Antibody-Based Immunotoxins for the Treatment of Cancer

Abstract
:1. Introduction
2. General Features of Immunotoxins
3. Immunotoxins in Clinical Evaluation
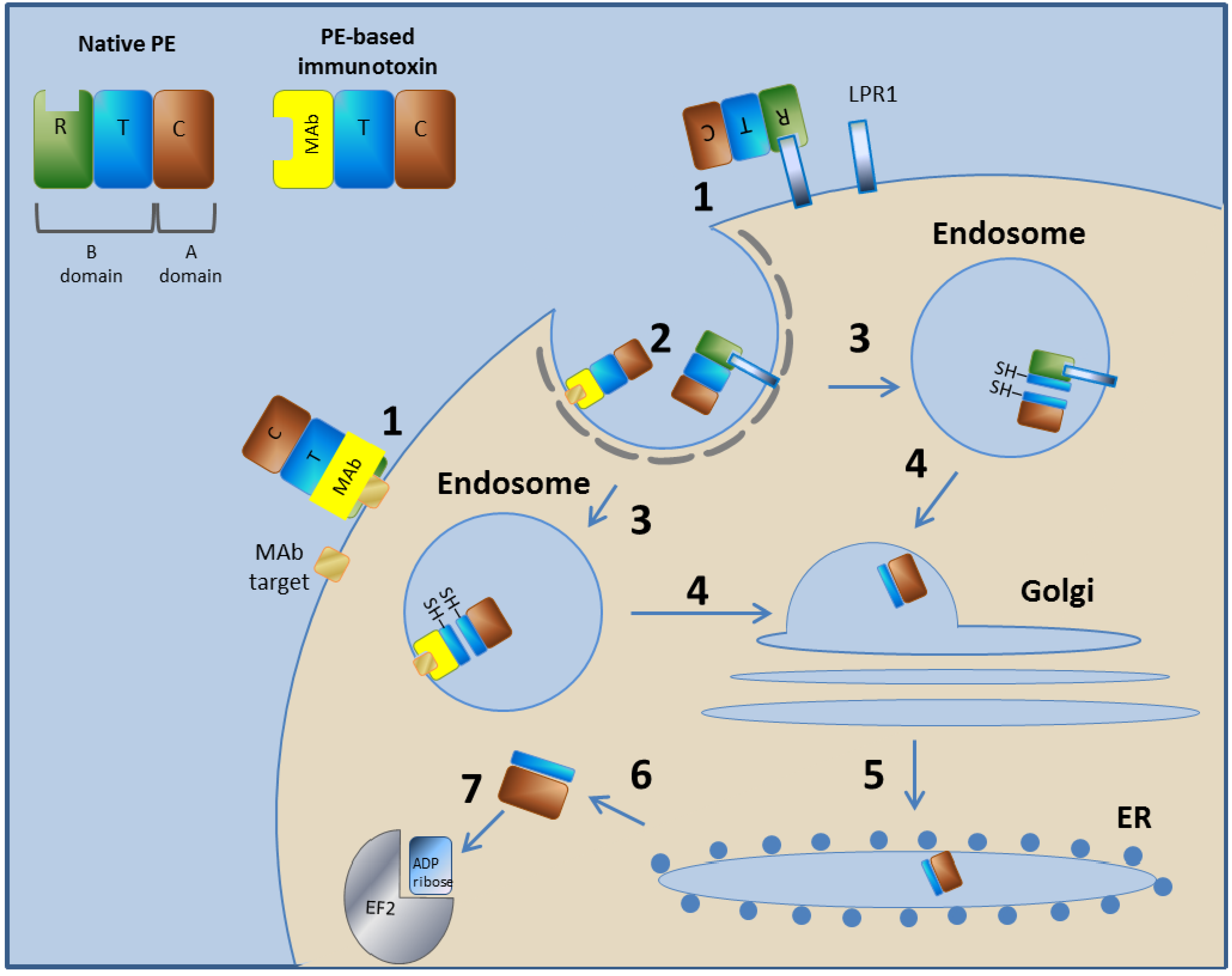
3.1. Pseudomonas Exotoxin A-Based Immunotoxins

{kind=link}
{kind=link}
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| LMB-2 * | CD25 | PE38 | Leukemia, lymphoma | II (ongoing) | [39,40] |
| RFB4(dsFv)-PE38 (BL22/CAT3888) | CD22 | PE38 | NHL, CLL, HCL, ALL | I,II (2008) | [25,41,42,43] |
| Mutated RFB4(dsFv)-PE38 (HA22/CAT-8015) | CD22 | PE38 | HCL, ALL, NHL, CLL, PLL, SLL | I (2012 **) | [44,45] |
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| LMB-2 * | CD25 | PE38 | Metastatic melanoma | I (2008) | [50] |
| OVB3-PE | Ovarian Antigen | Full length PE | Ovarian cancer | I (1991 **) | [22] |
| ERB-38 | erbB2/HER2 | PE38 | Breast, esophageal cancers | I (1999 **) | [51] |
| SS1(dsFv)PE38 (SS1P) | Mesothelin | PE38 | Mesothelioma, ovarian, pancreatic cancers | I (Ongoing) | [52,53] |
| LMB-1 | Lewis Y | PE38 | Adenoca | I (1996) | [54] |
| B3(Fv)-PE38 (LMB-7) | Lewis Y | PE38 | Adenoca | I (2011) | [15] |
| B3(dsFv)-PE38 (LMB-9) | Lewis Y | PE38 | Adenoca | I (2009) | [15] |
| BR96sFv-PE40 (SGN-10) | Lewis Y | PE40 | Adenoca | I (2002 **) | [55] |
| scFv(FRP5)-ETA | erbB2/HER2 | PE40 | Melanoma, breast, colon cancers | I (2005 **) | [56,57] |
3.1.1. Pseudomonas Exotoxin A—Mechanism of Action
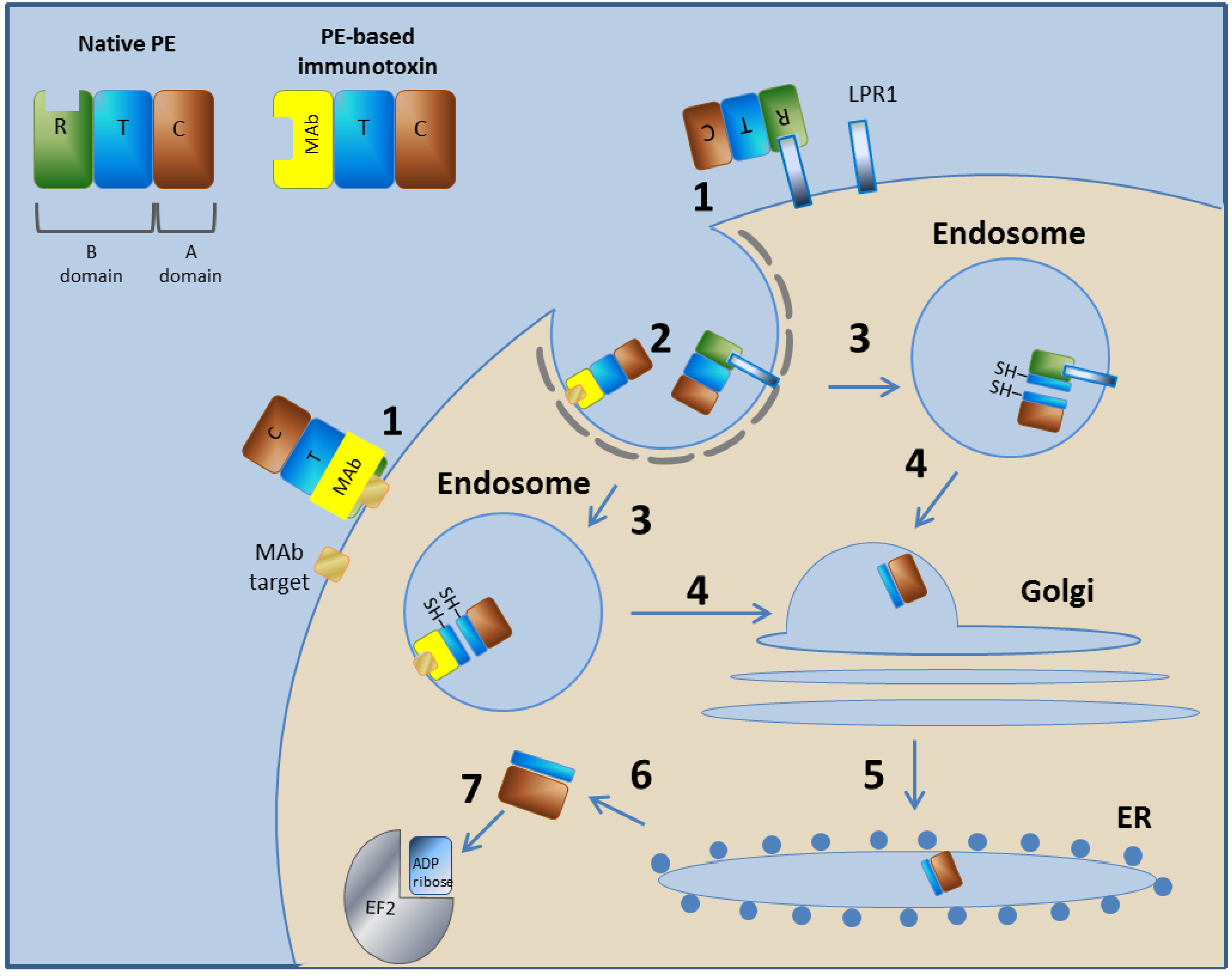
3.1.2. LMB-2
3.1.3. RFB4(dsFv)-PE38 (BL22/CAT3888)
3.1.4. Mutated RFB4(dsFv)-PE38 (HA22/CAT-8015)
3.1.5. OVB3-PE
3.1.6. ERB-38
3.1.7. SS1(dsFv)PE38 (SS1P)
3.1.8. LMB-1
3.1.9. B3(Fv)-PE38 (LMB-7)
3.1.10. LMB-9
3.1.11. BR96sFv-PE40 (SGN-10)
3.1.12. scFv(FRP5)-ETA
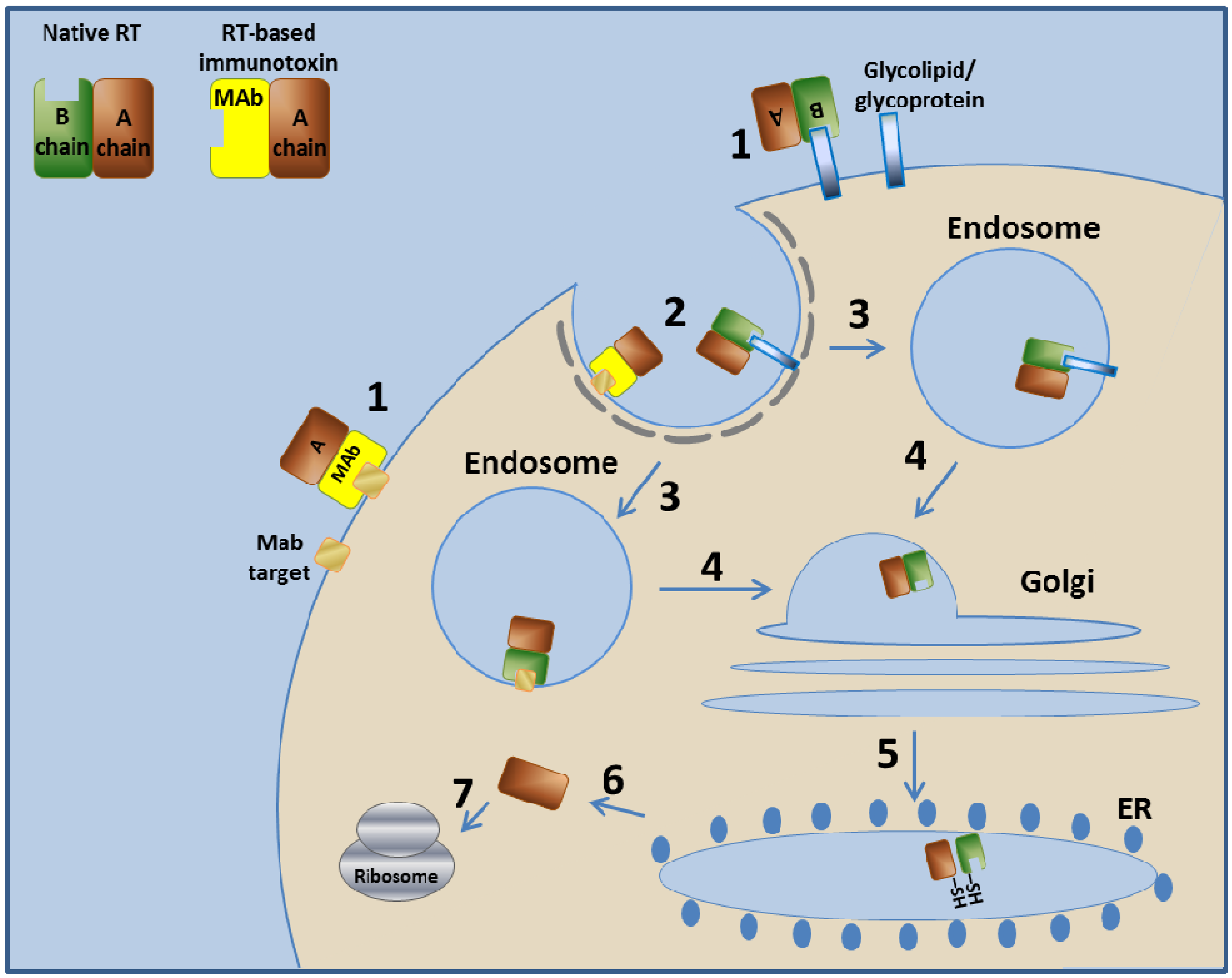
3.2. Ricin Toxin-Based Immunotoxins
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| RFB4-Fab'-dgA | CD22 | dgA | B-NHL | I (1991 **) | [64] |
| RFB4-dgA (IMTOX-22) | CD22 | dgA | B-NHL, CLL | I (2001) | [104,105] |
| HD37-dgA (IMTOX-19) | CD19 | dgA | NHL | I (1996 **) | [106,107] |
| RFB4-dgA + HD37-dgA (Combotox) | CD22, CD19 | dgA | NHL, ALL | I (Ongoing) | [108,109] |
| RFT5-dgA (IMTOX-25) * | CD25 | dgA | HD, CTCL, GVHD, haploidentical SCT | I,II (Ongoing) | [24,110,111,112,113] |
| Ki-4.dgA | CD30 | dgA | HD, NHL | I (2002 **) | [112,114] |
| Anti-B4-bR | CD19 | Blocked ricin | B-NHL | II,III (1992-2011 **) | [115,116,117,118,119,120] |
| Anti-CD7-dgA (DA7) | CD7 | dgA | T-NHL | I (1997 **) | [121] |
| H65-RTA | CD5 | RTA | CTCL, GVHD | I, I/II (1991 **) | [122,123,124] |
| T101-RTA | CD5 | RTA | CLL | I (1989 **) | [125,126,127] |
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| RFT5-dgA (IMTOX-25) * | CD25 | dgA | Melanoma | I,II (Ongoing) | [128] |
| Anti-CEA-bR | CEA | Blocked ricin | Colorectal cancer | I/II (1994 **) | [129] |
| N901-bR | CD56 | Blocked ricin | SCLC | I,II (2002 **) | [130,131,132,133] |
| XomaZyme-Mel (XMMME-001-RTA) | Melanoma antigen | RTA | Melanoma | I
II with CTL I/II with cyclosporine (1993 **) | [134,135,136,137,138,139] |
| XomaZyme-791 (79IT/36-RTA) | 72 kDa TAA | RTA | Colorectal cancer | I (1995 **) | [140,141,142] |
| 454A12-rRA | TfR | RTA | Leptomeningeal neoplasia | I (1997 **) | [143] |
| 260F9-rRTA | 55 kDa Breast cancer antigen | RTA | Breast cancer | I (1989) | [23,144] |
3.2.1. Ricin Toxin—Mechanism of Action
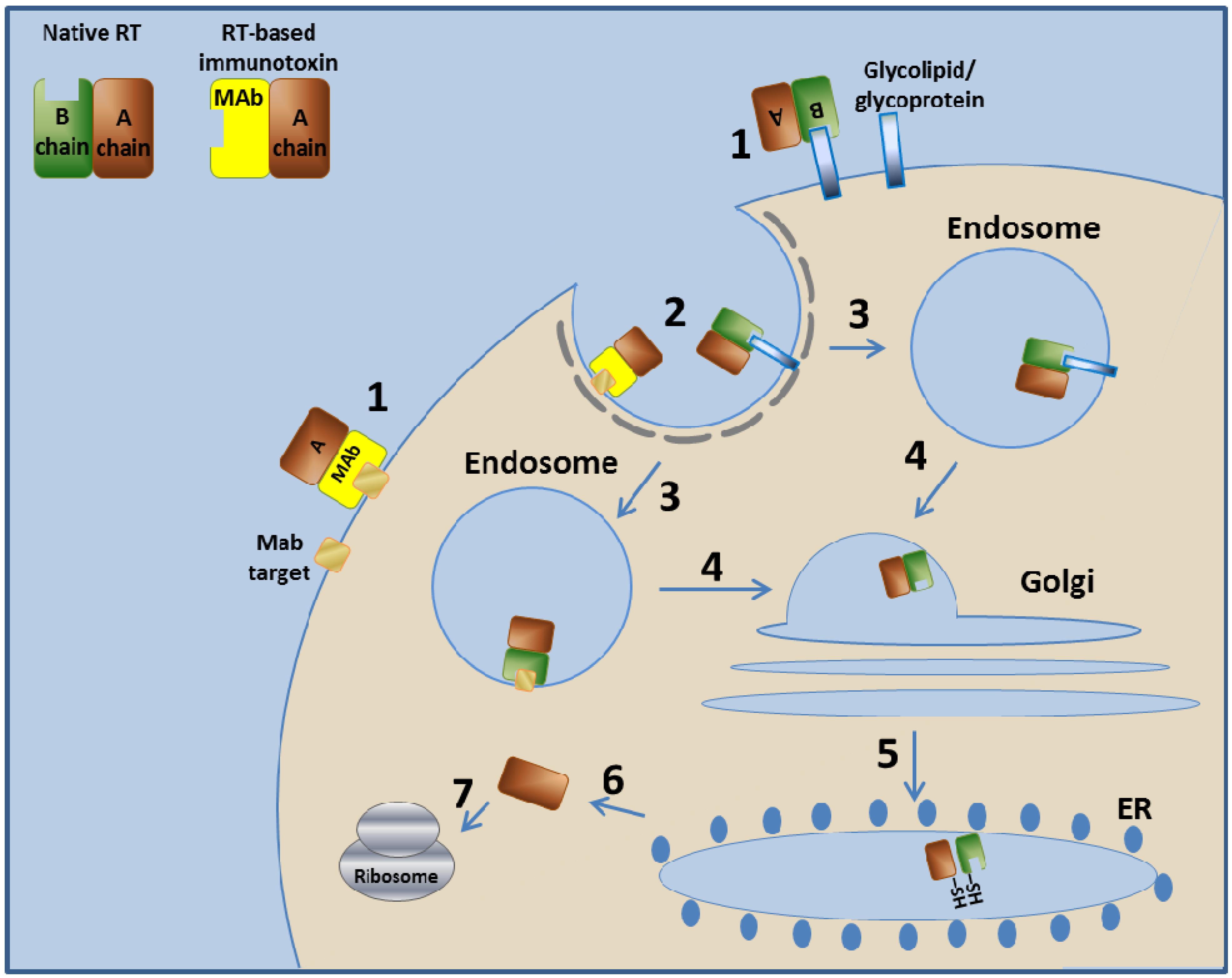
3.2.2. RFB4-Fab'-dgA
3.2.3. RFB4-dgA (IMTOX-22)
3.2.4. HD37-dgA (IMTOX-19)
3.2.5. RFB4-dgA + HD37-dgA (Combotox)
3.2.6. RFT5-dgA (IMTOX-25)
3.2.7. Ki-4.dgA
3.2.8. Anti-B4-bR
3.2.9. Anti-CD7-dgA (DA7)
3.2.10. H65-RTA
3.2.11. T101-RTA
3.2.12. Anti-CEA-bR
3.2.13. N901-bR
3.2.14. XomaZyme-Mel (XMMME-001-RTA)
3.2.15. XomaZyme-791 (79IT/36-RTA)
3.2.16. 454A12-rRA
3.2.17. 260F9-rRTA
3.3. Additional Toxin-Based Immunotoxins Evaluated/Under Clinical Evaluation
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| A-dmDT390- bisFV (UCHT1) | CD3ε | DT390 | T-cell lymphoma/leukemia | I/II (2010 **) | [183,185] |
| B43-PAP | CD19 | PAP | ALL | I (1993 **) | [187] |
| BER-H2-Sap6 | CD30 | Saporin | HD | I (1992 **) | [188] |
| HUM-195/rGel | CD33 | Gelonin | AML, CML | I (2010 **) | [189] |
3.3.1. A-dmDT390- bisFv (UCHT1)
3.3.2. B43-PAP
3.3.3. BER-H2-Sap6
3.3.4. HUM-195/rGel
4. Advantages and Disadvantages of Immunotoxins
5. Conclusions
References
- Society, A.c. Cancer facts and figures 2010. In Atlanta: American cancer society. 2010. Available online: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-026238.pdf (accessed on 9 May 2012).
