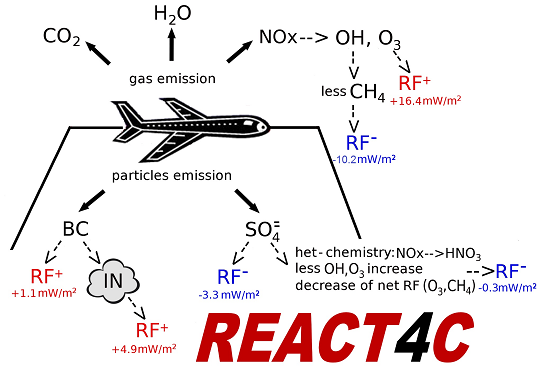
3.4.3. Radiative Forcing Results
The global annual average RFs calculated off-line with the radiative transfer models (ULAQ, Oslo), for the three numerical simulations (NA, AE, AE*) and all three models (ULAQ, CTM2 and CTM3) are presented in
Table 3 and
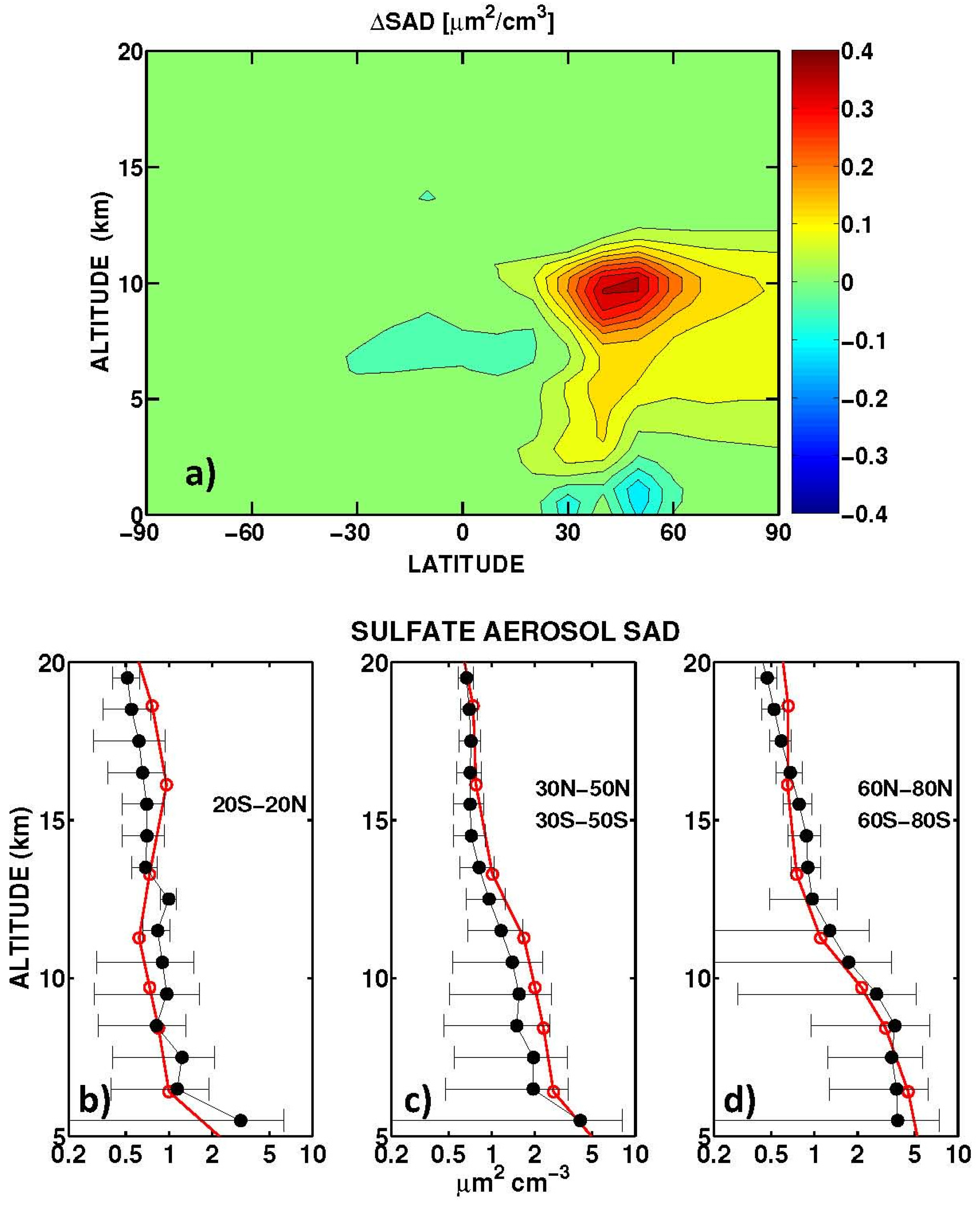
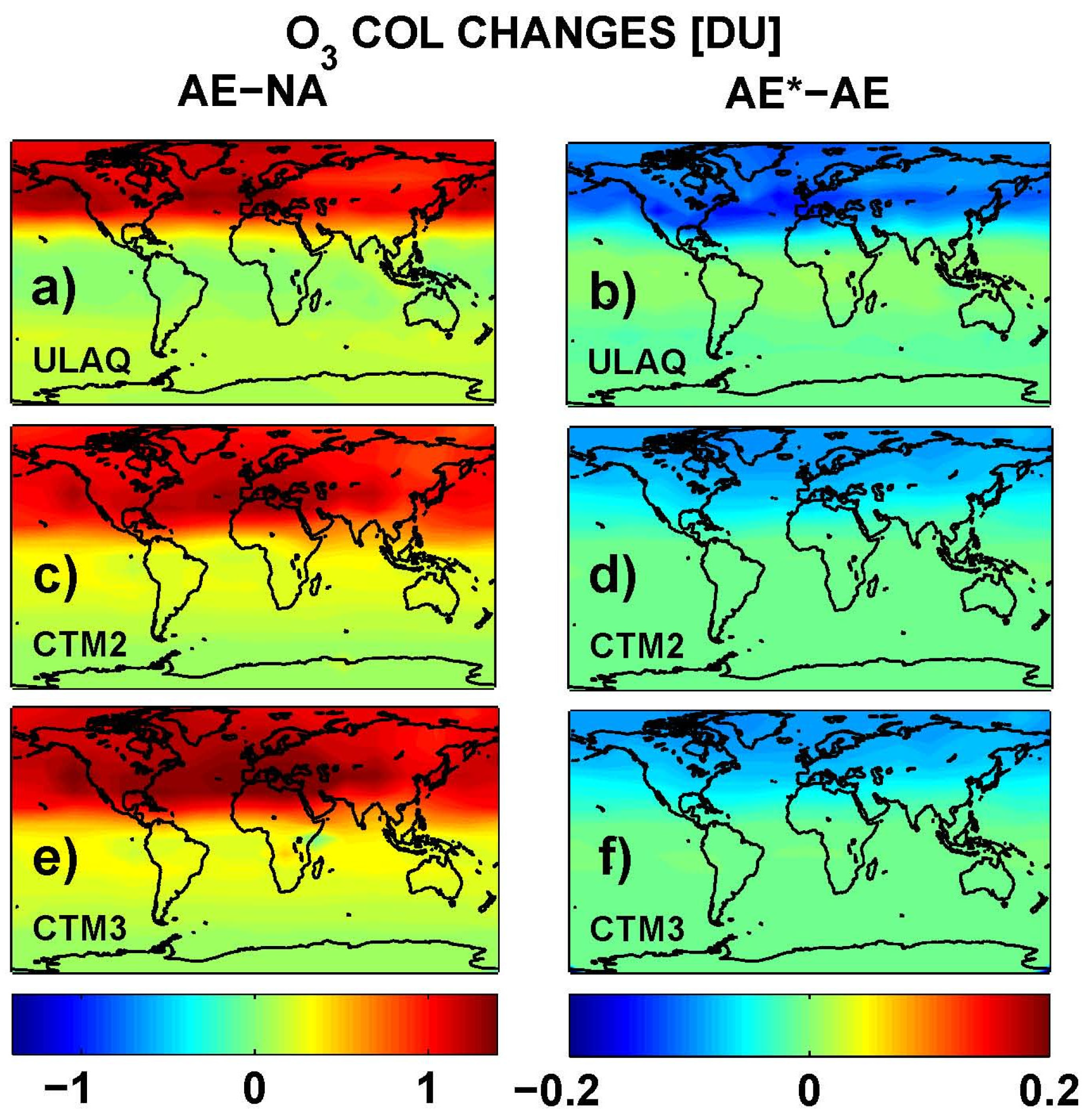
Table 4. On average, the three models calculate an ozone column difference due to the aircraft induced increase in aerosol SAD of −5%, which is consistent with the −7% calculated by Unger [
21]. The ULAQ estimate is closer to this independent value, because it includes the hydrolysis of BrONO
2 in addition to that of N
2O
5, in contrast to CTM2 and CTM3. The tables give RF values for clear and total sky conditions, with stratospheric temperature adjustment applied for the longwave forcing in the case of total sky conditions. A good consistency between the radiative transfer models is clear from the instantaneous longwave forcing in
Table 3: with a 0.5% difference found between the two radiative models. A 15% difference for SW and 5%–10% for total sky LW RFs (
Table 4) are due to slight differences in surface albedo and cloud cover adopted in the two models. Results in
Table 4 also indicate the importance of stratospheric temperature adjustment, whereby the aircraft induced O
3 increase in the lower stratosphere, warming this region both in the UV and in the planetary infrared 9.6 μm band. This produced an indirect negative forcing from well-mixed greenhouse gases (e.g., CO
2, CH
4, N
2O) [
81].
