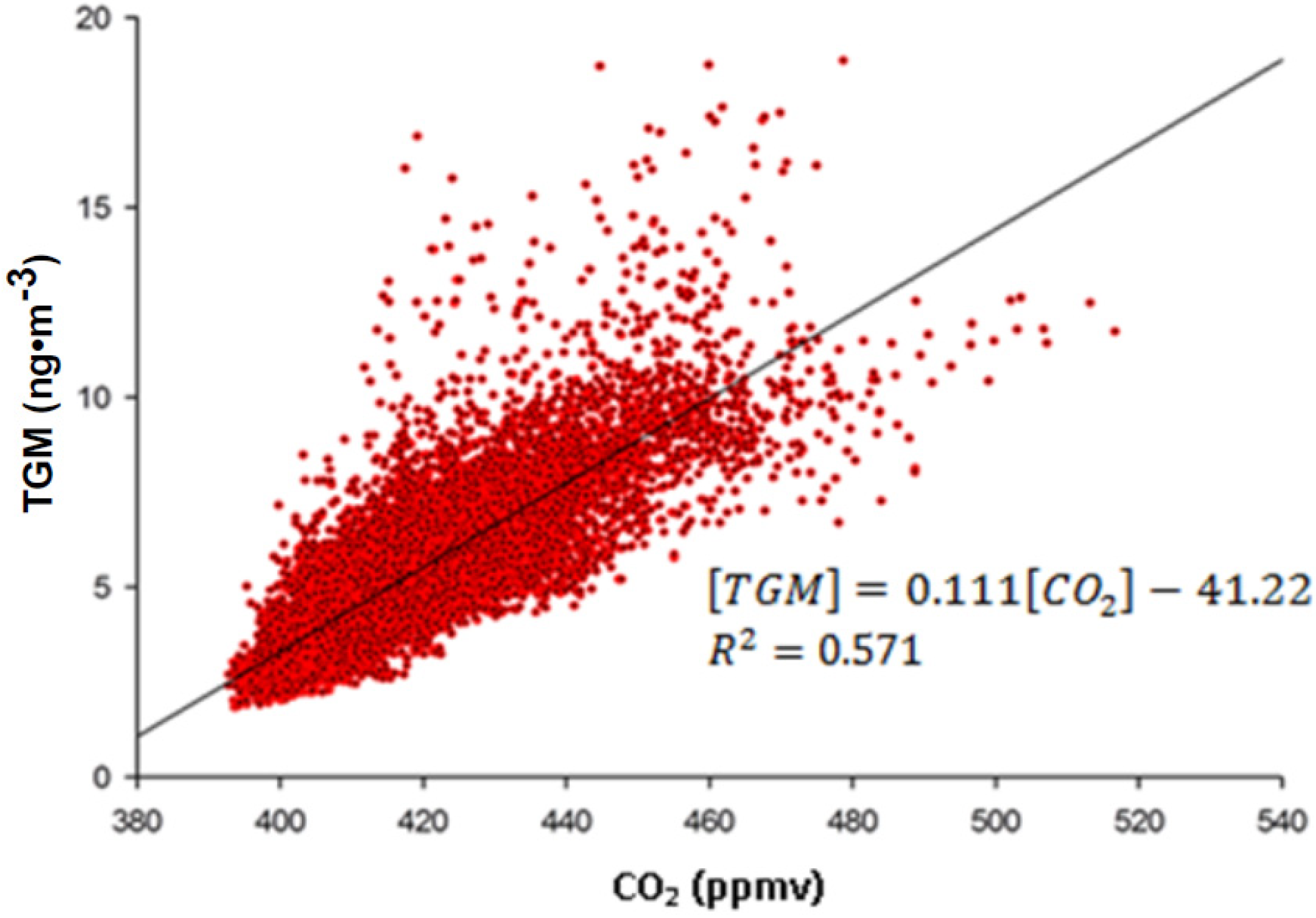
2.2. Diurnal Variation