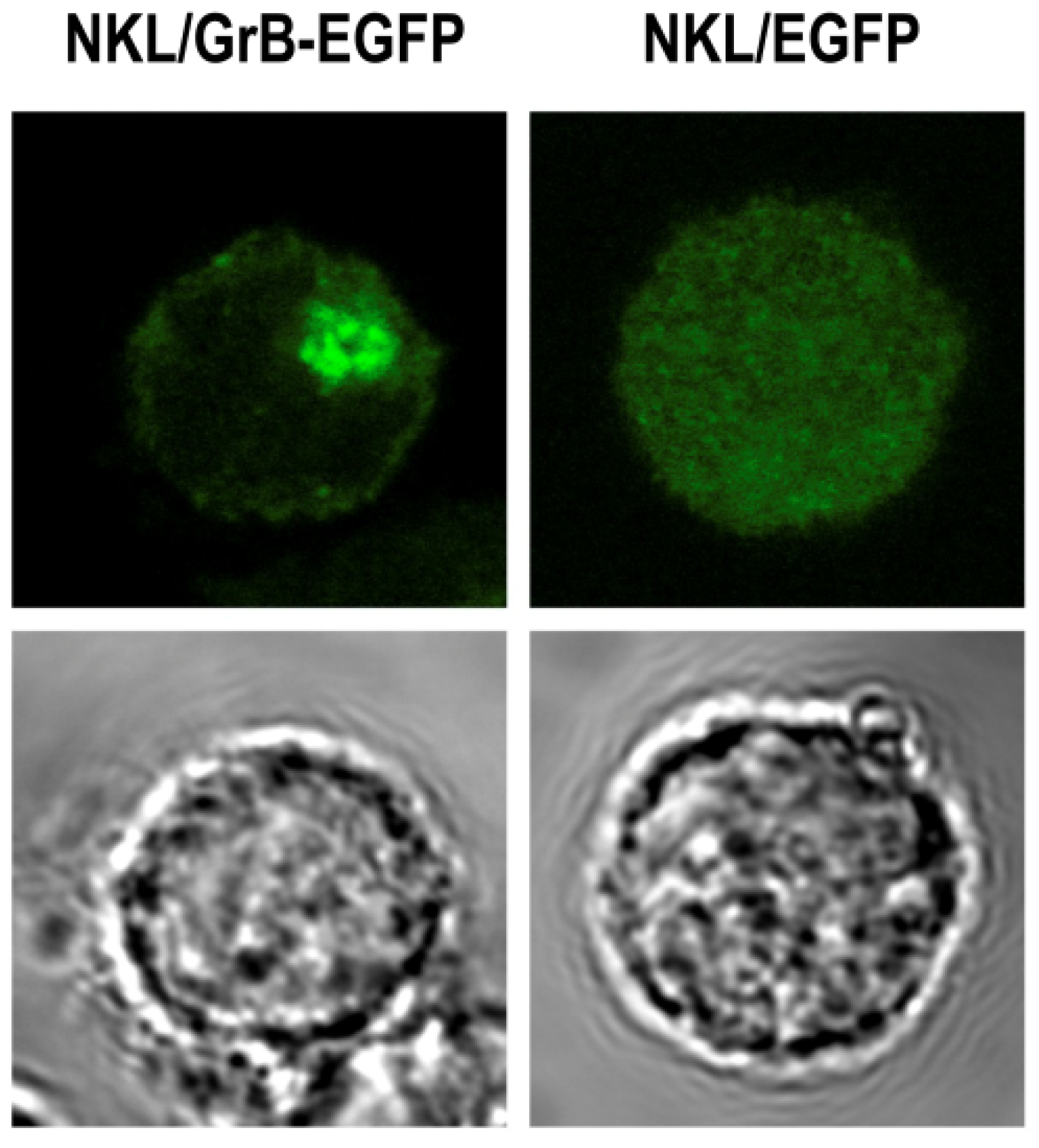
3.4. Intracellular Routing and Cytosolic Delivery of Granzyme B
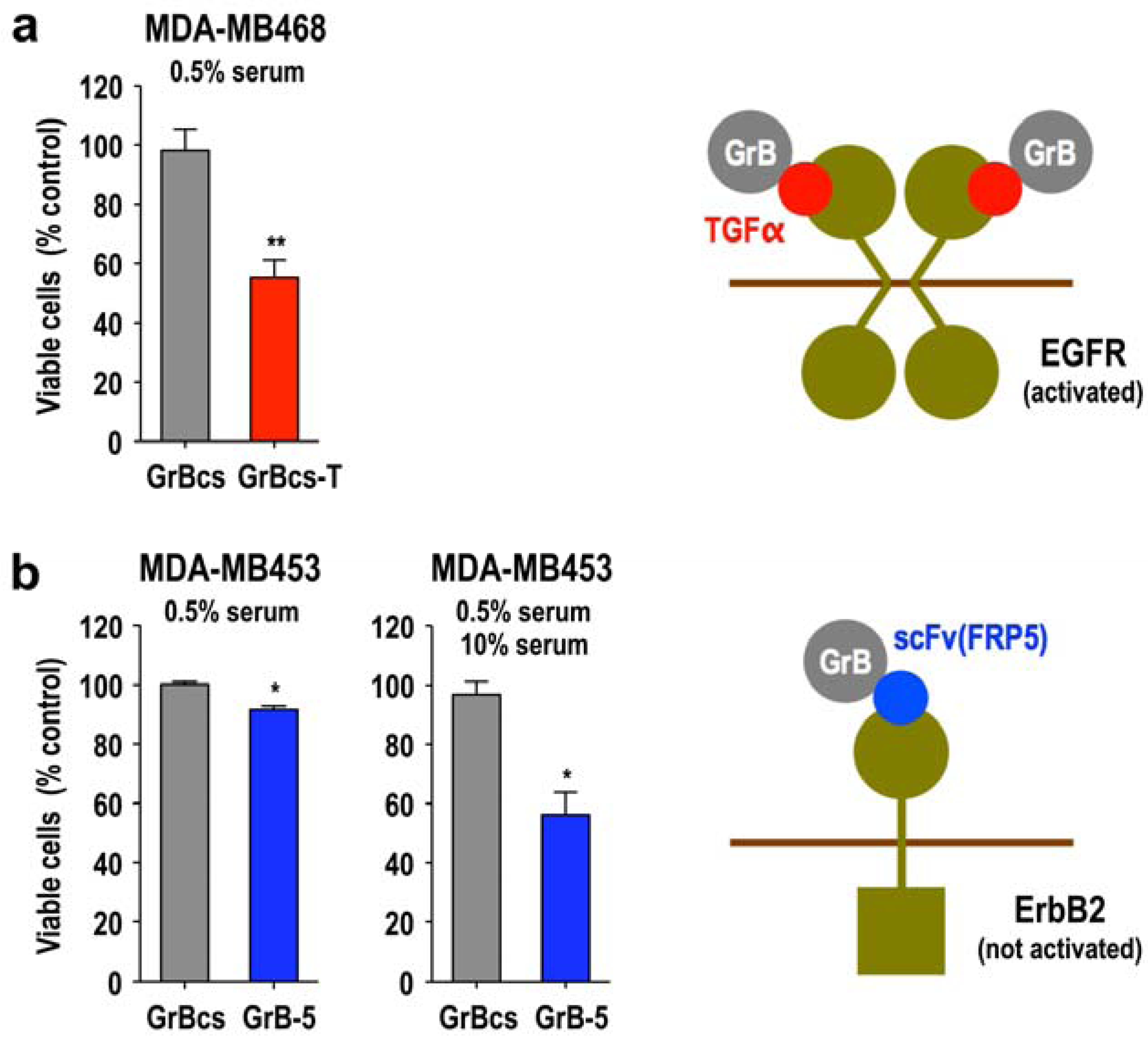
3.5. Activity of Granzyme Fusion Proteins against Resting Cancer Cells
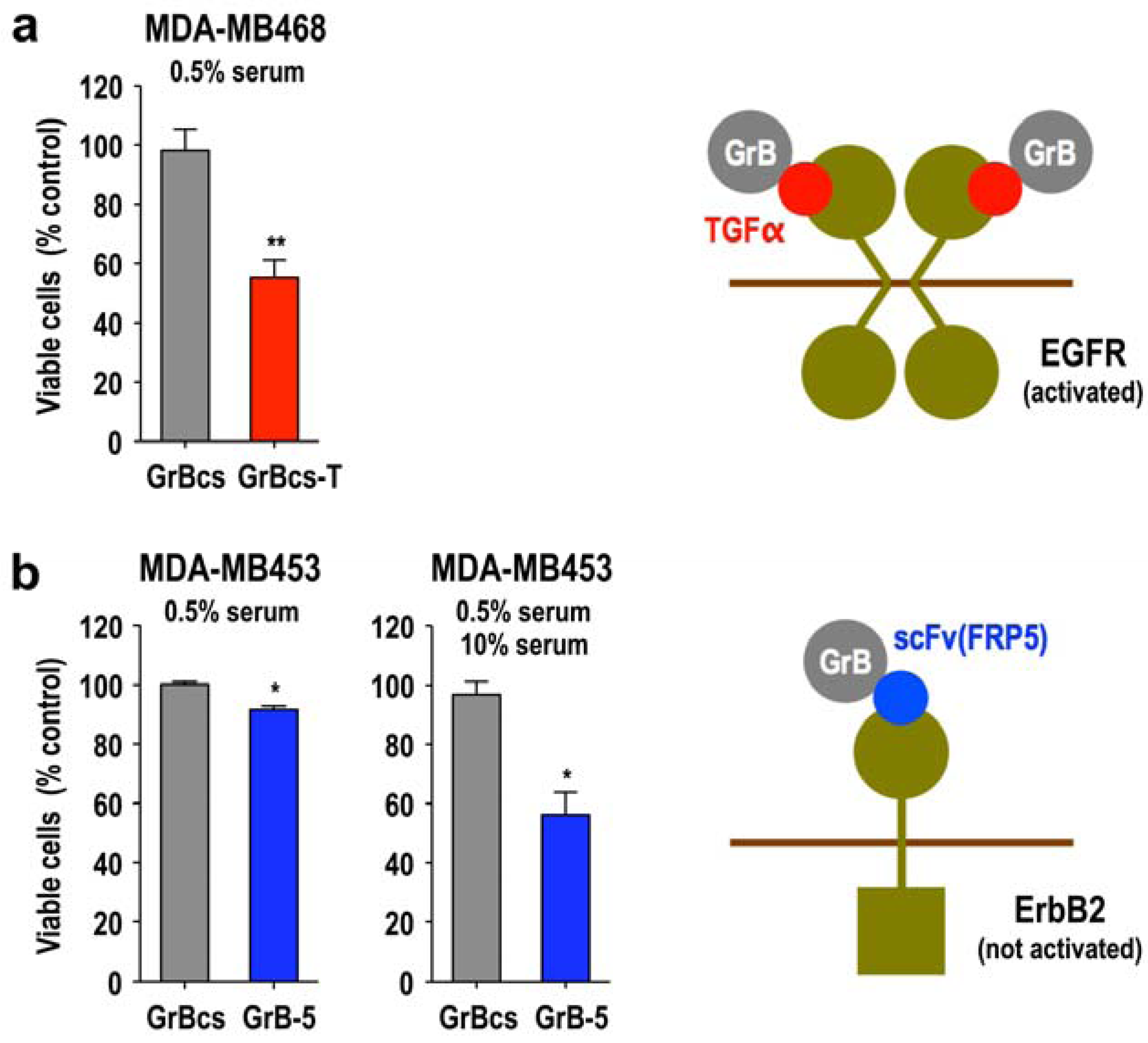
3.6. Local Delivery of Targeted Granzyme B by Genetically Modified Lymphocytes
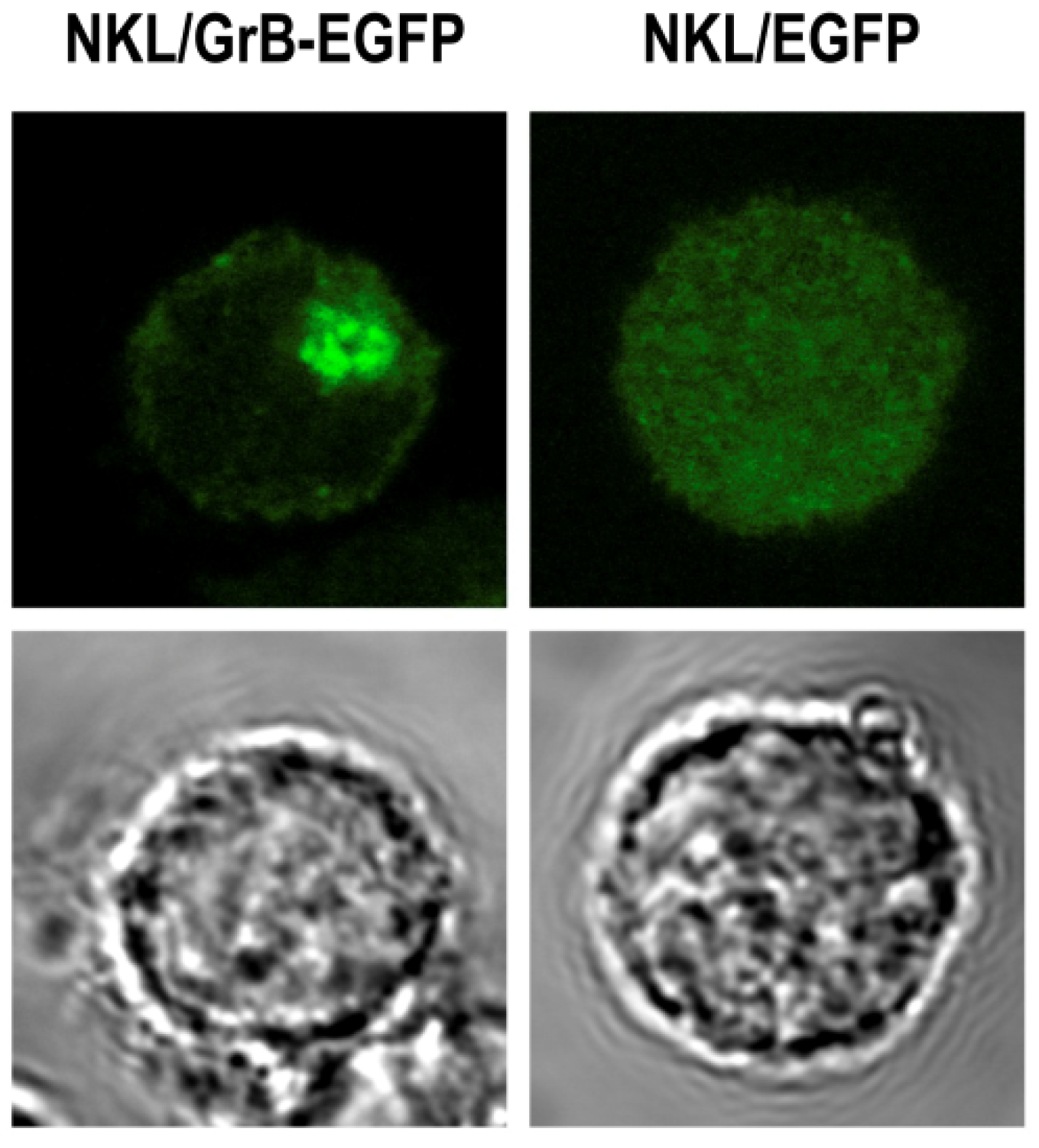
4. Conclusions
Acknowledgments
References and Notes
- Maloney, D.G. Immunotherapy for non-Hodgkin's lymphoma: Monoclonal antibodies and vaccines. J. Clin. Oncol. 2005, 23, 6421–6428. [Google Scholar] [CrossRef]
- Groner, B.; Hartmann, C.; Wels, W. Therapeutic antibodies. Curr. Mol. Med. 2004, 4, 539–547. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Lan, K.H.; Lu, C.H.; Yu, D. Mechanisms of trastuzumab resistance and their clinical implications. Ann. NY Acad. Sci. 2005, 1059, 70–75. [Google Scholar] [CrossRef]
- Nahta, R.; Esteva, F.J. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006, 8, 215. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Wels, W.; Biburger, M.; Müller, T.; Dälken, B.; Giesübel, U.; Tonn, T.; Uherek, C. Recombinant immunotoxins and retargeted killer cells: employing engineered antibody fragments for tumor-specific targeting of cytotoxic effectors. Cancer Immunol. Immunother. 2004, 53, 217–226. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jäger, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005, 7, R617–R626. [Google Scholar]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef]
- Kelly, R.J.; Sharon, E.; Pastan, I.; Hassan, R. Mesothelin-targeted agents in clinical trials and in preclinical development. Mol. Cancer Ther. 2012, 11, 517–525. [Google Scholar] [CrossRef]
- Pai, L.H.; Wittes, R.; Setser, A.; Willingham, M.C.; Pastan, I. Treatment of advanced solid tumors with immunotoxin LMB-1: An antibody linked to Pseudomonas exotoxin. Nat. Med. 1996, 2, 350–353. [Google Scholar] [CrossRef]
- Azemar, M.; Djahansouzi, S.; Jäger, E.; Solbach, C.; Schmidt, M.; Maurer, A.B.; Mross, K.; Unger, C.; von Minckwitz, G.; Dall, P.; et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res. Treat. 2003, 82, 155–164. [Google Scholar]
- Keppler-Hafkemeyer, A.; Kreitman, R.J.; Pastan, I. Apoptosis induced by immunotoxins used in the treatment of hematologic malignancies. Int. J. Cancer 2000, 87, 86–94. [Google Scholar] [CrossRef]
- Schmidt, M.; McWatters, A.; White, R.A.; Groner, B.; Wels, W.; Fan, Z.; Bast, R.C., Jr. Synergistic interaction between an anti-p185HER-2 pseudomonas exotoxin fusion protein [scFv(FRP5)-ETA] and ionizing radiation for inhibiting growth of ovarian cancer cells that overexpress HER-2. Gynecol. Oncol. 2001, 80, 145–155. [Google Scholar] [CrossRef]