The results of additional ULAQ-CTM simulations (
i.e., model version labeled ULAQ_FLX) have been included in both tables. In this case, the CH
4 prediction is made using a flux boundary condition instead of a fixed mixing ratio at the surface (as in ULAQ, CTM2 and CTM3). Gridded fluxes of CH
4 are used at the surface for anthropogenic and natural sources [
82,
83]. Due to the long CH
4 lifetime, the ULAQ_FLX numerical simulations are extended up to 50 years, to allow the model to reach equilibrium between the predicted CH
4 field and O
3 (via OH and NO
x). This means that the O
3 RF from ULAQ_FLX includes both the short-term ozone response and its long-term response (
i.e., the so-called primary mode O
3 RF) [
84].
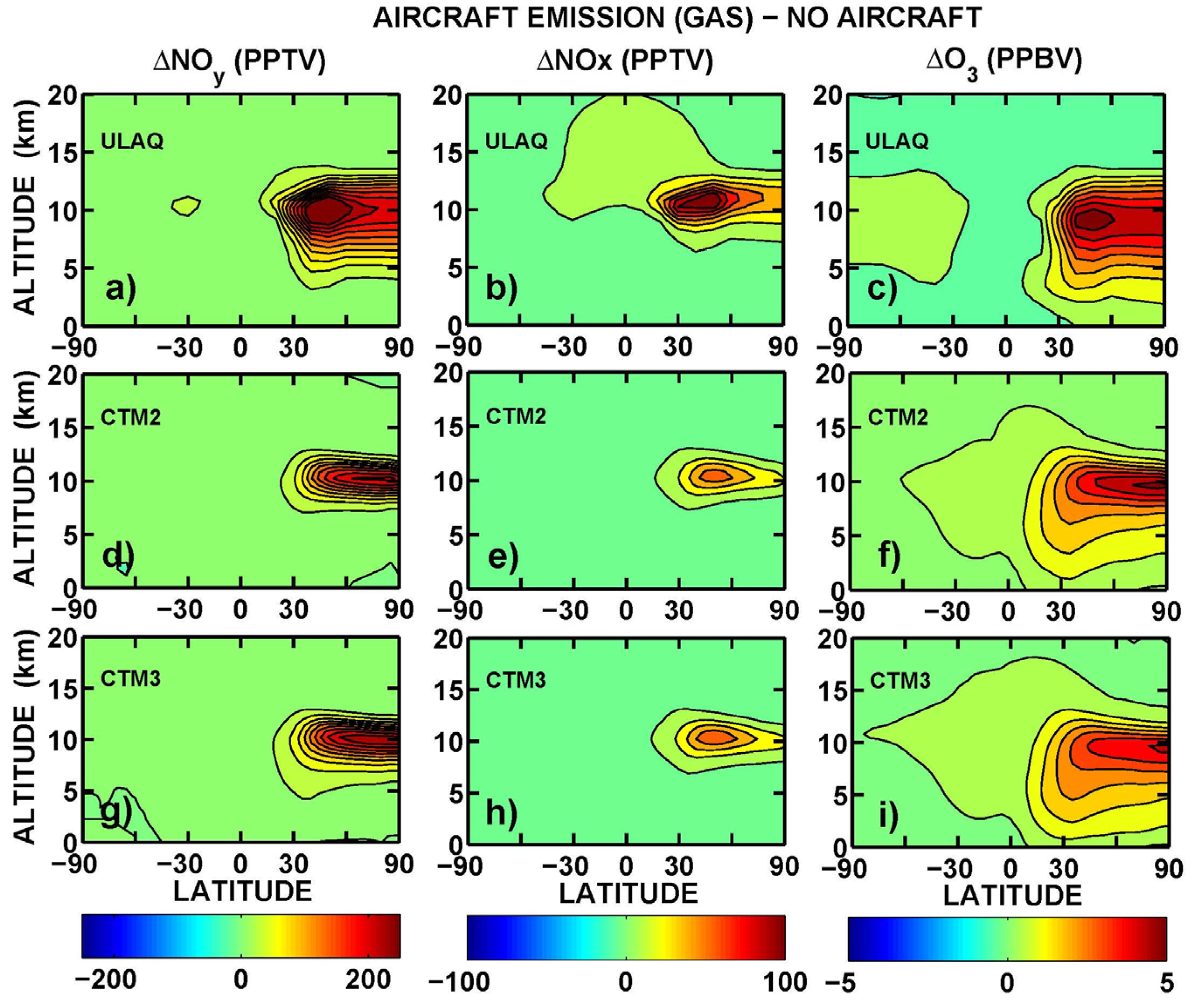
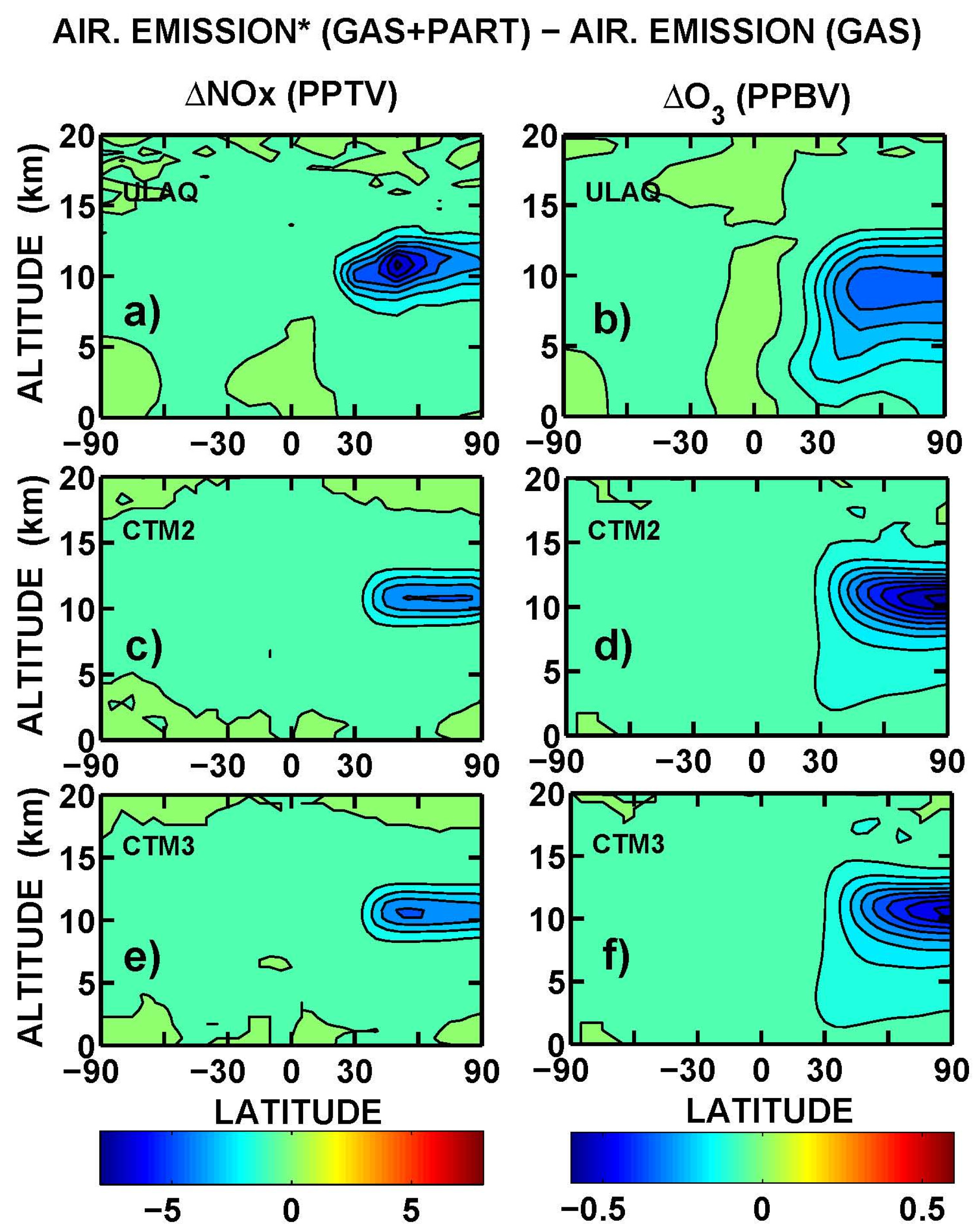
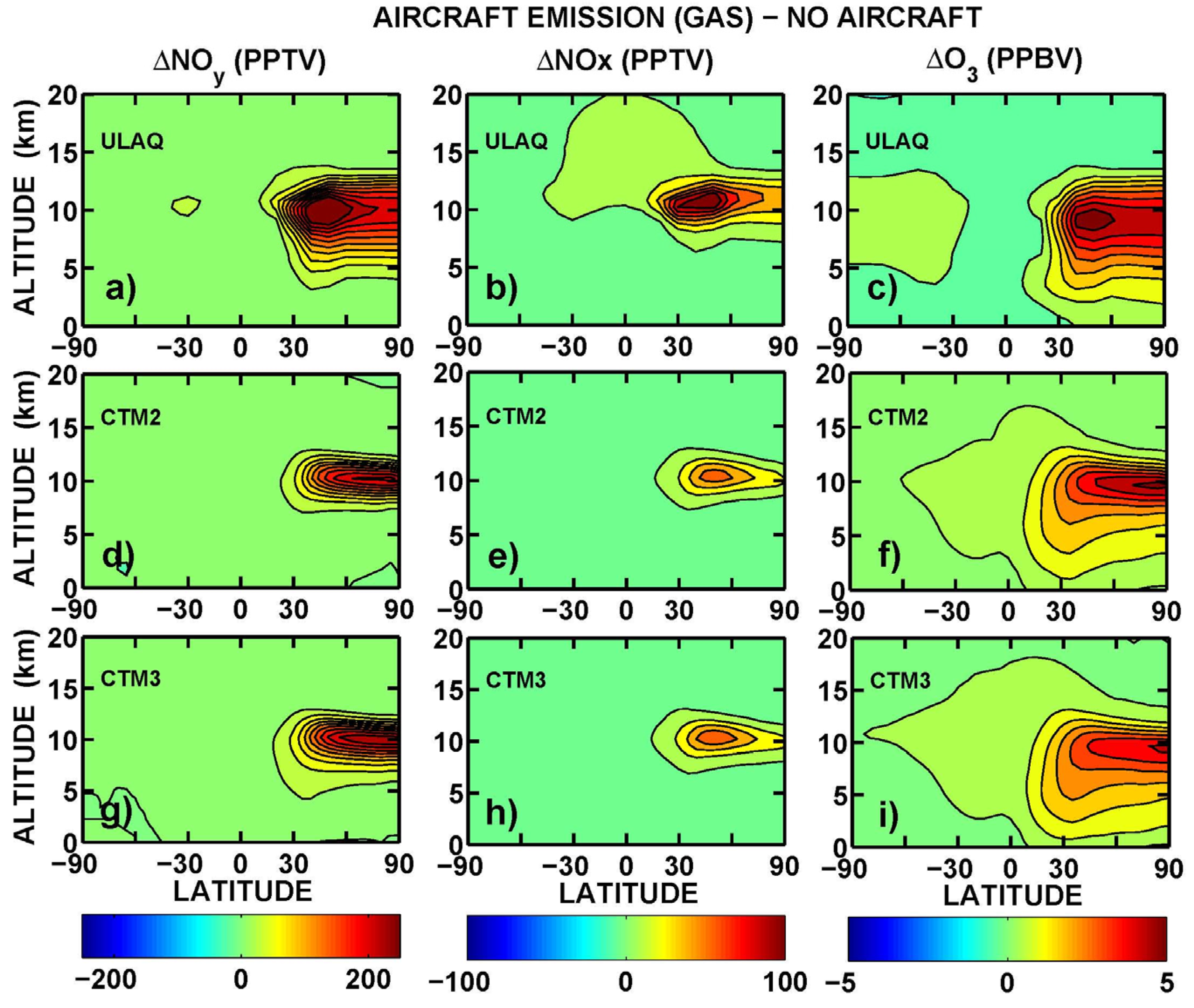
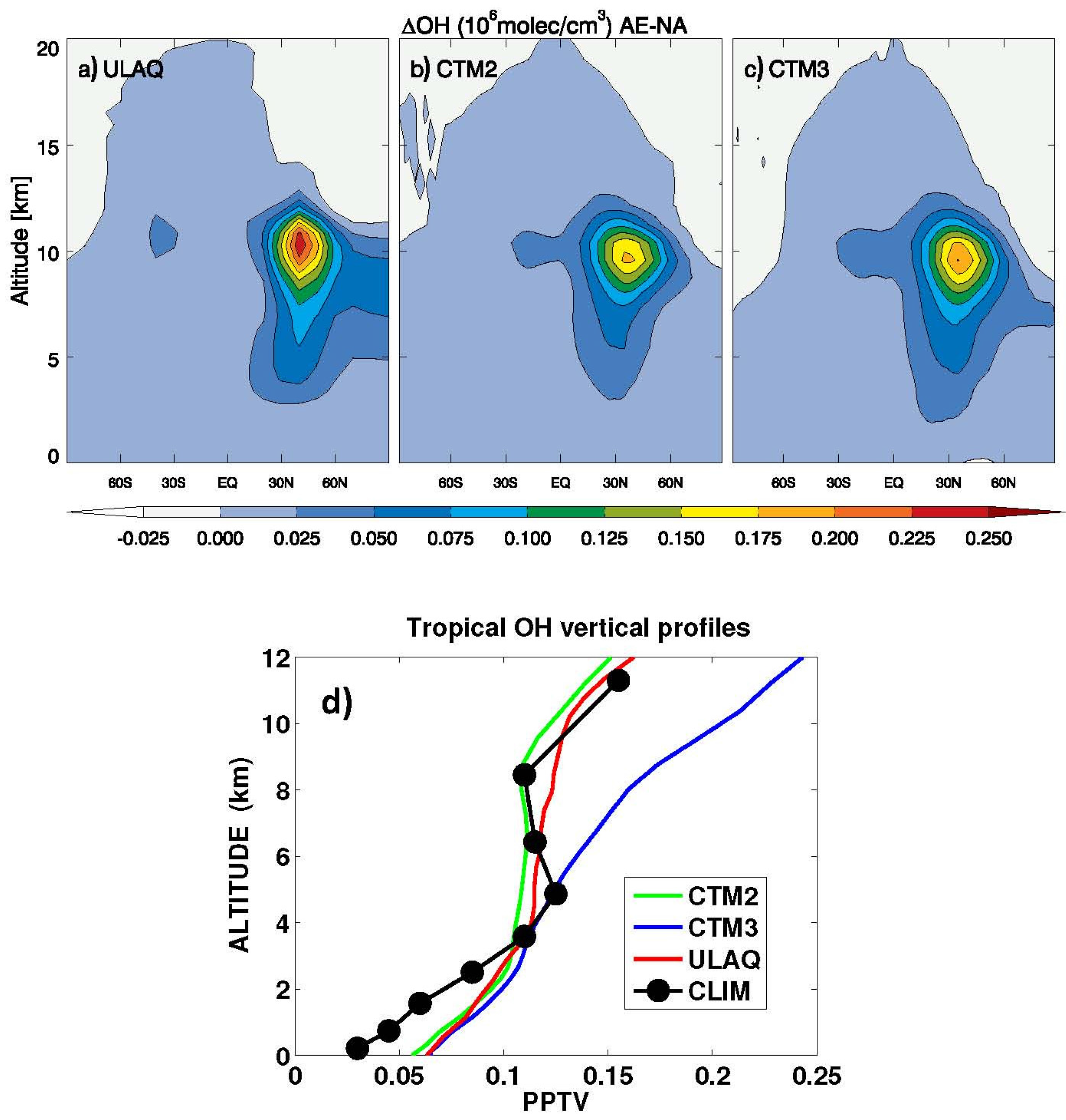
Latitude-longitude maps of the short-term O
3 RF are presented in
Figure 11, in terms of average values over the three models (ULAQ, CTM2 and CTM3) and the two off-line radiative calculations (ULAQ, Oslo). Globally, the aviation aerosol effect on O
3 photochemistry is quantified as −4% of the aviation NO
x effect, in terms of radiative forcing. As expected, the geographical patterns of the calculated radiative forcing are closely linked to those of the ozone column changes in
Figure 10.