of TGM and Unaccounted for Sources
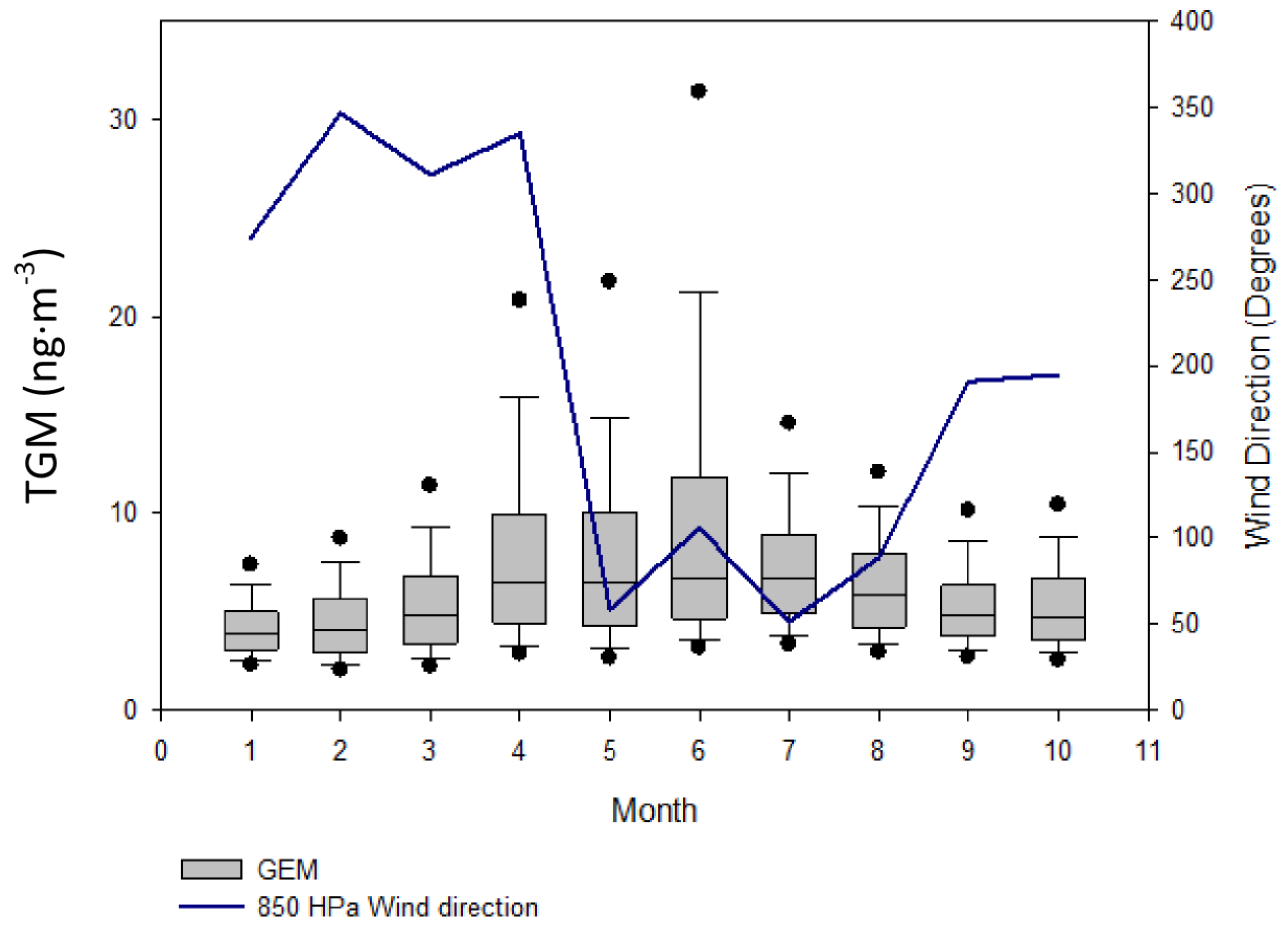