- Weidle, U.H.; Georges, G.; Brinkmann, U. Fully human targeted cytotoxic fusion proteins: New anticancer agents on the horizon. Cancer Genomics Proteomics 2012, 9, 119–133. [Google Scholar]
- Gerspach, J.; Wajant, H.; Pfizenmaier, K. Death ligands designed to kill: development and application of targeted cancer therapeutics based on proapoptotic TNF family ligands. Results Probl. Cell Differ. 2009, 49, 241–273. [Google Scholar] [CrossRef]
- Antignani, A.; Youle, R.J. A chimeric protein induces tumor cell apoptosis by delivering the human Bcl-2 family BH3-only protein Bad. Biochemistry 2005, 44, 4074–4082. [Google Scholar] [CrossRef]
- Lorberboum-Galski, H. Human toxin-based recombinant immunotoxins/chimeric proteins as a drug delivery system for targeted treatment of human diseases. Expert Opin. Drug Deliv. 2011, 8, 605–621. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Mahmud, H.; Dälken, B.; Wels, W.S. Induction of programmed cell death in ErbB2/HER2-expressing cancer cells by targeted delivery of apoptosis-inducing factor. Mol. Cancer Ther. 2009, 8, 1526–1535. [Google Scholar] [CrossRef]
- Liu, Y.; Cheung, L.H.; Thorpe, P.; Rosenblum, M.G. Mechanistic studies of a novel human fusion toxin composed of vascular endothelial growth factor (VEGF)121 and the serine protease granzyme B: Directed apoptotic events in vascular endothelial cells. Mol. Cancer Ther. 2003, 2, 949–959. [Google Scholar]
- Dälken, B.; Giesübel, U.; Knauer, S.K.; Wels, W.S. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006, 13, 576–585. [Google Scholar] [CrossRef]
- Rosenblum, M.G.; Barth, S. Development of novel, highly cytotoxic fusion constructs containing granzyme B: unique mechanisms and functions. Curr. Pharm. Des. 2009, 15, 2676–2692. [Google Scholar] [CrossRef]
- Kurschus, F.C.; Jenne, D.E. Delivery and therapeutic potential of human granzyme B. Immunol. Rev. 2010, 235, 159–171. [Google Scholar]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in cancer and immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef]
- Pham, C.T.; Ley, T.J. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 8627–8632. [Google Scholar] [CrossRef]
- Chowdhury, D.; Lieberman, J. Death by a thousand cuts: Granzyme pathways of programmed cell death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef]
- Bromley, S.K.; Burack, W.R.; Johnson, K.G.; Somersalo, K.; Sims, T.N.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse. Annu. Rev. Immunol. 2001, 19, 375–396. [Google Scholar] [CrossRef]
- Huse, M.; Quann, E.J.; Davis, M.M. Shouts, whispers and the kiss of death: Directional secretion in T cells. Nat. Immunol. 2008, 9, 1105–1111. [Google Scholar] [CrossRef]
- Afonina, I.S.; Cullen, S.P.; Martin, S.J. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev. 2010, 235, 105–116. [Google Scholar]