Table 3.
Summary of O3 RF mean values for clear sky conditions. Differences are calculated between NA (experiment with no aircraft emission), AE (experiment with only gaseous emission [NOx+H2O]) and AE* (experiment with gaseous and particle emission [NOx+H2O+SO4]). RF values are calculated with Oslo and ULAQ radiative code at ECMWF and National Center for Environmental Prediction (NCEP) tropopause level, respectively. LW, SW, NET are longwave, shortwave and net RFs, respectively. Only LW instantaneous values are considered here.
Table 3.
Summary of O3 RF mean values for clear sky conditions. Differences are calculated between NA (experiment with no aircraft emission), AE (experiment with only gaseous emission [NOx+H2O]) and AE* (experiment with gaseous and particle emission [NOx+H2O+SO4]). RF values are calculated with Oslo and ULAQ radiative code at ECMWF and National Center for Environmental Prediction (NCEP) tropopause level, respectively. LW, SW, NET are longwave, shortwave and net RFs, respectively. Only LW instantaneous values are considered here.
| | | | Oslo | ULAQ |
|---|
| Model | EXP | O3 col [DU] | LW-inst [mW/m2] | SW [mW/m2] | NET-inst [mW/m2] | LW-inst [mW/m2] | SW [mW/m2] | NET-inst [mW/m2] |
|---|
| ULAQ | AE-NA | 0.463 | 20.65 | 3.43 | 24.08 | 20.48 | 2.84 | 23.52 |
| AE*-NA | 0.430 | 19.73 | 3.27 | 23.00 | 19.53 | 2.72 | 22.25 |
| AE*-AE | −0.033 | −0.92 | −0.16 | −1.08 | −0.95 | −0.12 | −1.07 |
| CTM2 | AE-NA | 0.576 | 25.88 | 3.13 | 29.02 | 25.97 | 2.56 | 28.53 |
| AE*-NA | 0.549 | 25.02 | 3.09 | 28.11 | 25.14 | 2.47 | 27.61 |
| AE*-AE | −0.026 | −0.86 | −0.04 | −0.91 | −0.83 | −0.09 | −0.92 |
| CTM3 | AE-NA | 0.632 | 27.46 | 3.49 | 30.95 | 27.56 | 2.84 | 30.40 |
| AE*-NA | 0.606 | 26.63 | 3.42 | 30.05 | 26.79 | 2.78 | 29.57 |
| AE*-AE | −0.026 | −0.83 | −0.07 | −0.90 | −0.77 | −0.06 | −0.83 |