- Choudhary, S.; Mathew, M.; Verma, R.S. Therapeutic potential of anticancer immunotoxins. Drug Discov. Today 2011, 16, 495–503. [Google Scholar]
- Li, J.; Zhu, Z. Research and development of next generation of antibody-based therapeutics. Acta Pharma. Sin. 2010, 31, 1198–1207. [Google Scholar]
- Reichert, J.M. Antibody-based therapeutics to watch in 2011. mAbs 2011, 3, 76–99. [Google Scholar]
- Reichert, J.M. Monoclonal antibodies as innovative therapeutics. Curr. Pharm. Biotechnol. 2008, 9, 423–430. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar]
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins (Basel) 2011, 3, 848–883. [Google Scholar]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar]
- Govindan, S.V.; Goldenberg, D.M. New antibody conjugates in cancer therapy. TheScientificWorldJo 2010, 10, 2070–2089. [Google Scholar]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar]
- Lambert, J.M.; Goldmacher, V.S.; Collinson, A.R.; Nadler, L.M.; Blattler, W.A. An immunotoxin prepared with blocked ricin: A natural plant toxin adapted for therapeutic use. Cancer Res. 1991, 51, 6236–6242. [Google Scholar]
- Lambert, J.M.; McIntyre, G.; Gauthier, M.N.; Zullo, D.; Rao, V.; Steeves, R.M.; Goldmacher, V.S.; Blattler, W.A. The galactose-binding sites of the cytotoxic lectin ricin can be chemically blocked in high yield with reactive ligands prepared by chemical modification of glycopeptides containing triantennary N-linked oligosaccharides. Biochemistry 1991, 30, 3234–3247. [Google Scholar]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar]
- Chaudhary, V.K.; Queen, C.; Junghans, R.P.; Waldmann, T.A.; FitzGerald, D.J.; Pastan, I. A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature 1989, 339, 394–397. [Google Scholar]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar]
- Kreitman, R.J.; Wilson, W.H.; Robbins, D.; Margulies, I.; Stetler-Stevenson, M.; Waldmann, T.A.; Pastan, I. Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood 1999, 94, 3340–3348. [Google Scholar]
- Liu, W.; Onda, M.; Kim, C.; Xiang, L.; Weldon, J.E.; Lee, B.; Pastan, I. A recombinant immunotoxin engineered for increased stability by adding a disulfide bond has decreased immunogenicity. Protein Eng. Des. Sel. 2012, 25, 1–6. [Google Scholar]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar]
- Pai, L.H.; Bookman, M.A.; Ozols, R.F.; Young, R.C.; Smith, J.W., 2nd; Longo, D.L.; Gould, B.; Frankel, A.; McClay, E.F.; Howell, S.; et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J. Clin Oncol. 1991, 9, 2095–2103. [Google Scholar]
- Weiner, L.M.; O'Dwyer, J.; Kitson, J.; Comis, R.L.; Frankel, A.E.; Bauer, R.J.; Konrad, M.S.; Groves, E.S. Phase I evaluation of an anti-breast carcinoma monoclonal antibody 260F9-recombinant ricin A chain immunoconjugate. Cancer Res. 1989, 49, 4062–4067. [Google Scholar]
- Martin, P.J.; Pei, J.; Gooley, T.; Anasetti, C.; Appelbaum, F.R.; Deeg, J.; Hansen, J.A.; Nash, R.A.; Petersdorf, E.W.; Storb, R.; et al. Evaluation of a CD25-specific immunotoxin for prevention of graft-versus-host disease after unrelated marrow transplantation. Biol. Blood Marrow Transplant. 2004, 10, 552–560. [Google Scholar] [CrossRef]
- Wayne, A.S.; Kreitman, R.J.; Findley, H.W.; Lew, G.; Delbrook, C.; Steinberg, S.M.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Pastan, I. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: Preclinical studies and phase I clinical trial. Clin. Cancer Res. 2010, 16, 1894–1903. [Google Scholar]
- Deng, Q.; Barbieri, J.T. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. Microbiol. 2008, 62, 271–288. [Google Scholar]
- Saelinger, C.B.; Morris, R.E. Intracellular trafficking of Pseudomonas exotoxin A. Antibiot. Chemother. 1987, 39, 149–159. [Google Scholar]
- FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Pseudomonas exotoxin—Immunotoxins. Cancer Treat. Res. 1988, 37, 161–173. [Google Scholar]
- Collier, R.J. Structure-activity relationships in diphtheria toxin and Pseudomonas aeruginosa exotoxin A. Cancer Treat. Res. 1988, 37, 25–35. [Google Scholar]
- Wick, M.J.; Frank, D.W.; Storey, D.G.; Iglewski, B.H. Structure, function, and regulation of Pseudomonas aeruginosa exotoxin A. Ann. Rev. Microbiol. 1990, 44, 335–363. [Google Scholar] [CrossRef]
- Wilson, B.A.; Collier, R.J. Diphtheria toxin and Pseudomonas aeruginosa exotoxin A: Active-site structure and enzymic mechanism. Curr. Top. Microbiol. Immunol. 1992, 175, 27–41. [Google Scholar]