- Baran, K.; Dunstone, M.; Chia, J.; Ciccone, A.; Browne, K.A.; Clarke, C.J.; Lukoyanova, N.; Saibil, H.; Whisstock, J.C.; Voskoboinik, I.; et al. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity 2009, 30, 684–695. [Google Scholar] [CrossRef]
- Law, R.H.; Lukoyanova, N.; Voskoboinik, I.; Caradoc-Davies, T.T.; Baran, K.; Dunstone, M.A.; D'Angelo, M.E.; Orlova, E.V.; Coulibaly, F.; Verschoor, S.; et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 2010, 468, 447–451. [Google Scholar]
- Thiery, J.; Keefe, D.; Saffarian, S.; Martinvalet, D.; Walch, M.; Boucrot, E.; Kirchhausen, T.; Lieberman, J. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 2010, 115, 1582–1593. [Google Scholar]
- Thiery, J.; Keefe, D.; Boulant, S.; Boucrot, E.; Walch, M.; Martinvalet, D.; Goping, I.S.; Bleackley, R.C.; Kirchhausen, T.; Lieberman, J. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 2011, 12, 770–777. [Google Scholar]
- Gross, C.; Koelch, W.; DeMaio, A.; Arispe, N.; Multhoff, G. Cell surface-bound heat shock protein 70 (Hsp70) mediates perforin-independent apoptosis by specific binding and uptake of granzyme B. J. Biol. Chem. 2003, 278, 41173–41181. [Google Scholar]
- Gehrmann, M.; Stangl, S.; Kirschner, A.; Foulds, G.A.; Sievert, W.; Doss, B.T.; Walch, A.; Pockley, A.G.; Multhoff, G. Immunotherapeutic targeting of membrane Hsp70-expressing tumors using recombinant human granzyme B. PLoS One 2012, 7, e41341. [Google Scholar]
- Jabulowsky, R.A.; Oberoi, P.; Bähr-Mahmud, H.; Dälken, B.; Wels, W.S. Surface charge-modification prevents sequestration and enhances tumor-cell specificity of a recombinant granzyme B-TGFalpha fusion protein. Bioconjug. Chem. 2012, 23, 1567–1576. [Google Scholar] [CrossRef]
- Boivin, W.A.; Cooper, D.M.; Hiebert, P.R.; Granville, D.J. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab. Invest. 2009, 89, 1195–1220. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar]
- Adrain, C.; Murphy, B.M.; Martin, S.J. Molecular ordering of the caspase activation cascade initiated by the cytotoxic T lymphocyte/natural killer (CTL/NK) protease granzyme B. J. Biol. Chem. 2005, 280, 4663–4673. [Google Scholar] [CrossRef]
- Barry, M.; Heibein, J.A.; Pinkoski, M.J.; Lee, S.F.; Moyer, R.W.; Green, D.R.; Bleackley, R.C. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol. Cell. Biol. 2000, 20, 3781–3794. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sedelies, K.A.; Browne, K.A.; Wowk, M.E.; Newbold, A.; Sutton, V.R.; Clarke, C.J.; Oliaro, J.; Lindemann, R.K.; Bird, P.I.; et al. A central role for Bid in granzyme B-induced apoptosis. J. Biol. Chem. 2005, 280, 4476–4482. [Google Scholar]
- Thomas, D.A.; Du, C.; Xu, M.; Wang, X.; Ley, T.J. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 2000, 12, 621–632. [Google Scholar] [CrossRef]