| ULAQ_FLX | AE-NA | 0.264 | 14.12 | 3.52 | 17.64 | 14.10 | 2.93 | 17.03 |
| AE*-NA | 0.251 | 13.66 | 3.37 | 17.03 | 13.64 | 2.84 | 16.48 |
| AE*-AE | −0.013 | −0.46 | −0.15 | −0.61 | −0.46 | −0.09 | −0.55 |
Table 4.
As in
Table 3, but for total sky conditions. LW-RF and NET-RF with stratospheric temperature adjustment are also included here.
Table 4.
As in Table 3, but for total sky conditions. LW-RF and NET-RF with stratospheric temperature adjustment are also included here.
| | | | Oslo | ULAQ |
|---|
| model | EXP | O3 col [DU] | LW-inst [mW/m2] | LW-adj [mW/m2] | SW [mW/m2] | NET-adj [mW/m2] | LW-inst [mW/m2] | LW-adj [mW/m2] | SW [mW/m2] | NET-adj [mW/m2] |
|---|
| ULAQ | AE-NA | 0.463 | 14.82 | 12.59 | 5.26 | 17.85 | 16.43 | 13.64 | 4.52 | 18.16 |
| AE*-NA | 0.430 | 14.16 | 12.03 | 5.02 | 17.05 | 15.72 | 13.03 | 4.32 | 17.35 |
| AE*-AE | −0.033 | −0.66 | −0.56 | −0.24 | −0.80 | −0.71 | −0.61 | −0.20 | −0.81 |
| CTM2 | AE-NA | 0.576 | 18.70 | 16.19 | 4.75 | 20.94 | 21.65 | 17.99 | 4.01 | 22.00 |
| AE*-NA | 0.549 | 18.06 | 15.50 | 4.63 | 20.13 | 20.95 | 17.26 | 3.89 | 21.15 |
| AE*-AE | −0.026 | −0.64 | −0.69 | −0.12 | −0.81 | −0.70 | −0.73 | −0.12 | −0.85 |
| CTM3 | AE-NA | 0.632 | 19.77 | 17.11 | 5.27 | 22.38 | 22.80 | 19.16 | 4.44 | 23.60 |
| AE*-NA | 0.606 | 19.18 | 16.59 | 5.14 | 21.63 | 22.25 | 18.52 | 4.35 | 22.87 |
| AE*-AE | −0.026 | −0.59 | −0.62 | −0.13 | −0.75 | −0.64 | −0.64 | −0.09 | −0.73 |
| ULAQ_FLX | AE-NA | 0.264 | 10.09 | 7.25 | 4.73 | 11.98 | 11.05 | 8.12 | 4.14 | 12.26 |
| AE*-NA | 0.251 | 9.77 | 7.06 | 4.54 | 11.60 | 10.73 | 7.90 | 3.95 | 11.85 |
| AE*-AE | −0.013 | −0.32 | −0.19 | −0.19 | −0.38 | −0.32 | −0.22 | −0.19 | −0.41 |
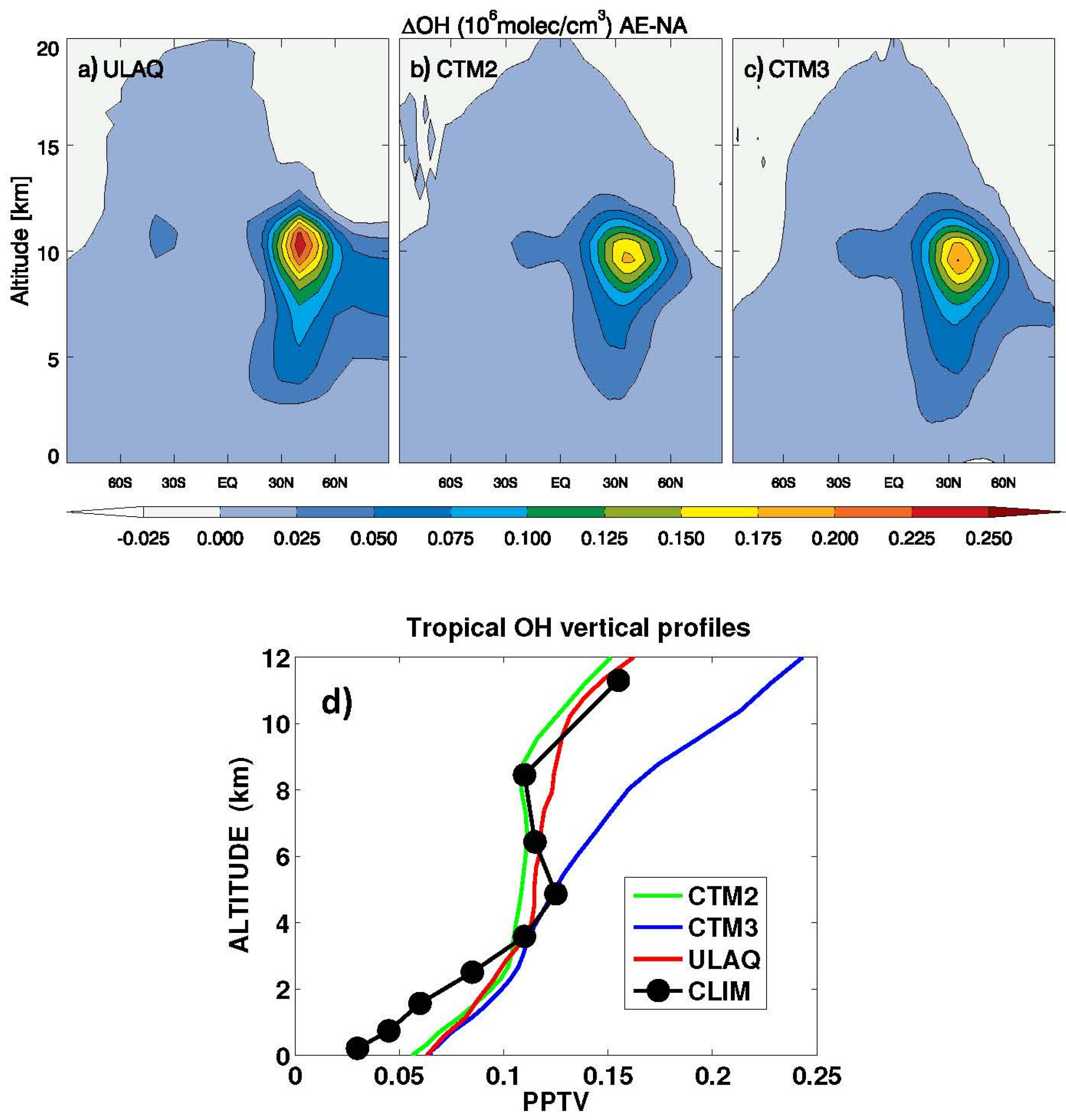
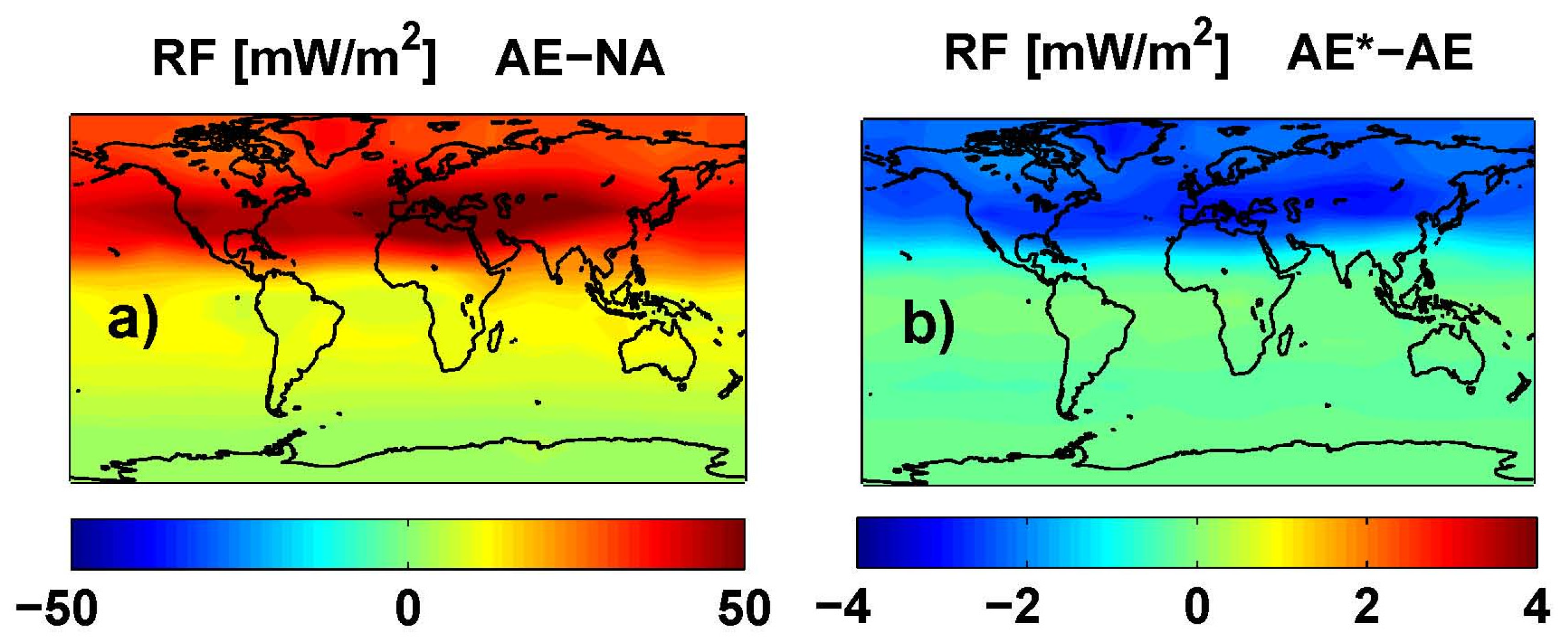
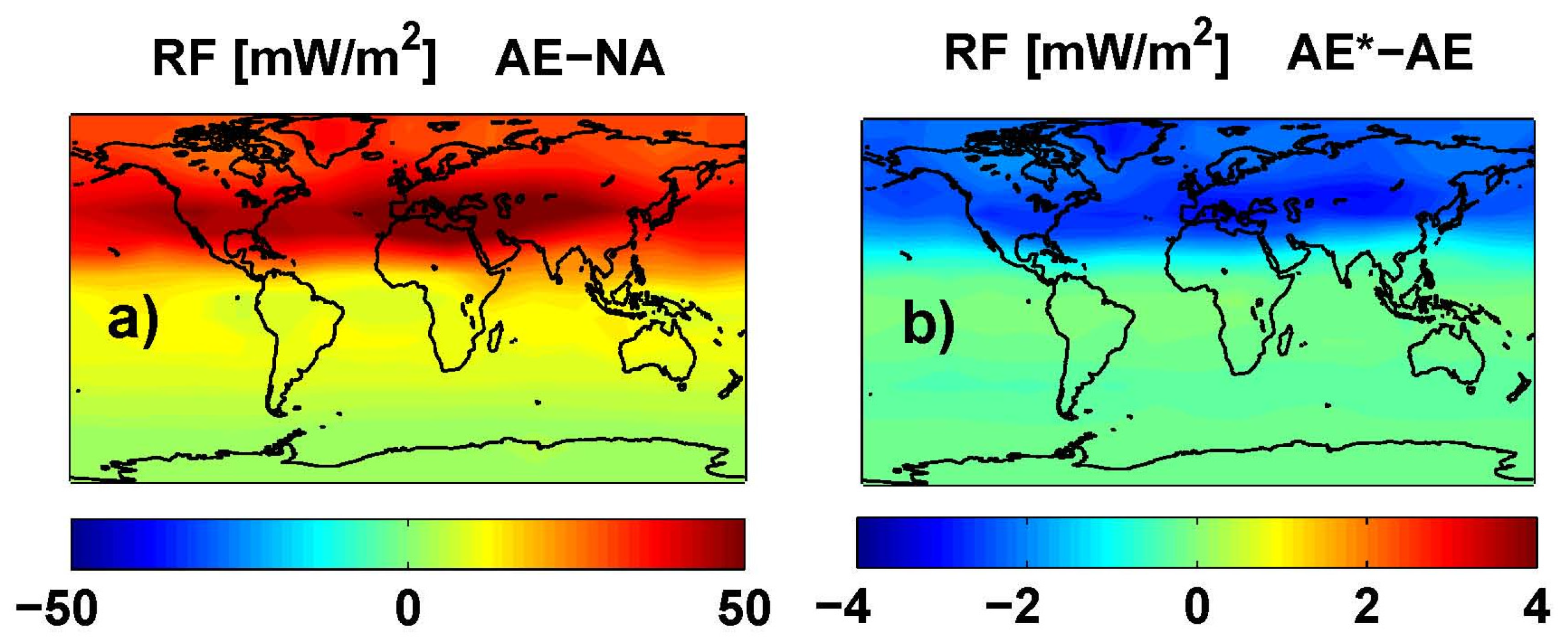
Figure 11.