- Mrsny, R.J.; Daugherty, A.L.; McKee, M.L.; FitzGerald, D.J. Bacterial toxins as tools for mucosal vaccination. Drug Discov. Today 2002, 7, 247–258. [Google Scholar]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar]
- Chaudhary, V.K.; Jinno, Y.; Gallo, M.G.; FitzGerald, D.; Pastan, I. Mutagenesis of Pseudomonas exotoxin in identification of sequences responsible for the animal toxicity. J. Biol. Chem. 1990, 265, 16306–16310. [Google Scholar]
- Brinkmann, U.; Pai, L.H.; FitzGerald, D.J.; Pastan, I. Alteration of a protease-sensitive region of Pseudomonas exotoxin prolongs its survival in the circulation of mice. Proc. Natl. Acad. Sci. USA 1992, 89, 3065–3069. [Google Scholar]
- Kasturi, S.; Kihara, A.; FitzGerald, D.; Pastan, I. Alanine scanning mutagenesis identifies surface amino acids on domain II of Pseudomonas exotoxin required for cytotoxicity, proper folding, and secretion into periplasm. J. Biol. Chem. 1992, 267, 23427–23433. [Google Scholar]
- Kuan, C.T.; Wang, Q.C.; Pastan, I. Pseudomonas exotoxin A mutants. Replacement of surface exposed residues in domain II with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J. Biol. Chem. 1994, 269, 7610–7616. [Google Scholar]
- Benhar, I.; Wang, Q.C.; FitzGerald, D.; Pastan, I. Pseudomonas exotoxin A mutants. Replacement of surface-exposed residues in domain III with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J. Biol. Chem. 1994, 269, 13398–13404. [Google Scholar]
- Kreitman, R.J.; Wilson, W.H.; White, J.D.; Stetler-Stevenson, M.; Jaffe, E.S.; Giardina, S.; Waldmann, T.A.; Pastan, I. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J. Clin. Oncol. 2000, 18, 1622–1636. [Google Scholar]
- Kreitman, R.J.; Arons, E.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Recombinant immunotoxins and other therapies for relapsed/refractory hairy cell leukemia. Leuk. Lymphoma 2011, 52 Suppl. 2, 82–86. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Wilson, W.H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N. Engl. J. Med. 2001, 345, 241–247. [Google Scholar]
- Kreitman, R.J.; Squires, D.R.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.; Wilson, W.H.; Pastan, I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J. Clin. Oncol. 2005, 23, 6719–6729. [Google Scholar]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar]
- Kreitman, R.J.; Tallman, M.S.; Robak, T.; Coutre, S.; Wilson, W.H.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Lechleider, R.; Pastan, I. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Mussai, F.; Campana, D.; Bhojwani, D.; Stetler-Stevenson, M.; Steinberg, S.M.; Wayne, A.S.; Pastan, I. Cytotoxicity of the anti-CD22 immunotoxin HA22 (CAT-8015) against paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 352–358. [Google Scholar]
- Fitzgerald, D.J.; Fryling, C.M.; Zdanovsky, A.; Saelinger, C.B.; Kounnas, M.; Strickland, D.K.; Leppla, S. Selection of Pseudomonas exotoxin-resistant cells with altered expression of alpha 2MR/LRP. Ann. NY Acad. Sci. 1994, 737, 138–144. [Google Scholar]
- Kounnas, M.Z.; Morris, R.E.; Thompson, M.R.; FitzGerald, D.J.; Strickland, D.K.; Saelinger, C.B. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 1992, 267, 12420–12423. [Google Scholar]
- FitzGerald, D.; Morris, R.E.; Saelinger, C.B. Receptor-mediated internalization of Pseudomonas toxin by mouse fibroblasts. Cell 1980, 21, 867–873. [Google Scholar]
- Smith, D.C.; Spooner, R.A.; Watson, P.D.; Murray, J.L.; Hodge, T.W.; Amessou, M.; Johannes, L.; Lord, J.M.; Roberts, L.M. Internalized Pseudomonas exotoxin A can exploit multiple pathways to reach the endoplasmic reticulum. Traffic 2006, 7, 379–393. [Google Scholar]
- Powell, D.J., Jr.; Felipe-Silva, A.; Merino, M.J.; Ahmadzadeh, M.; Allen, T.; Levy, C.; White, D.E.; Mavroukakis, S.; Kreitman, R.J.; Rosenberg, S.A.; Pastan, I. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J. Immunol. 2007, 179, 4919–4928. [Google Scholar]
- Pai-Scherf, L.H.; Villa, J.; Pearson, D.; Watson, T.; Liu, E.; Willingham, M.C.; Pastan, I. Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin. Cancer Res. 1999, 5, 2311–2315. [Google Scholar]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar]
- Kreitman, R.J.; Hassan, R.; Fitzgerald, D.J.; Pastan, I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin. Cancer Res. 2009, 15, 5274–5279. [Google Scholar]