- Sharif-Askari, E.; Alam, A.; Rheaume, E.; Beresford, P.J.; Scotto, C.; Sharma, K.; Lee, D.; DeWolf, W.E.; Nuttall, M.E.; Lieberman, J.; et al. Direct cleavage of the human DNA fragmentation factor-45 by granzyme B induces caspase-activated DNase release and DNA fragmentation. EMBO J. 2001, 20, 3101–3113. [Google Scholar]
- Adrain, C.; Duriez, P.J.; Brumatti, G.; Delivani, P.; Martin, S.J. The cytotoxic lymphocyte protease, granzyme B, targets the cytoskeleton and perturbs microtubule polymerization dynamics. J. Biol. Chem. 2006, 281, 8118–8125. [Google Scholar]
- Zhang, D.; Beresford, P.J.; Greenberg, A.H.; Lieberman, J. Granzymes A and B directly cleave lamins and disrupt the nuclear lamina during granule-mediated cytolysis. Proc. Natl. Acad. Sci. USA 2001, 98, 5746–5751. [Google Scholar]
- Froelich, C.J.; Hanna, W.L.; Poirier, G.G.; Duriez, P.J.; D'Amours, D.; Salvesen, G.S.; Alnemri, E.S.; Earnshaw, W.C.; Shah, G.M. Granzyme B/perforin-mediated apoptosis of Jurkat cells results in cleavage of poly(ADP-ribose) polymerase to the 89-kDa apoptotic fragment and less abundant 64-kDa fragment. Biochem. Biophys. Res. Commun. 1996, 227, 658–665. [Google Scholar]
- Giesübel, U.; Dälken, B.; Mahmud, H.; Wels, W.S. Cell binding, internalization and cytotoxic activity of human granzyme B expressed in the yeast Pichia pastoris. Biochem. J. 2006, 394, 563–573. [Google Scholar] [CrossRef]
- Liu, Y.; Cheung, L.H.; Hittelman, W.N.; Rosenblum, M.G. Targeted delivery of human pro-apoptotic enzymes to tumor cells: In vitro studies describing a novel class of recombinant highly cytotoxic agents. Mol. Cancer Ther. 2003, 2, 1341–1350. [Google Scholar]
- Kurschus, F.C.; Kleinschmidt, M.; Fellows, E.; Dornmair, K.; Rudolph, R.; Lilie, H.; Jenne, D.E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004, 562, 87–92. [Google Scholar] [CrossRef]
- Lorentsen, R.H.; Fynbo, C.H.; Thogersen, H.C.; Etzerodt, M.; Holtet, T.L. Expression, refolding, and purification of recombinant human granzyme B. Protein Expr. Purif. 2005, 39, 18–26. [Google Scholar] [CrossRef]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef]
- Gehrmann, M.; Doss, B.T.; Wagner, M.; Zettlitz, K.A.; Kontermann, R.E.; Foulds, G.; Pockley, A.G.; Multhoff, G. A novel expression and purification system for the production of enzymatic and biologically active human granzyme B. J. Immunol. Methods 2011, 371, 8–17. [Google Scholar] [CrossRef]
- Cereghino, J.L.; Cregg, J.M. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 2000, 24, 45–66. [Google Scholar] [CrossRef]
- Pham, C.T.; Thomas, D.A.; Mercer, J.D.; Ley, T.J. Production of fully active recombinant murine granzyme B in yeast. J. Biol. Chem. 1998, 273, 1629–1633. [Google Scholar]