O3 RF (adjusted) (mW/m2) from an average of ULAQ, CTM2 and CTM3 model results and both radiative transfer codes. Left panel (a) is for AE-NA (globally averaged value: 20.82 mW/m2); right panel (b) is for AE*-AE (globally averaged value: −0.79 mW/m2).
Figure 11.
O3 RF (adjusted) (mW/m2) from an average of ULAQ, CTM2 and CTM3 model results and both radiative transfer codes. Left panel (a) is for AE-NA (globally averaged value: 20.82 mW/m2); right panel (b) is for AE*-AE (globally averaged value: −0.79 mW/m2).
A summary of the radiative forcing due to the aviation NO
x perturbation is presented in
Table 5. We note that the short-term O
3 RF is close to, or slightly above, the mean value reported by Holmes
et al. [
5]. Further discussion on short-term O
3 RF from our models can be found in Søvde
et al. [
24]. Also listed in
Table 5 is CH
4 radiative forcing. The latter is calculated as a function of the lifetime percentage reduction (∆τ-CH
4) due to the OH increase produced by the aircraft-induced NO
x increase (via NO + HO
2 → NO
2 + OH), with the following formula: CH
4-RF(mW/m
2) = χ-CH
4(ppbv) × 0.37 × ∆τ-CH
4(%)/100 × 1.4 [
77], where τ is the CH
4 lifetime. A magnification factor 1.4 is used to take into account that the CH
4 prediction in the models is usually made using a fixed mixing ratio boundary condition at the surface, so that the CH
4 adjustment to the upper tropospheric OH perturbation is underestimated. As explained in IPCC [
85], the factor 1.4 may be used as a best estimate to adjust the calculated CH
4 lifetime change to the missing feedback of the upper tropospheric OH perturbation on the calculated lower tropospheric CH
4 mixing ratios. An exception to this is for the ULAQ_FLX experiment, where the CH
4 adjustment to the changing OH field is accounted for by the use of a flux boundary condition at the surface. Incidentally, we note that the ULAQ_FLX to ULAQ ratio of the CH
4 lifetime change is 1.39 for both AE-NA and AE*-NA, which is very close to the 1.4 factor suggested in IPCC [
85].
The uncertainty in the calculated CH4 RF is estimated to be approximately ±10% [
86], which would not significantly change our result. In the NO
x-related RF, we also include the effect of CH
4 on stratospheric water vapour. This is estimated to give an additional RF of 15% of the CH
4 RF [
87], although the uncertainty is large, about 70% [
86]. The CH
4 RF equation is a simplification of the detailed formula reported by IPCC [
86]; those formulas are the basis for calculating the specific forcing, which is linked to the unperturbed CH
4 concentrations so that a higher CH
4 concentration yields a lower specific forcing. Our approach is often used when assessing impacts of small CH4 perturbations, and we find that our CH
4 RF differs by less than 1% compared to the full equations using CH
4 and N
2O. Scaling up our lifetime changes linearly to match 1 Tg(N)/year, we get about 1.1%–1.5%, which is in the lower range of Holmes
et al. [
5]. Gottschaldt
et al. [
23] finds a similar value of 1.5%, which is in agreement with our higher values. Correspondingly, our CH
4 RF is also in the lower range of Holmes
et al. [
5].
Table 5.
Summary of NOx-related RF mean value terms: O3, CH4 and PMO (primary mode ozone, i.e., long-term ozone response). Ozone RF values are obtained as a mean from Oslo and ULAQ radiative code calculations. Differences are calculated between NA (experiment with no aircraft emission), AE (experiment with only gaseous emission [NOx+H2O]) and AE* (experiment with gaseous and particle emission [NOx+H2O+SO4]). See text for details on the calculations of CH4 RF, PMO RF and the stratospheric H2O RF induced by CH4 changes. Note that for ULAQ_FLX, O3-RF includes both short-term and long-term (primary mode) effects, whereas for the other models, only short-term O3 RF is included (long-term effects are then included in “PMO RF”).
Table 5.
Summary of NOx-related RF mean value terms: O3, CH4 and PMO (primary mode ozone, i.e., long-term ozone response). Ozone RF values are obtained as a mean from Oslo and ULAQ radiative code calculations. Differences are calculated between NA (experiment with no aircraft emission), AE (experiment with only gaseous emission [NOx+H2O]) and AE* (experiment with gaseous and particle emission [NOx+H2O+SO4]). See text for details on the calculations of CH4 RF, PMO RF and the stratospheric H2O RF induced by CH4 changes. Note that for ULAQ_FLX, O3-RF includes both short-term and long-term (primary mode) effects, whereas for the other models, only short-term O3 RF is included (long-term effects are then included in “PMO RF”).
| Species | EXP | ULAQ | CTM2 | CTM3 | ULAQ_FLX |
|---|
| ΔO3 col [DU] | AE-NA | 0.463 | 0.576 | 0.632 | 0.264 |
| AE*-NA | 0.430 | 0.549 | 0.606 | 0.251 |
| AE*-AE | −0.033 | −0.026 | −0.026 | −0.013 |
| O3 RF [mW/m2] | AE-NA | 18.00 | 21.47 | 22.99 | 12.12 |
| AE*-NA | 17.20 | 20.64 | 22.25 | 11.72 |
| AE*-AE | −0.80 | −0.83 | −0.74 | −0.40 |
| Δτ-CH4 [mo] | AE-NA | −0.82 | −1.24 | −1.13 | −1.14 |
| AE*-NA | −0.76 | −1.21 | −1.09 | −1.06 |
| AE*-AE | +0.06 | +0.03 | +0.04 | +0.08 |
| Δτ-CH4 [%] | AE-NA | −0.81 | −1.03 | −1.09 | −1.17 |
| AE*-NA | −0.75 | −1.01 | −1.05 | −1.09 |
| AE*-AE | +0.06 | +0.02 | +0.04 | +0.08 |
| CH4 RF [mW/m2] | AE-NA | −7.30 | −9.33 | −9.88 | −7.60 |
| AE*-NA | −6.86 | −9.14 | −9.57 | −7.08 |
| AE*-AE | +0.44 | +0.18 | +0.32 | +0.52 |
| Stratospheric H2O RF [mW/m2] from CH4 changes | AE-NA | −1.10 | −1.40 | −1.48 | −1.14 |
| AE*-NA | −1.03 | −1.37 | −1.43 | −1.06 |
| AE*-AE | +0.07 | +0.03 | +0.05 | +0.08 |
| PMO RF [mW/m2] | AE-NA | −3.65 | −4.66 | −4.94 | – |
| AE*-NA | −3.43 | −4.57 | −4.78 | – |
| AE*-AE | +0.22 | +0.09 | +0.16 | -- |
| TOTAL RF from NOx [mW/m2] | AE-NA | 5.96 | 6.08 | 6.69 | 3.38 |
| AE*-NA | 5.88 | 5.56 | 6.47 | 3.58 |
| AE*-AE | −0.08 | −0.52 | −0.22 | +0.20 |
Ozone changes produced by tropospheric NO
x emissions are not only associated with a short-term response of the NO
x and HO
x perturbations (see
Table 3 and
Table 4). The long-term response to CH
4 changes driven by the OH perturbation has to be considered as well. As discussed above, CTMs are normally run using a fixed mixing ratio boundary condition at the surface and this does not allow lower tropospheric CH
4 to adjust to the upper tropospheric OH perturbation produced by aviation NO
x emissions. In this case, a parametric formula, such as the one adopted here,
i.e., PMO-RF = 0.5 × CH
4-RF [
86], has to be used in order to estimate the long-term “primary mode” effect. The O
3 RF reported in
Table 5 for ULAQ_FLX includes both the short-and long-term responses of ozone. The difference between ULAQ and ULAQ_FLX represents in a first approximation the long-term O
3 response in ULAQ_FLX, which is approximately 77% of the ULAQ_FLX CH
4-RF. This is a larger value with respect to the 50% used in the parametric formula above, simply because the calculated difference between ULAQ and ULAQ_FLX in the short-term O
3 RF includes not only the PMO RF, but also the feedback in ULAQ_FLX of the changing CH
4 on stratospheric water vapour and finally on HO
x and NO
x catalytic cycles for ozone destruction.