- Pai, L.H.; Wittes, R.; Setser, A.; Willingham, M.C.; Pastan, I. Treatment of advanced solid tumors with immunotoxin LMB-1: An antibody linked to Pseudomonas exotoxin. Nat. Med. 1996, 2, 350–353. [Google Scholar]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin. Cancer Res. 2002, 8, 3092–3099. [Google Scholar]
- Azemar, M.; Djahansouzi, S.; Jager, E.; Solbach, C.; Schmidt, M.; Maurer, A.B.; Mross, K.; Unger, C.; von Minckwitz, G.; Dall, P.; et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res. Treat. 2003, 82, 155–164. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jager, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005, 7, R617–R626. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Minami, Y. The IL-2/IL-2 receptor system: A current overview. Cell 1993, 73, 5–8. [Google Scholar]
- Diamantstein, T.; Osawa, H. The interleukin-2 receptor, its physiology and a new approach to a selective immunosuppressive therapy by anti-interleukin-2 receptor monoclonal antibodies. Immunol. Rev. 1986, 92, 5–27. [Google Scholar]
- Anti-Tac(Fv)-PE38 (LMB-2) to Treat Chronic Lymphocytic Leukemia. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00077922?term=LMB2&rank=2 (accessed on 31 March 2012).
- LMB-2 to Treat Hairy Cell Leukemia. Available online: http://clinicaltrials.gov/ct2/show/NCT00321555?term=LMB-2&rank=5 (accessed on 31 March 2012).
- Phase II Trial of LMB-2, Fludarabine and Cyclophosphamide for Adult T-Cell Leukemia. Available online: http://clinicaltrials.gov/ct2/show/NCT00924170?term=LMB-2&rank=1 (accessed on 31 March 2012).
- Li, J.L.; Shen, G.L.; Ghetie, M.A.; May, R.D.; Till, M.; Ghetie, V.; Uhr, J.W.; Janossy, G.; Thorpe, P.E.; Amlot, P.; et al. The epitope specificity and tissue reactivity of four murine monoclonal anti-CD22 antibodies. Cell. Immunol. 1989, 118, 85–99. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Stone, M.; Amlot, P.; Fay, J.; May, R.; Till, M.; Newman, J.; Clark, P.; Collins, R.; Cunningham, D.; et al. Phase I immunotoxin trial in patients with B-cell lymphoma. Cancer Res. 1991, 51, 4052–4058. [Google Scholar]
- Clark, E.A. CD22, a B cell-specific receptor, mediates adhesion and signal transduction. J. Immunol. 1993, 150, 4715–4718. [Google Scholar]
- Robbins, B.A.; Ellison, D.J.; Spinosa, J.C.; Carey, C.A.; Lukes, R.J.; Poppema, S.; Saven, A.; Piro, L.D. Diagnostic application of two-color flow cytometry in 161 cases of hairy cell leukemia. Blood 1993, 82, 1277–1287. [Google Scholar]
- Cordone, I.; Annino, L.; Masi, S.; Pescarmona, E.; Rahimi, S.; Ferrari, A.; Giubilei, E.; Pignoloni, P.; Faraggiana, T.; Mandelli, F. Diagnostic relevance of peripheral blood immunocytochemistry in hairy cell leukaemia. J. Clin. Pathol. 1995, 48, 955–960. [Google Scholar]
- Robbins, D.H.; Margulies, I.; Stetler-Stevenson, M.; Kreitman, R.J. Hairy cell leukemia, a B-cell neoplasm that is particularly sensitive to the cytotoxic effect of anti-Tac(Fv)-PE38 (LMB-2). Clin. Cancer Res. 2000, 6, 693–700. [Google Scholar]
- Anderson, K.C.; Bates, M.P.; Slaughenhoupt, B.L.; Pinkus, G.S.; Schlossman, S.F.; Nadler, L.M. Expression of human B cell-associated antigens on leukemias and lymphomas: A model of human B cell differentiation. Blood 1984, 63, 1424–1433. [Google Scholar]
- Salvatore, G.; Beers, R.; Margulies, I.; Kreitman, R.J.; Pastan, I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin. Cancer Res. 2002, 8, 995–1002. [Google Scholar]
- Batra, J.K.; Kasprzyk, P.G.; Bird, R.E.; Pastan, I.; King, C.R. Recombinant anti-erbB2 immunotoxins containing Pseudomonas exotoxin. Proc. Natl. Acad. Sci. USA 1992, 89, 5867–5871. [Google Scholar]
- Reiter, Y.; Brinkmann, U.; Jung, S.H.; Lee, B.; Kasprzyk, P.G.; King, C.R.; Pastan, I. Improved binding and antitumor activity of a recombinant anti-erbB2 immunotoxin by disulfide stabilization of the Fv fragment. J. Biol. Chem. 1994, 269, 18327–18331. [Google Scholar]
- Chowdhury, P.S.; Pastan, I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat. Biotechnol. 1999, 17, 568–572. [Google Scholar] [CrossRef]
- Hassan, R.; Bera, T.; Pastan, I. Mesothelin: A new target for immunotherapy. Clin. Cancer Res. 2004, 10, 3937–3942. [Google Scholar]
- Chowdhury, P.S.; Viner, J.L.; Beers, R.; Pastan, I. Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc. Natl. Acad. Sci. USA 1998, 95, 669–674. [Google Scholar]
- Ricart, A.D. Immunoconjugates against solid tumors: Mind the gap. Clin. Pharmacol. Ther. 2011, 89, 513–523. [Google Scholar]
- SS1P and Pentostatin Plus Cyclophosphamide for Mesothelioma. Available online: http://clinicaltrials.gov/ct2/show/NCT01362790?term=SS1P&rank=1 (accessed on 31 March 2012).