- Sun, J.; Bird, C.H.; Buzza, M.S.; McKee, K.E.; Whisstock, J.C.; Bird, P.I. Expression and purification of recombinant human granzyme B from Pichia pastoris. Biochem. Biophys. Res. Commun. 1999, 261, 251–255. [Google Scholar] [CrossRef]
- Dälken, B.; Jabulowsky, R.A.; Oberoi, P.; Benhar, I.; Wels, W.S. Maltose-binding protein enhances secretion of recombinant human granzyme B accompanied by in vivo processing of a precursor MBP fusion protein. PLoS One 2010, 5, e14404. [Google Scholar]
- Liu, Y.; Zhang, W.; Niu, T.; Cheung, L.H.; Munshi, A.; Meyn, R.E.; Rosenblum, M.G. Targeted apoptosis activation with GrB/scFvMEL modulates melanoma growth, metastatic spread, chemosensitivity, and radiosensitivity. Neoplasia 2006, 8, 125–135. [Google Scholar] [CrossRef]
- Wels, W.; Harwerth, I.M.; Mueller, M.; Groner, B.; Hynes, N.E. Selective inhibition of tumor cell growth by a recombinant single-chain antibody-toxin specific for the erbB-2 receptor. Cancer Res. 1992, 52, 6310–6317. [Google Scholar]
- Wels, W.; Beerli, R.; Hellmann, P.; Schmidt, M.; Marte, B.M.; Kornilova, E.S.; Hekele, A.; Mendelsohn, J.; Groner, B.; Hynes, N.E. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int. J. Cancer 1995, 60, 137–144. [Google Scholar] [CrossRef]
- Schmidt, M.; Wels, W. Targeted inhibition of tumour cell growth by a bispecific single-chain toxin containing an antibody domain and TGF alpha. Br. J. Cancer 1996, 74, 853–862. [Google Scholar] [CrossRef]
- Zenke, M.; Steinlein, P.; Wagner, E.; Cotten, M.; Beug, H.; Birnstiel, M.L. Receptor-mediated endocytosis of transferrin-polycation conjugates: an efficient way to introduce DNA into hematopoietic cells. Proc. Natl. Acad. Sci. USA 1990, 87, 3655–3659. [Google Scholar]
- Maurer-Gebhard, M.; Schmidt, M.; Azemar, M.; Altenschmidt, U.; Stöcklin, E.; Wels, W.; Groner, B. Systemic treatment with a recombinant erbB-2 receptor-specific tumor toxin efficiently reduces pulmonary metastases in mice injected with genetically modified carcinoma cells. Cancer Res. 1998, 58, 2661–2666. [Google Scholar]
- Kanatani, I.; Lin, X.; Yuan, X.; Manorek, G.; Shang, X.; Cheung, L.H.; Rosenblum, M.G.; Howell, S.B. Targeting granzyme B to tumor cells using a yoked human chorionic gonadotropin. Cancer Chemother. Pharmacol. 2011, 68, 979–990. [Google Scholar] [CrossRef]
- Bird, C.H.; Sun, J.; Ung, K.; Karambalis, D.; Whisstock, J.C.; Trapani, J.A.; Bird, P.I. Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol. Cell. Biol. 2005, 25, 7854–7867. [Google Scholar] [CrossRef]
- Shi, L.; Keefe, D.; Durand, E.; Feng, H.; Zhang, D.; Lieberman, J. Granzyme B binds to target cells mostly by charge and must be added at the same time as perforin to trigger apoptosis. J. Immunol. 2005, 174, 5456–5461. [Google Scholar]