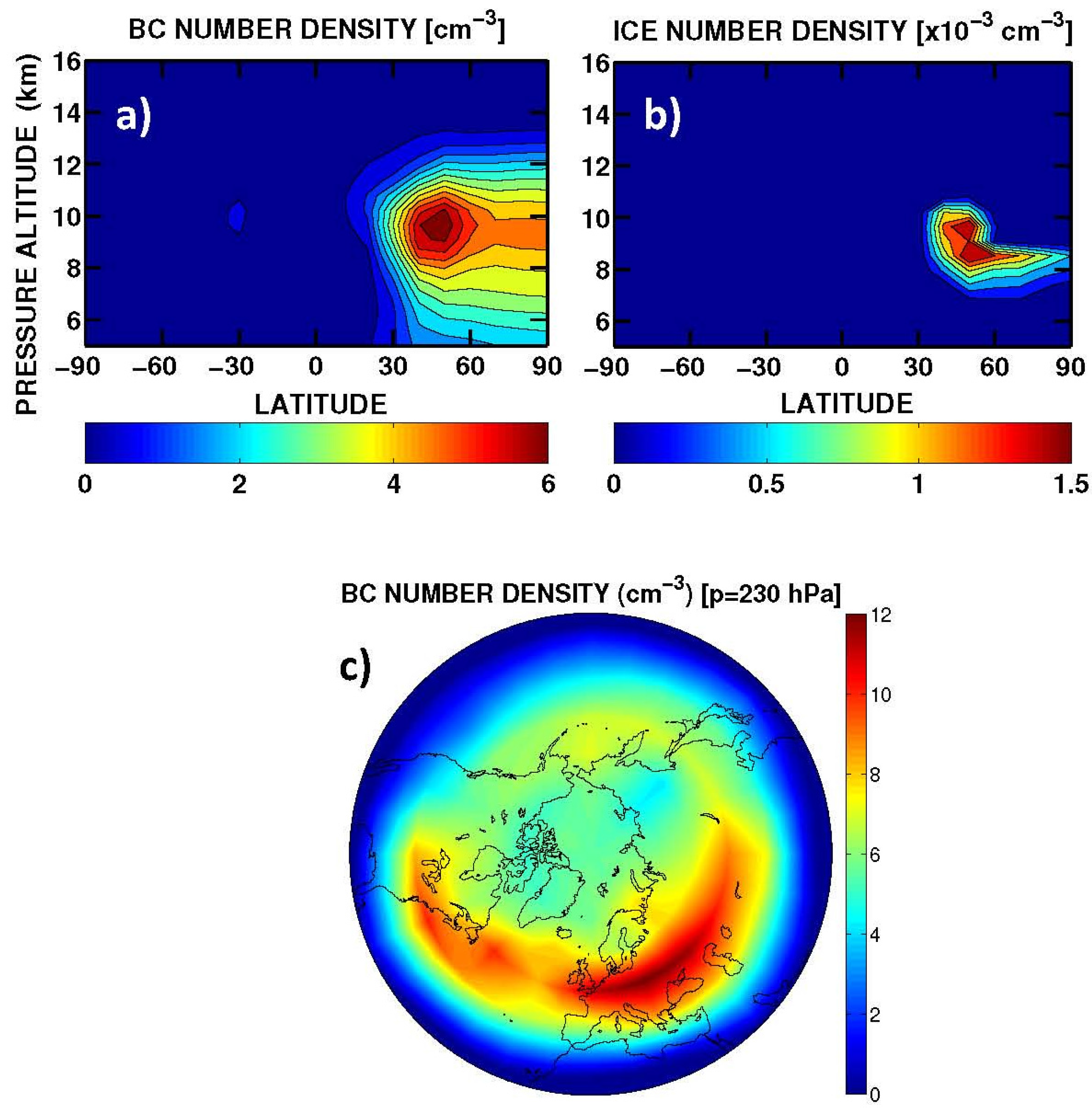
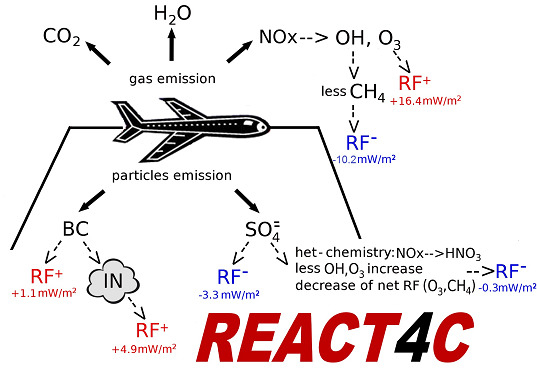
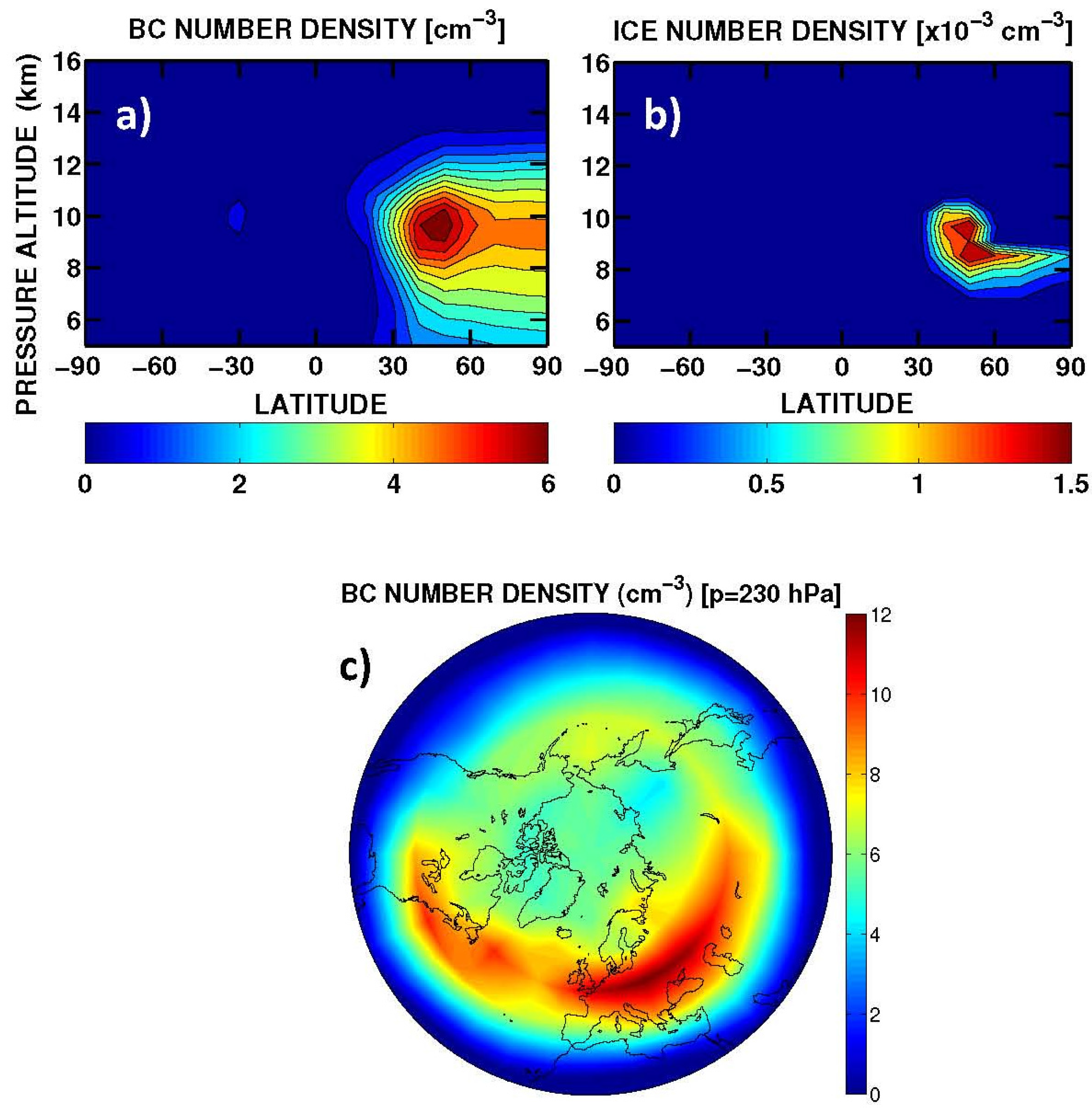
A summary of the RF from aviation aerosol is presented in
Table 6. The calculated normalized direct forcings for sulphate and BC (−140 and 1700 W/g, respectively) are comparable with those reported in IPCC [
85] (
i.e., −125 to −214 and 1100–3000 W/g, respectively). The BC RF (+0.8 mW/m
2, globally) is due to direct soot emission from the aircraft (with an effective radius of 0.14 μm). The sulphate RF (−3.5 mW/m
2, globally) is due to aircraft emissions of SO
2 (EI = 0.8 g-SO
2/kg-fuel), which is oxidized by OH to SO
4 with additional condensation in the accumulation mode of the sulphate aerosol size distribution (with a calculated effective radius of 0.16 μm). The RF signs of BC (positive) and SO
4 (negative) are due to the dominant solar radiation absorption and scattering, respectively. The indirect RF of aviation soot through formation of upper tropospheric cirrus-ice particles via heterogeneous freezing (
i.e., the soot-cirrus RF) is obtained on the basis of a particle size distribution with a calculated effective radius of 5 μm (+4.9 mW/m
2, globally). The largest uncertainty on this soot-cirrus RF comes from the BC fraction assumed to act as ice nuclei (0.1% in our case). Values in the range 0.1%–1.0% (or even smaller) are suggested by Hendricks
et al. [
68]. For soot-cirrus, the longwave contribution dominates over the shortwave forcing, due to the size of these particles, whereas for BC and SO
4 the solar forcing dominates.