- Bigner, D.D.; Archer, G.E.; McLendon, R.E.; Friedman, H.S.; Fuchs, H.E.; Pai, L.H.; Herndon, J.E., 2nd; Pastan, I.H. Efficacy of compartmental administration of immunotoxin LMB-1 (B3-LysPE38) in a rat model of carcinomatous meningitis. Clin. Cancer Res. 1995, 1, 1545–1555. [Google Scholar]
- Pai, L.H.; Batra, J.K.; FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Anti-tumor activities of immunotoxins made of monoclonal antibody B3 and various forms of Pseudomonas exotoxin. Proc. Natl. Acad. Sci. USA 1991, 88, 3358–3362. [Google Scholar]
- Friedman, P.N.; McAndrew, S.J.; Gawlak, S.L.; Chace, D.; Trail, P.A.; Brown, J.P.; Siegall, C.B. BR96 sFv-PE40, a potent single-chain immunotoxin that selectively kills carcinoma cells. Cancer Res. 1993, 53, 334–339. [Google Scholar]
- Siegall, C.B.; Chace, D.; Mixan, B.; Garrigues, U.; Wan, H.; Paul, L.; Wolff, E.; Hellstrom, I.; Hellstrom, K.E. In vitro and in vivo characterization of BR96 sFv-PE40. A single-chain immunotoxin fusion protein that cures human breast carcinoma xenografts in athymic mice and rats. J. Immunol. 1994, 152, 2377–2384. [Google Scholar]
- Wels, W.; Harwerth, I.M.; Mueller, M.; Groner, B.; Hynes, N.E. Selective inhibition of tumor cell growth by a recombinant single-chain antibody-toxin specific for the erbB-2 receptor. Cancer Res. 1992, 52, 6310–6317. [Google Scholar]
- Wels, W.; Beerli, R.; Hellmann, P.; Schmidt, M.; Marte, B.M.; Kornilova, E.S.; Hekele, A.; Mendelsohn, J.; Groner, B.; Hynes, N.E. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int. J. Cancer 1995, 60, 137–144. [Google Scholar]
- Nielsen, K.; Boston, R.S. RIBOSOME-INACTIVATING PROTEINS: A Plant Perspective. Annu. Rev. Plant Phys. 2001, 52, 785–816. [Google Scholar]
- Frankel, A.E.; FitzGerald, D.; Siegall, C.; Press, O.W. Advances in immunotoxin biology and therapy: A summary of the Fourth International Symposium on Immunotoxins. Cancer Res. 1996, 56, 926–932. [Google Scholar]
- Pastan, I.; FitzGerald, D. Recombinant toxins for cancer treatment. Science 1991, 254, 1173–1177. [Google Scholar]
- Fulton, R.J.; Uhr, J.W.; Vitetta, E.S. In vivo therapy of the BCL1 tumor: Effect of immunotoxin valency and deglycosylation of the ricin A chain. Cancer Res. 1988, 48, 2626–2631. [Google Scholar]
- Fulton, R.J.; Tucker, T.F.; Vitetta, E.S.; Uhr, J.W. Pharmacokinetics of tumor-reactive immunotoxins in tumor-bearing mice: Effect of antibody valency and deglycosylation of the ricin A chain on clearance and tumor localization. Cancer Res. 1988, 48, 2618–2625. [Google Scholar]
- Bourrie, B.J.; Casellas, P.; Blythman, H.E.; Jansen, F.K. Study of the plasma clearance of antibody—Ricin-A-chain immunotoxins. Evidence for specific recognition sites on the A chain that mediate rapid clearance of the immunotoxin. Eur. J. Biochem. 1986, 155, 1–10. [Google Scholar] [CrossRef]
- Blakey, D.C.; Watson, G.J.; Knowles, P.P.; Thorpe, P.E. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987, 47, 947–952. [Google Scholar]
- Ramakrishnan, S.; Bjorn, M.J.; Houston, L.L. Recombinant ricin A chain conjugated to monoclonal antibodies: Improved tumor cell inhibition in the presence of lysosomotropic compounds. Cancer Res. 1989, 49, 613–617. [Google Scholar]
- Kreitman, R.J. Immunotoxins in cancer therapy. Curr. Opin. Immunol. 1999, 11, 570–578. [Google Scholar]
- Baluna, R.; Rizo, J.; Gordon, B.E.; Ghetie, V.; Vitetta, E.S. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 3957–3962. [Google Scholar]
- Hirao, I.; Madin, K.; Endo, Y.; Yokoyama, S.; Ellington, A.D. RNA aptamers that bind to and inhibit the ribosome-inactivating protein, pepocin. J. Biol. Chem. 2000, 275, 4943–4948. [Google Scholar]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins (Basel) 2010, 2, 2519–2583. [Google Scholar]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar]
- Endo, Y.; Tsurugi, K.; Lambert, J.M. The site of action of six different ribosome-inactivating proteins from plants on eukaryotic ribosomes: The RNA N-glycosidase activity of the proteins. Biochem. Biophys. Res. Commun. 1988, 150, 1032–1036. [Google Scholar]
- Stirpe, F.; Bailey, S.; Miller, S.P.; Bodley, J.W. Modification of ribosomal RNA by ribosome-inactivating proteins from plants. Nucleic Acids Res. 1988, 16, 1349–1357. [Google Scholar]
- Montanaro, L.; Sperti, S.; Mattioli, A.; Testoni, G.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro. Inhibition of the binding of elongation factor 2 and of adenosine diphosphate-ribosylated elongation factor 2 to ribosomes. Biochem. J. 1975, 146, 127–131. [Google Scholar]
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J. Pathol. 1987, 151, 221–229. [Google Scholar]
- Bolognesi, A.; Tazzari, P.L.; Olivieri, F.; Polito, L.; Falini, B.; Stirpe, F. Induction of apoptosis by ribosome-inactivating proteins and related immunotoxins. Int. J. Cancer 1996, 68, 349–355. [Google Scholar]
- Narayanan, S.; Surolia, A.; Karande, A.A. Ribosome-inactivating protein and apoptosis: Abrin causes cell death via mitochondrial pathway in Jurkat cells. Biochem. J. 2004, 377, 233–240. [Google Scholar]
- Amlot, P.L.; Stone, M.J.; Cunningham, D.; Fay, J.; Newman, J.; Collins, R.; May, R.; McCarthy, M.; Richardson, J.; Ghetie, V.; et al. A phase I study of an anti-CD22-deglycosylated ricin A chain immunotoxin in the treatment of B-cell lymphomas resistant to conventional therapy. Blood 1993, 82, 2624–2633. [Google Scholar]
- Sausville, E.A.; Headlee, D.; Stetler-Stevenson, M.; Jaffe, E.S.; Solomon, D.; Figg, W.D.; Herdt, J.; Kopp, W.C.; Rager, H.; Steinberg, S.M.; et al. Continuous infusion of the anti-CD22 immunotoxin IgG-RFB4-SMPT-dgA in patients with B-cell lymphoma: A phase I study. Blood 1995, 85, 3457–3465. [Google Scholar]
- Stone, M.J.; Sausville, E.A.; Fay, J.W.; Headlee, D.; Collins, R.H.; Figg, W.D.; Stetler-Stevenson, M.; Jain, V.; Jaffe, E.S.; Solomon, D.; et al. A phase I study of bolus versus continuous infusion of the anti-CD19 immunotoxin, IgG-HD37-dgA, in patients with B-cell lymphoma. Blood 1996, 88, 1188–1197. [Google Scholar]
- Conry, R.M.; Khazaeli, M.B.; Saleh, M.N.; Ghetie, V.; Vitetta, E.S.; Liu, T.; LoBuglio, A.F. Phase I trial of an anti-CD19 deglycosylated ricin A chain immunotoxin in non-Hodgkin's lymphoma: Effect of an intensive schedule of administration. J. Immunother. Emphasis Tumor Immunol. 1995, 18, 231–241. [Google Scholar]
- Herrera, L.; Bostrom, B.; Gore, L.; Sandler, E.; Lew, G.; Schlegel, P.G.; Aquino, V.; Ghetie, V.; Vitetta, E.S.; Schindler, J. A phase 1 study of Combotox in pediatric patients with refractory B-lineage acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2009, 31, 936–941. [Google Scholar]