- Kurschus, F.C.; Fellows, E.; Stegmann, E.; Jenne, D.E. Granzyme B delivery via perforin is restricted by size, but not by heparan sulfate-dependent endocytosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13799–13804. [Google Scholar]
- Metkar, S.S.; Wang, B.; Aguilar-Santelises, M.; Raja, S.M.; Uhlin-Hansen, L.; Podack, E.; Trapani, J.A.; Froelich, C.J. Cytotoxic cell granule-mediated apoptosis: Perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity 2002, 16, 417–428. [Google Scholar] [CrossRef]
- Raja, S.M.; Metkar, S.S.; Honing, S.; Wang, B.; Russin, W.A.; Pipalia, N.H.; Menaa, C.; Belting, M.; Cao, X.; Dressel, R.; et al. A novel mechanism for protein delivery: Granzyme B undergoes electrostatic exchange from serglycin to target cells. J. Biol. Chem. 2005, 280, 20752–20761. [Google Scholar]
- Kurschus, F.C.; Bruno, R.; Fellows, E.; Falk, C.S.; Jenne, D.E. Membrane receptors are not required to deliver granzyme B during killer cell attack. Blood 2005, 105, 2049–2058. [Google Scholar] [CrossRef]
- Estebanez-Perpina, E.; Fuentes-Prior, P.; Belorgey, D.; Braun, M.; Kiefersauer, R.; Maskos, K.; Huber, R.; Rubin, H.; Bode, W. Crystal structure of the caspase activator human granzyme B, a proteinase highly specific for an Asp-P1 residue. Biol. Chem. 2000, 381, 1203–1214. [Google Scholar]
- Buzza, M.S.; Zamurs, L.; Sun, J.; Bird, C.H.; Smith, A.I.; Trapani, J.A.; Froelich, C.J.; Nice, E.C.; Bird, P.I. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J. Biol. Chem. 2005, 280, 23549–23558. [Google Scholar]
- Tak, P.P.; Spaeny-Dekking, L.; Kraan, M.C.; Breedveld, F.C.; Froelich, C.J.; Hack, C.E. The levels of soluble granzyme A and B are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA). Clin. Exp. Immunol. 1999, 116, 366–370. [Google Scholar] [CrossRef]
- Ronday, H.K.; van der Laan, W.H.; Tak, P.P.; de Roos, J.A.; Bank, R.A.; TeKoppele, J.M.; Froelich, C.J.; Hack, C.E.; Hogendoorn, P.C.; Breedveld, F.C.; et al. Human granzyme B mediates cartilage proteoglycan degradation and is expressed at the invasive front of the synovium in rheumatoid arthritis. Rheumatology (Oxford) 2001, 40, 55–61. [Google Scholar] [CrossRef]
- Skjelland, M.; Michelsen, A.E.; Krohg-Sorensen, K.; Tennoe, B.; Dahl, A.; Bakke, S.; Brosstad, F.; Damas, J.K.; Russell, D.; Halvorsen, B.; et al. lasma levels of granzyme B are increased in patients with lipid-rich carotid plaques as determined by echogenicity. Atherosclerosis 2007, 195, e142–146. [Google Scholar] [CrossRef]
- Saito, Y.; Kondo, H.; Hojo, Y. Granzyme B as a novel factor involved in cardiovascular diseases. J. Cardiol. 2011, 57, 141–147. [Google Scholar] [CrossRef]
- Boivin, W.A.; Shackleford, M.; Vanden Hoek, A.; Zhao, H.; Hackett, T.L.; Knight, D.A.; Granville, D.J. Granzyme B cleaves decorin, biglycan and soluble betaglycan, releasing active transforming growth factor-beta1. PLoS One 2012, 7, e33163. [Google Scholar]