Table 7 shows that the indirect BC results are consistent with Gettelman and Chen [
20], where an aviation impact <10 mW/m
2 was observed when a fixed nucleating efficiency of 0.1% for BC was applied. Furthermore, the RFs per unit BC emission from both studies are within 2.5% of each other. However, it should be noted that the treatment of background ice nucleation, together with the uncertainties in the ice nucleation potential of “pre-activated” aircraft soot, could produce large uncertainties surrounding the indirect effect of aircraft soot, resulting in either negative or positive RF, as shown by Zhou and Penner [
88] and also summarized in
Table 7.
Table 8 presents an overall summary of the aviation RFs calculated in the present study, excluding CO
2, contrails and contrail-cirrus, which are not explicitly considered in the present study. The contribution of aviation water vapour accumulating above the tropopause is also considered, and the calculated value (0.6 mW/m
2) is consistent with that obtained and discussed in Wilcox
et al. [
89] (0.9 ± 0.5 mW/m
2). According to our calculations, aircraft emitted aerosols tend to decrease the O
3 production and its positive RF (by −0.27±0.2 mW/m
2), but on the other hand the net contribution of direct and indirect soot forcings (positive) plus the direct sulphate forcing (negative) (+2.7 mW/m
2) tends to increase the overall aviation RF. The small and negative SO4 forcing (−0.2 mW/m
2) in the AE-NA case is due to the OH increase produced by aviation NO
x emissions, which favours the SO
2 oxidation into sulphate. According to our calculations, the net NO
x-induced RF appears to be the dominant term (excluding CO
2 and contrails), although it might be somewhat overestimated, because the effect of NOx emissions on nitrate aerosols is not included in this study. However, the existence of such an effect (from aviation emissions) and its magnitude are not well established. An increasing contribution to solar radiation scattering would add a negative term to the NO
x-related RF, as demonstrated by Unger
et al. [
90]. However, there are not enough studies whereby nitrates are included in CTM simulations and therefore, a reliable RF estimate that is comparable to the rest of the aviation RFs reported in the study is not available. In addition, a rather marginal effect of aircraft emissions on nitrate aerosol could be expected when taking into account that the formation of nitrate aerosols (NH4
+NO3
−) requires a significant presence of ammonium, which may be the case only in the lower troposphere. Holmes
et al. [
5] have shown a net NO
x effect amounting to 4.5 ± 4.5 mW/m
2 for 1 Tg(N)/year aircraft emissions. Our results are consistent with that estimate.
Table 6.
Summary of aerosol Radiative Forcing mean values: direct contributions from sulphate and soot (BC) and indirect BC forcing (soot-cirrus) (AE*-NA): ULAQ-CTM values and ULAQ radiative code. Here Δτ is the particle optical thickness perturbation at λ = 0.55 μm. The final column shows the normalized RF (i.e., absolute value of the net-RF divided by the globally averaged Δ-load).
Table 6.
Summary of aerosol Radiative Forcing mean values: direct contributions from sulphate and soot (BC) and indirect BC forcing (soot-cirrus) (AE*-NA): ULAQ-CTM values and ULAQ radiative code. Here Δτ is the particle optical thickness perturbation at λ = 0.55 μm. The final column shows the normalized RF (i.e., absolute value of the net-RF divided by the globally averaged Δ-load).
| Species | Δ-load TOT [μg/m2] | Δ-load STRAT [μg/m2] | Δτ [λ = 0.55 μm] | SW [mW/m2] | LW-inst [mW/m2] | LW-adj [mW/m2] | NET-adj [mW/m2] | NRF [W/g] |
|---|
| SO4 | 25.0 | 6.6 | 2.4E-04 | −5.2 | 1.8 | 1.7 | −3.5 | 140 |
| BC | 0.46 | 0.10 | 3.0E-06 | 0.85 | 0.01 | −0.08 | 0.78 | 1700 |
| Soot Cirrus | 320 | 0 | 1.0E-04 | −1.7 | 6.6 | 6.6 | 4.9 | 15 |
Table 7.
RF from other soot-cirrus studies. Relevant experiments not directly comparable to this study are also included for illustrative purposes.
Table 7.
RF from other soot-cirrus studies. Relevant experiments not directly comparable to this study are also included for illustrative purposes.
| | This study | Gettelman and Chen [20] | Zhou and Penner [88] |
|---|
| Model | ULAQ-CTM | CAM5/MAM | CAM5.2/IMPACT |
| Experimental setup | Fixed nucleating efficiency (0.1%), with aviation NOx and SO4, no contrails. | Fixed nucleating efficiency (0.1%), no aviation NOx/SO4/contrails. [Also with SO4 and contrails] | [Varying nucleating efficiencies to include pre-activated soot and sensitivity to background. No aviation NOx, with contrails] |
| Aircraft BC emission (Tg-BC/year) | 0.00407 | 0.00681 | 0.00681 |
| Soot-cirrus TOA RF (mW/m2) | 4.9 | 8.0 [−21 ± 11] | [−350 to 90] |
| RF per unit BC emission (Wm−2/Tg-BC/year) | 1.20 | 1.17 | |
Table 8.
Summary of Radiative Forcing (RF) mean values relevant for aviation emissions, excluding CO2, contrails and contrail-cirrus, which are not explicitly considered in the present study. RF values from NOx are obtained as a mean from the CTMs (ULAQ, CTM2 and CTM3) and Oslo and ULAQ radiative code calculations. The uncertainty interval is calculated as the range of the net NOx-related RF from the three models.
Table 8.
Summary of Radiative Forcing (RF) mean values relevant for aviation emissions, excluding CO2, contrails and contrail-cirrus, which are not explicitly considered in the present study. RF values from NOx are obtained as a mean from the CTMs (ULAQ, CTM2 and CTM3) and Oslo and ULAQ radiative code calculations. The uncertainty interval is calculated as the range of the net NOx-related RF from the three models.
| species | RF [mW/m2] AE-NA | RF [mW/m2] AE*-NA | RF [mW/m2] AE*-AE |
|---|
| TOTAL RF from NOx | 6.2 ± 0.4 | 6.0 ± 0.4 | −0.27 ± 0.2 |
| Direct SO4 RF | −0.2 | −3.5 | −3.3 |
| Direct BC RF | 0.0 | 0.8 | 0.8 |
| Indirect BC RF (soot cirrus) | 0.0 | 4.9 | 4.9 |
| Stratospheric H2O | 0.6 | 0.6 | 0.0 |
| TOTAL RF | 6.6 | 8.8 | 2.2 |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}