- Messmann, R.A.; Vitetta, E.S.; Headlee, D.; Senderowicz, A.M.; Figg, W.D.; Schindler, J.; Michiel, D.F.; Creekmore, S.; Steinberg, S.M.; Kohler, D.; et al. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(+), CD22(+) B cell lymphoma. Clin. Cancer Res. 2000, 6, 1302–1313. [Google Scholar]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schon, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin's lymphoma. Blood 1997, 89, 403–410. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Barth, S.; Winkler, U.; Borchmann, P.; Hansmann, M.L.; Diehl, V.; Ghetie, V.; Engert, A. Clinical trials with an anti-CD25 ricin A-chain experimental and immunotoxin (RFT5-SMPT-dgA) in Hodgkin's lymphoma. Leuk. Lymphoma 1998, 30, 525–537. [Google Scholar]
- Schnell, R.; Borchmann, P.; Staak, J.O.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Engert, A. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin's lymphoma. Ann. Oncol. 2003, 14, 729–736. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Borchmann, P.; Barth, S.; Ghetie, V.; Hell, K.; Drillich, S.; Diehl, V.; Engert, A. Treatment of refractory Hodgkin's lymphoma patients with an anti-CD25 ricin A-chain immunotoxin. Leukemia 2000, 14, 129–135. [Google Scholar]
- Schnell, R.; Staak, O.; Borchmann, P.; Schwartz, C.; Matthey, B.; Hansen, H.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Diehl, V.; Engert, A. A Phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4.dgA) in patients with refractory CD30+ Hodgkin's and non-Hodgkin's lymphoma. Clin. Cancer Res. 2002, 8, 1779–1786. [Google Scholar]
- Multani, P.S.; O'Day, S.; Nadler, L.M.; Grossbard, M.L. Phase II clinical trial of bolus infusion anti-B4 blocked ricin immunoconjugate in patients with relapsed B-cell non-Hodgkin's lymphoma. Clin. Cancer Res. 1998, 4, 2599–2604. [Google Scholar]
- Grossbard, M.L.; Gribben, J.G.; Freedman, A.S.; Lambert, J.M.; Kinsella, J.; Rabinowe, S.N.; Eliseo, L.; Taylor, J.A.; Blattler, W.A.; Epstein, C.L.; et al. Adjuvant immunotoxin therapy with anti-B4-blocked ricin after autologous bone marrow transplantation for patients with B-cell non-Hodgkin's lymphoma. Blood 1993, 81, 2263–2271. [Google Scholar]
- Grossbard, M.L.; Lambert, J.M.; Goldmacher, V.S.; Spector, N.L.; Kinsella, J.; Eliseo, L.; Coral, F.; Taylor, J.A.; Blattler, W.A.; Epstein, C.L.; et al. Anti-B4-blocked ricin: A phase I trial of 7-day continuous infusion in patients with B-cell neoplasms. J. Clin. Oncol. 1993, 11, 726–737. [Google Scholar]
- Grossbard, M.L.; Multani, P.S.; Freedman, A.S.; O'Day, S.; Gribben, J.G.; Rhuda, C.; Neuberg, D.; Nadler, L.M. A Phase II study of adjuvant therapy with anti-B4-blocked ricin after autologous bone marrow transplantation for patients with relapsed B-cell non-Hodgkin's lymphoma. Clin. Cancer Res. 1999, 5, 2392–2398. [Google Scholar]
- Furman, R.R.; Grossbard, M.L.; Johnson, J.L.; Pecora, A.L.; Cassileth, P.A.; Jung, S.H.; Peterson, B.A.; Nadler, L.M.; Freedman, A.; Bayer, R.L.; et al. A phase III study of anti-B4-blocked ricin as adjuvant therapy post-autologous bone marrow transplant: CALGB 9254. Leuk. Lymphoma 2011, 52, 587–596. [Google Scholar]
- Grossbard, M.L.; Freedman, A.S.; Ritz, J.; Coral, F.; Goldmacher, V.S.; Eliseo, L.; Spector, N.; Dear, K.; Lambert, J.M.; Blattler, W.A.; et al. Serotherapy of B-cell neoplasms with anti-B4-blocked ricin: A phase I trial of daily bolus infusion. Blood 1992, 79, 576–585. [Google Scholar]
- Frankel, A.E.; Laver, J.H.; Willingham, M.C.; Burns, L.J.; Kersey, J.H.; Vallera, D.A. Therapy of patients with T-cell lymphomas and leukemias using an anti-CD7 monoclonal antibody-ricin A chain immunotoxin. Leuk. Lymphoma 1997, 26, 287–298. [Google Scholar]
- Kernan, N.A.; Byers, V.; Scannon, P.J.; Mischak, R.P.; Brochstein, J.; Flomenberg, N.; Dupont, B.; O'Reilly, R.J. Treatment of steroid-resistant acute graft-vs-host disease by in vivo administration of an anti-T-cell ricin A chain immunotoxin. JAMA 1988, 259, 3154–3157. [Google Scholar]
- Byers, V.S.; Henslee, P.J.; Kernan, N.A.; Blazar, B.R.; Gingrich, R.; Phillips, G.L.; LeMaistre, C.F.; Gilliland, G.; Antin, J.H.; Martin, P.; et al. Use of an anti-pan T-lymphocyte ricin a chain immunotoxin in steroid-resistant acute graft-versus-host disease. Blood 1990, 75, 1426–1432. [Google Scholar]
- LeMaistre, C.F.; Rosen, S.; Frankel, A.; Kornfeld, S.; Saria, E.; Meneghetti, C.; Drajesk, J.; Fishwild, D.; Scannon, P.; Byers, V. Phase I trial of H65-RTA immunoconjugate in patients with cutaneous T-cell lymphoma. Blood 1991, 78, 1173–1182. [Google Scholar]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Laurent, G.; Jansen, F.K.; Schmidt, C.; Frankel, A.E. A phase I study of T101-ricin A chain immunotoxin in refractory chronic lymphocytic leukemia. J. Biol. Response Mod. 1988, 7, 97–113. [Google Scholar]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Blythman, H.E.; Casellas, P.; Frankel, A.E. An anti-CD5 immunotoxin for chronic lymphocytic leukemia: Enhancement of cytotoxicity with human serum albumin-monensin. Int. J. Cancer 1989, 43, 215–219. [Google Scholar]
- Laurent, G.; Pris, J.; Farcet, J.P.; Carayon, P.; Blythman, H.; Casellas, P.; Poncelet, P.; Jansen, F.K. Effects of therapy with T101 ricin A-chain immunotoxin in two leukemia patients. Blood 1986, 67, 1680–1687. [Google Scholar]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar]
- Zalcberg, J.R.; Pietersz, G.; Toohey, B.; Laird, J.; Huggins, R.; Zimet, A.S.; Hennessy, O.; McKenzie, A.; McKenzie, I.F. A phase I/II study of the intralesional injection of ricin-monoclonal antibody conjugates in patients with hepatic metastases. Eur. J. Cancer 1994, 30A, 1227–1231. [Google Scholar]
- Lynch, T.J., Jr. Immunotoxin therapy of small-cell lung cancer. N901-blocked ricin for relapsed small-cell lung cancer. Chest 1993, 103, 436S–439S. [Google Scholar] [CrossRef]
- Lynch, T.J., Jr.; Lambert, J.M.; Coral, F.; Shefner, J.; Wen, P.; Blattler, W.A.; Collinson, A.R.; Ariniello, P.D.; Braman, G.; Cook, S.; et al. Immunotoxin therapy of small-cell lung cancer: A phase I study of N901-blocked ricin. J. Clin. Oncol. 1997, 15, 723–734. [Google Scholar]
- Epstein, C.; Lynch, T.; Shefner, J.; Wen, P.; Maxted, D.; Braman, V.; Ariniello, P.; Coral, F.; Ritz, J. Use of the immunotoxin N901-blocked ricin in patients with small-cell lung cancer. Int. J. Cancer Suppl. 1994, 8, 57–59. [Google Scholar]
- Fidias, P.; Grossbard, M.; Lynch, T.J., Jr. A phase II study of the immunotoxin N901-blocked ricin in small-cell lung cancer. Clin. Lung Cancer 2002, 3, 219–222. [Google Scholar]
- Hertler, A.A.; Spitler, L.E.; Frankel, A.E. Humoral immune response to a ricin A chain immunotoxin in patients with metastatic melanoma. Cancer Drug Deliv. 1987, 4, 245–253. [Google Scholar]
- Spitler, L.E.; del Rio, M.; Khentigan, A.; Wedel, N.I.; Brophy, N.A.; Miller, L.L.; Harkonen, W.S.; Rosendorf, L.L.; Lee, H.M.; Mischak, R.P.; et al. Therapy of patients with malignant melanoma using a monoclonal antimelanoma antibody-ricin A chain immunotoxin. Cancer Res. 1987, 47, 1717–1723. [Google Scholar]