- Wang, T.; Lee, M.H.; Choi, E.; Pardo-Villamizar, C.A.; Lee, S.B.; Yang, I.H.; Calabresi, P.A.; Nath, A. Granzyme B-induced neurotoxicity is mediated via activation of PAR-1 receptor and Kv1.3 channel. PLoS One 2012, 7, e43950. [Google Scholar]
- Buzza, M.S.; Bird, P.I. Extracellular granzymes: Current perspectives. Biol. Chem. 2006, 387, 827–837. [Google Scholar]
- Sun, J.; Bird, C.H.; Sutton, V.; McDonald, L.; Coughlin, P.B.; De Jong, T.A.; Trapani, J.A.; Bird, P.I. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J. Biol. Chem. 1996, 271, 27802–27809. [Google Scholar]
- Bird, C.H.; Sutton, V.R.; Sun, J.; Hirst, C.E.; Novak, A.; Kumar, S.; Trapani, J.A.; Bird, P.I. Selective regulation of apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 1998, 18, 6387–6398. [Google Scholar]
- Classen, C.F.; Bird, P.I.; Debatin, K.M. Modulation of the granzyme B inhibitor proteinase inhibitor 9 (PI-9) by activation of lymphocytes and monocytes in vitro and by Epstein-Barr virus and bacterial infection. Clin. Exp. Immunol. 2006, 143, 534–542. [Google Scholar] [CrossRef]
- Hirst, C.E.; Buzza, M.S.; Bird, C.H.; Warren, H.S.; Cameron, P.U.; Zhang, M.; Ashton-Rickardt, P.G.; Bird, P.I. The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J. Immunol. 2003, 170, 805–815. [Google Scholar]
- Bots, M.; de Bruin, E.; Rademaker-Koot, M.T.; Medema, J.P. Proteinase inhibitor-9 expression is induced by maturation in dendritic cells via p38 MAP kinase. Hum. Immunol. 2007, 68, 959–964. [Google Scholar] [CrossRef]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar]
- Rousalova, I.; Krepela, E.; Prochazka, J.; Cermak, J.; Benkova, K. Expression of proteinase inhibitor-9/serpinB9 in non-small cell lung carcinoma cells and tissues. Int. J. Oncol. 2010, 36, 275–283. [Google Scholar]
- ten Berge, R.L.; Meijer, C.J.; Dukers, D.F.; Kummer, J.A.; Bladergroen, B.A.; Vos, W.; Hack, C.E.; Ossenkoppele, G.J.; Oudejans, J.J. Expression levels of apoptosis-related proteins predict clinical outcome in anaplastic large cell lymphoma. Blood 2002, 99, 4540–4546. [Google Scholar] [CrossRef]
- van Houdt, I.S.; Oudejans, J.J.; van den Eertwegh, A.J.; Baars, A.; Vos, W.; Bladergroen, B.A.; Rimoldi, D.; Muris, J.J.; Hooijberg, E.; Gundy, C.M.; et al. Expression of the apoptosis inhibitor protease inhibitor 9 predicts clinical outcome in vaccinated patients with stage III and IV melanoma. Clin. Cancer Res. 2005, 11, 6400–6407. [Google Scholar]
- Soriano, C.; Mukaro, V.; Hodge, G.; Ahern, J.; Holmes, M.; Jersmann, H.; Moffat, D.; Meredith, D.; Jurisevic, C.; Reynolds, P.N.; et al. Increased proteinase inhibitor-9 (PI-9) and reduced granzyme B in lung cancer: mechanism for immune evasion? Lung Cancer 2012, 77, 38–45. [Google Scholar] [CrossRef]