- Mischak, R.P.; Foxall, C.; Rosendorf, L.L.; Knebel, K.; Scannon, P.J.; Spitler, L.E. Human antibody responses to components of the monoclonal antimelanoma antibody ricin A chain immunotoxin XomaZyme-MEL. Mol. Biother. 1990, 2, 104–109. [Google Scholar]
- Oratz, R.; Speyer, J.L.; Wernz, J.C.; Hochster, H.; Meyers, M.; Mischak, R.; Spitler, L.E. Antimelanoma monoclonal antibody-ricin A chain immunoconjugate (XMMME-001-RTA) plus cyclophosphamide in the treatment of metastatic malignant melanoma: Results of a phase II trial. J. Biol. Response Mod. 1990, 9, 345–354. [Google Scholar]
- Gonzalez, R.; Salem, P.; Bunn, P.A., Jr.; Zukiwski, A.A.; Lamb, R.; Benjamin, R.S.; Spitler, L.; Wedel, N.; Robinson, W.A. Single-dose murine monoclonal antibody ricin A chain immunotoxin in the treatment of metastatic melanoma: A phase I trial. Mol. Biother. 1991, 3, 192–196. [Google Scholar]
- Selvaggi, K.; Saria, E.A.; Schwartz, R.; Vlock, D.R.; Ackerman, S.; Wedel, N.; Kirkwood, J.M.; Jones, H.; Ernstoff, M.S. Phase I/II study of murine monoclonal antibody-ricin A chain (XOMAZYME-Mel) immunoconjugate plus cyclosporine A in patients with metastatic melanoma. J. Immunother. Emphasis Tumor Immunol. 1993, 13, 201–207. [Google Scholar]
- Durrant, L.G.; Byers, V.S.; Scannon, P.J.; Rodvien, R.; Grant, K.; Robins, R.A.; Marksman, R.A.; Baldwin, R.W. Humoral immune responses to XMMCO-791-RTA immunotoxin in colorectal cancer patients. Clin. Exp. Immunol. 1989, 75, 258–264. [Google Scholar]
- LoRusso, P.M.; Lomen, P.L.; Redman, B.G.; Poplin, E.; Bander, J.J.; Valdivieso, M. Phase I study of monoclonal antibody-ricin A chain immunoconjugate Xomazyme-791 in patients with metastatic colon cancer. Am. J. Clin. Oncol. 1995, 18, 307–312. [Google Scholar]
- Byers, V.S.; Rodvien, R.; Grant, K.; Durrant, L.G.; Hudson, K.H.; Baldwin, R.W.; Scannon, P.J. Phase I study of monoclonal antibody-ricin A chain immunotoxin XomaZyme-791 in patients with metastatic colon cancer. Cancer Res. 1989, 49, 6153–6160. [Google Scholar]
- Laske, D.W.; Muraszko, K.M.; Oldfield, E.H.; DeVroom, H.L.; Sung, C.; Dedrick, R.L.; Simon, T.R.; Colandrea, J.; Copeland, C.; Katz, D.; et al. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 1997, 41, 1039–1049, discussion 1049–1051. [Google Scholar]
- Gould, B.J.; Borowitz, M.J.; Groves, E.S.; Carter, P.W.; Anthony, D.; Weiner, L.M.; Frankel, A.E. Phase I study of an anti-breast cancer immunotoxin by continuous infusion: Report of a targeted toxic effect not predicted by animal studies. J. Natl. Cancer Inst. 1989, 81, 775–781. [Google Scholar]
- Campana, D.; Janossy, G.; Bofill, M.; Trejdosiewicz, L.K.; Ma, D.; Hoffbrand, A.V.; Mason, D.Y.; Lebacq, A.M.; Forster, H.K. Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. J. Immunol. 1985, 134, 1524–1530. [Google Scholar]
- Thorpe, P.E.; Detre, S.I.; Foxwell, B.M.; Brown, A.N.; Skilleter, D.N.; Wilson, G.; Forrester, J.A.; Stirpe, F. Modification of the carbohydrate in ricin with metaperiodate-cyanoborohydride mixtures. Effects on toxicity and in vivo distribution. Eur. J. Biochem. 1985, 147, 197–206. [Google Scholar] [CrossRef]
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.; Watson, G.J.; Blakey, D.C.; Newell, D.R. Improved antitumor effects of immunotoxins prepared with deglycosylated ricin A-chain and hindered disulfide linkages. Cancer Res. 1988, 48, 6396–6403. [Google Scholar]
- Dorken, B.; Moldenhauer, G.; Schwartz, R.; Pez-zutto, A.; Hammerling, G. B cell differentiation antigens identified by monoclonal antibodies (HD6, HD28, HD37, HD39). Immunobiology 1983, 165, 253–254. [Google Scholar]
- Ghetie, M.A.; Ghetie, V.; Vitetta, E.S. Immunotoxins for the treatment of B-cell lymphomas. Mol. Med. 1997, 3, 420–427. [Google Scholar]
- Ghetie, M.A.; Tucker, K.; Richardson, J.; Uhr, J.W.; Vitetta, E.S. The antitumor activity of an anti-CD22 immunotoxin in SCID mice with disseminated Daudi lymphoma is enhanced by either an anti-CD19 antibody or an anti-CD19 immunotoxin. Blood 1992, 80, 2315–2320. [Google Scholar]
- Phase 1 Study of Combotox With Cytarabine in Relapsed/Refractory B-lineage Acute Lymphoblastic Leukemia (ALL). Available online: http://clinicaltrials.gov/ct2/show/NCT01408160?term=combotox&rank=1 (accessed on 31 March 2012).
- RFT5-dgA. Available online: http://clinicaltrials.gov/ct2/results?term=RFT5-dgA (accessed on 31 March 2012).
- Schwab, U.; Stein, H.; Gerdes, J.; Lemke, H.; Kirchner, H.; Schaadt, M.; Diehl, V. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin's disease and a subset of normal lymphoid cells. Nature 1982, 299, 65–67. [Google Scholar]
- Scadden, D.T.; Schenkein, D.P.; Bernstein, Z.; Luskey, B.; Doweiko, J.; Tulpule, A.; Levine, A.M. Immunotoxin combined with chemotherapy for patients with AIDS-related non-Hodgkin's lymphoma. Cancer 1998, 83, 2580–2587. [Google Scholar]
- Longo, D.L.; Duffey, P.L.; Gribben, J.G.; Jaffe, E.S.; Curti, B.D.; Gause, B.L.; Janik, J.E.; Braman, V.M.; Esseltine, D.; Wilson, W.H.; et al. Combination chemotherapy followed by an immunotoxin (anti-B4-blocked ricin) in patients with indolent lymphoma: Results of a phase II study. Cancer J. 2000, 6, 146–150. [Google Scholar]
- Szatrowski, T.P.; Dodge, R.K.; Reynolds, C.; Westbrook, C.A.; Frankel, S.R.; Sklar, J.; Stewart, C.C.; Hurd, D.D.; Kolitz, J.E.; Velez-Garcia, E.; et al. Lineage specific treatment of adult patients with acute lymphoblastic leukemia in first remission with anti-B4-blocked ricin or high-dose cytarabine: Cancer and Leukemia Group B Study 9311. Cancer 2003, 97, 1471–1480. [Google Scholar]
- Aruffo, A.; Seed, B. Molecular cloning of two CD7 (T-cell leukemia antigen) cDNAs by a COS cell expression system. EMBO J. 1987, 6, 3313–3316. [Google Scholar]
- Haynes, B.F.; Eisenbarth, G.S.; Fauci, A.S. Human lymphocyte antigens: Production of a monoclonal antibody that defines functional thymus-derived lymphocyte subsets. Proc. Natl. Acad. Sci. USA 1979, 76, 5829–5833. [Google Scholar]
- Wood, G.S.; Deneau, D.G.; Miller, R.A.; Levy, R.; Hoppe, R.T.; Warnke, R.A. Subtypes of cutaneous T-cell lymphoma defined by expression of leu-1 and Ia. Blood 1982, 59, 876–882. [Google Scholar]
- Holden, C.A.; Staughton, R.C.; Campbell, M.A.; MacDonald, D.M. Differential loss of T lymphocyte markers in advanced cutaneous T cell lymphoma. J. Am. Acad. Dermatol. 1982, 6, 507–513. [Google Scholar]
- Royston, I.; Majda, J.A.; Baird, S.M.; Meserve, B.L.; Griffiths, J.C. Human T cell antigens defined by monoclonal antibodies: The 65,000-dalton antigen of T cells (T65) is also found on chronic lymphocytic leukemia cells bearing surface immunoglobulin. J. Immunol. 1980, 125, 725–731. [Google Scholar]
- Pietersz, G.A.; Kanellos, J.; McKenzie, I.F. Novel synthesis and in vitro characterization of disulfide-linked ricin-monoclonal antibody conjugates devoid of galactose binding activity. Cancer Res. 1988, 48, 4469–4476. [Google Scholar]
- Teh, J.G.; Thompson, C.H.; McKenzie, I.F. Production of monoclonal antibodies to serum antigens in colorectal carcinoma. J. Immunol. Methods 1988, 110, 101–109. [Google Scholar]
- Thomas, S.N.; Zhu, F.; Schnaar, R.L.; Alves, C.S.; Konstantopoulos, K. Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J. Biol. Chem. 2008, 283, 15647–15655. [Google Scholar]
- Campbell, D.G.; Price, M.R.; Baldwin, R.W. Analysis of a human osteogenic sarcoma antigen and its expression on various human tumour cell lines. Int. J. Cancer 1984, 34, 31–37. [Google Scholar]
- Price, M.R.; Campbell, D.G.; Robins, R.A.; Baldwin, R.W. Characteristics of a cell surface antigen defined by an anti-human osteogenic sarcoma monoclonal antibody. Eur. J. Cancer Clin. Oncol. 1983, 19, 81–90. [Google Scholar]
- Faulk, W.P.; Hsi, B.L.; Stevens, P.J. Transferrin and transferrin receptors in carcinoma of the breast. Lancet 1980, 2, 390–392. [Google Scholar]
- Galbraith, G.M.; Galbraith, R.M.; Faulk, W.P. Transferrin binding by human lymphoblastoid cell lines and other transformed cells. Cell. Immunol. 1980, 49, 215–222. [Google Scholar]
- Gatter, K.C.; Brown, G.; Trowbridge, I.S.; Woolston, R.E.; Mason, D.Y. Transferrin receptors in human tissues: Their distribution and possible clinical relevance. J. Clin. Pathol. 1983, 36, 539–545. [Google Scholar]
- Larrick, J.W.; Cresswell, P. Modulation of cell surface iron transferrin receptors by cellular density and state of activation. J. Supramol. Struct. 1979, 11, 579–586. [Google Scholar]
- Shindelman, J.E.; Ortmeyer, A.E.; Sussman, H.H. Demonstration of the transferrin receptor in human breast cancer tissue. Potential marker for identifying dividing cells. Int. J. Cancer 1981, 27, 329–334. [Google Scholar] [CrossRef]
- Trowbridge, I.S.; Domingo, D.L. Anti-transferrin receptor monoclonal antibody and toxin-antibody conjugates affect growth of human tumour cells. Nature 1981, 294, 171–173. [Google Scholar]
- Trowbridge, I.S.; Omary, M.B. Human cell surface glycoprotein related to cell proliferation is the receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 3039–3043. [Google Scholar]
- Trowbridge, I.S.; Newman, R.A.; Domingo, D.L.; Sauvage, C. Transferrin receptors: Structure and function. Biochem. Pharmacol. 1984, 33, 925–932. [Google Scholar]
- Bjorn, M.J.; Ring, D.; Frankel, A. Evaluation of monoclonal antibodies for the development of breast cancer immunotoxins. Cancer Res. 1985, 45, 1214–1221. [Google Scholar]
- Frankel, A.E.; Ring, D.B.; Tringale, F.; Hsieh-Ma, S.T. Tissue distribution of breast cancer-associated antigens defined by monoclonal antibodies. J. Biol. Response Mod. 1985, 4, 273–286. [Google Scholar]
- Frankel, A.E.; Powell, B.L.; Hall, P.D.; Case, L.D.; Kreitman, R.J. Phase I trial of a novel diphtheria toxin/granulocyte macrophage colony-stimulating factor fusion protein (DT388GMCSF) for refractory or relapsed acute myeloid leukemia. Clin. Cancer Res. 2002, 8, 1004–1013. [Google Scholar]
- LeMaistre, C.F.; Meneghetti, C.; Rosenblum, M.; Reuben, J.; Parker, K.; Shaw, J.; Deisseroth, A.; Woodworth, T.; Parkinson, D.R. Phase I trial of an interleukin-2 (IL-2) fusion toxin (DAB486IL-2) in hematologic malignancies expressing the IL-2 receptor. Blood 1992, 79, 2547–2554. [Google Scholar]
- Kuzel, T.M.; Rosen, S.T.; Gordon, L.I.; Winter, J.; Samuelson, E.; Kaul, K.; Roenigk, H.H.; Nylen, P.; Woodworth, T. Phase I trial of the diphtheria toxin/interleukin-2 fusion protein DAB486IL-2: Efficacy in mycosis fungoides and other non-Hodgkin's lymphomas. Leuk. Lymphoma 1993, 11, 369–377. [Google Scholar]
- LeMaistre, C.F.; Craig, F.E.; Meneghetti, C.; McMullin, B.; Parker, K.; Reuben, J.; Boldt, D.H.; Rosenblum, M.; Woodworth, T. Phase I trial of a 90-minute infusion of the fusion toxin DAB486IL-2 in hematological cancers. Cancer Res. 1993, 53, 3930–3934. [Google Scholar]
- Platanias, L.C.; Ratain, M.J.; O'Brien, S.; Larson, R.A.; Vardiman, J.W.; Shaw, J.P.; Williams, S.F.; Baron, J.M.; Parker, K.; Woodworth, T.G. Phase I trial of a genetically engineered interleukin-2 fusion toxin (DAB486IL-2) as a 6 hour intravenous infusion in patients with hematologic malignancies. Leuk. Lymphoma 1994, 14, 257–262. [Google Scholar]
- Tepler, I.; Schwartz, G.; Parker, K.; Charette, J.; Kadin, M.E.; Woodworth, T.G.; Schnipper, L.E. Phase I trial of an interleukin-2 fusion toxin (DAB486IL-2) in hematologic malignancies: Complete response in a patient with Hodgkin's disease refractory to chemotherapy. Cancer 1994, 73, 1276–1285. [Google Scholar]
- Woo, J.H.; Lee, Y.J.; Neville, D.M.; Frankel, A.E. Pharmacology of anti-CD3 diphtheria immunotoxin in CD3 positive T-cell lymphoma trials. Methods Mol. Biol. 2010, 651, 157–175. [Google Scholar]
- Thompson, J.; Hu, H.; Scharff, J.; Neville, D.M., Jr. An anti-CD3 single-chain immunotoxin with a truncated diphtheria toxin avoids inhibition by pre-existing antibodies in human blood. J. Biol. Chem. 1995, 270, 28037–28041. [Google Scholar]
- Frankel, A.E.; Zuckero, S.L.; Mankin, A.A.; Grable, M.; Mitchell, K.; Lee, Y.J.; Neville, D.M.; Woo, J.H. Anti-CD3 recombinant diphtheria immunotoxin therapy of cutaneous T cell lymphoma. Curr. Drug Targets 2009, 10, 104–109. [Google Scholar]
- A-dmDT390-bisFv (UCHT1) Immunotoxin Therapy for Patients With T-cell Diseases. Available online: http://clinicaltrials.gov/ct2/show/NCT00611208?term=UCHT1&rank=1 (accessed on 31 March 2012).
- Uckun, F.M. Immunotoxins for the treatment of leukaemia. Br. J. Haematol. 1993, 85, 435–438. [Google Scholar]
- Falini, B.; Bolognesi, A.; Flenghi, L.; Tazzari, P.L.; Broe, M.K.; Stein, H.; Durkop, H.; Aversa, F.; Corneli, P.; Pizzolo, G.; et al. Response of refractory Hodgkin's disease to monoclonal anti-CD30 immunotoxin. Lancet 1992, 339, 1195–1196. [Google Scholar]
- Dean, A.; Talpaz, M.; Kantarjian, H.; Faderl, S.; Jabbour, E.; Ravandi Kashani, F.; O'Brien, S.M.; Rosenblum, M.; Cortes, J.E. Phase I clinical trial of the anti-CD33 immunotoxin HuM195/rgel in patients (pts) with advanced myeloid malignancies. ASCO Meeting Abstracts 2010, 28, 6549. [Google Scholar]
- Uckun, F.M.; Chelstrom, L.M.; Irvin, J.D.; Finnegan, D.; Gunther, R.; Young, J.; Kuebelbeck, V.; Myers, D.E.; Houston, L.L. In vivo efficacy of B43 (anti-CD19)-pokeweed antiviral protein immunotoxin against BCL-1 murine B-cell leukemia. Blood 1992, 79, 2649–2661. [Google Scholar]
- Foyil, K.V.; Bartlett, N.L. Anti-CD30 Antibodies for Hodgkin lymphoma. Curr. Hematol. Malign. Rep. 2010, 5, 140–147. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Becker, N.; Benhar, I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies 2012, 1, 39-69. https://doi.org/10.3390/antib1010039
Becker N, Benhar I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies. 2012; 1(1):39-69. https://doi.org/10.3390/antib1010039
Chicago/Turabian StyleBecker, Nurit, and Itai Benhar. 2012. "Antibody-Based Immunotoxins for the Treatment of Cancer" Antibodies 1, no. 1: 39-69. https://doi.org/10.3390/antib1010039