- Losasso, V.; Schiffer, S.; Barth, S.; Carloni, P. Design of human granzyme B variants resistant to serpin B9. Proteins 2012, 80, 2514–2522. [Google Scholar]
- Sutton, V.R.; Sedelies, K.; Dewson, G.; Christensen, M.E.; Bird, P.I.; Johnstone, R.W.; Kluck, R.M.; Trapani, J.A.; Waterhouse, N.J. Granzyme B triggers a prolonged pressure to die in Bcl-2 overexpressing cells, defining a window of opportunity for effective treatment with ABT-737. Cell Death Dis. 2012, 3, e344. [Google Scholar] [CrossRef]
- Zhang, X.; Sawyer, G.J.; Dong, X.; Qiu, Y.; Collins, L.; Fabre, J.W. The in vivo use of chloroquine to promote non-viral gene delivery to the liver via the portal vein and bile duct. J. Gene Med. 2003, 5, 209–218. [Google Scholar] [CrossRef]
- Raynes, K. Bisquinoline antimalarials: their role in malaria chemotherapy. Int. J. Parasitol. 1999, 29, 367–379. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.H.; Jia, L.T.; Zhang, L.; Xu, Y.M.; Wang, Z.; Yu, C.J.; Peng, W.D.; Wen, W.H.; Wang, C.J.; et al. Secreted antibody/granzyme B fusion protein stimulates selective killing of HER2-overexpressing tumor cells. J. Biol. Chem. 2004, 279, 21343–21348. [Google Scholar]
- Fominaya, J.; Wels, W. Target cell-specific DNA transfer mediated by a chimeric multidomain protein. Novel non-viral gene delivery system. J. Biol. Chem. 1996, 271, 10560–10568. [Google Scholar] [CrossRef]
- Uherek, C.; Fominaya, J.; Wels, W. A modular DNA carrier protein based on the structure of diphtheria toxin mediates target cell-specific gene delivery. J. Biol. Chem. 1998, 273, 8835–8841. [Google Scholar] [CrossRef]
- Antignani, A.; Youle, R.J. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 2006, 18, 685–689. [Google Scholar] [CrossRef]
- Sampieri, K.; Fodde, R. Cancer stem cells and metastasis. Semin. Cancer Biol. 2012, 22, 187–193. [Google Scholar]
- Robertson, M.J.; Cochran, K.J.; Cameron, C.; Le, J.M.; Tantravahi, R.; Ritz, J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp. Hematol. 1996, 24, 406–415. [Google Scholar]
- Oberoi, P.; Jabulowsky, R.A.; Bähr-Mahmud, H.; Wels, W.S. Georg-Speyer-Haus: Frankfurt, Germany, 2013; unpublished work.
- Müller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oberoi, P.; Jabulowsky, R.A.; Wels, W.S. Selective Induction of Cancer Cell Death by Targeted Granzyme B. Antibodies 2013, 2, 130-151. https://doi.org/10.3390/antib2010130
Oberoi P, Jabulowsky RA, Wels WS. Selective Induction of Cancer Cell Death by Targeted Granzyme B. Antibodies. 2013; 2(1):130-151. https://doi.org/10.3390/antib2010130
Chicago/Turabian StyleOberoi, Pranav, Robert A. Jabulowsky, and Winfried S. Wels. 2013. "Selective Induction of Cancer Cell Death by Targeted Granzyme B" Antibodies 2, no. 1: 130-151. https://doi.org/10.3390/antib2010130