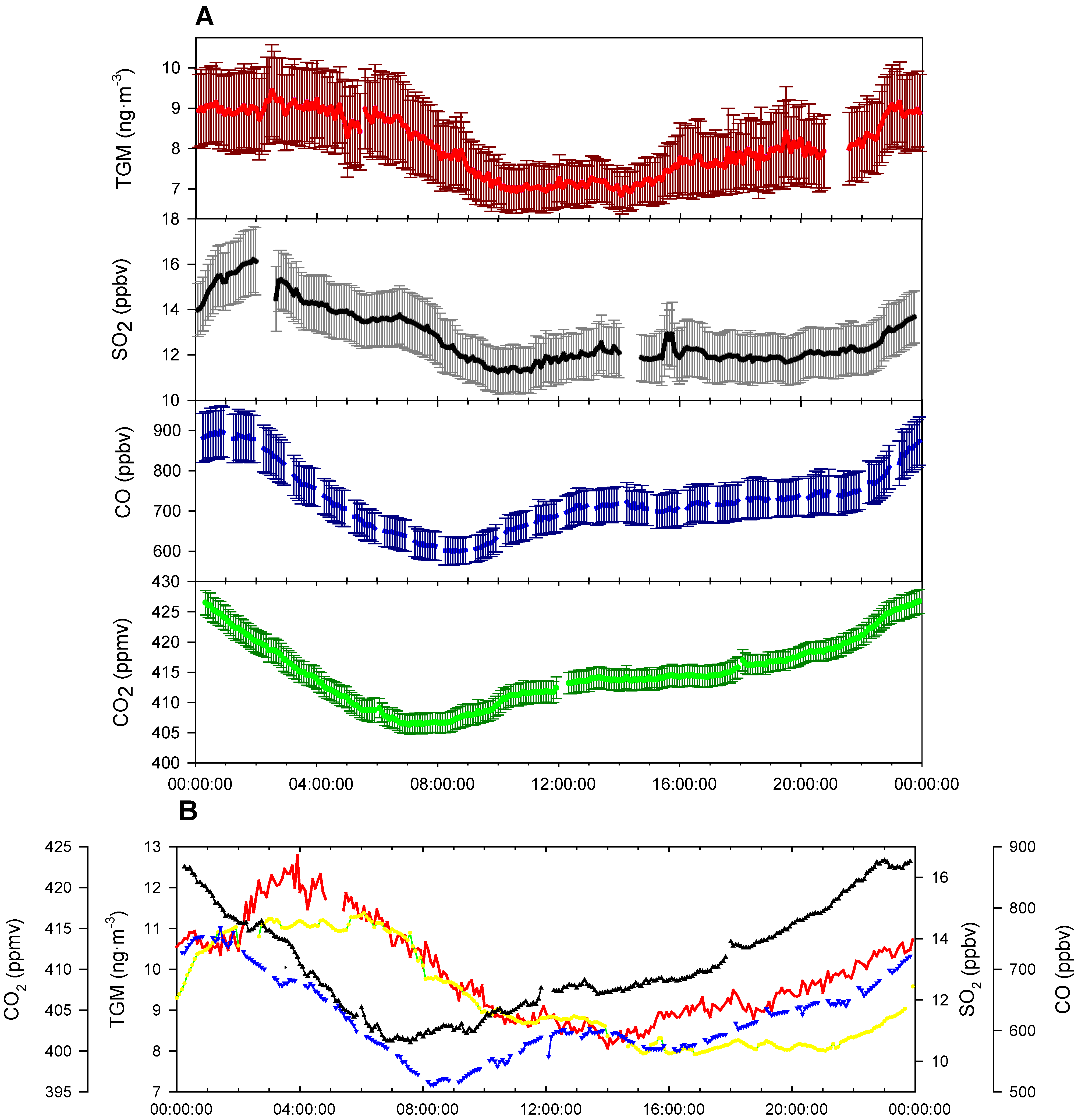
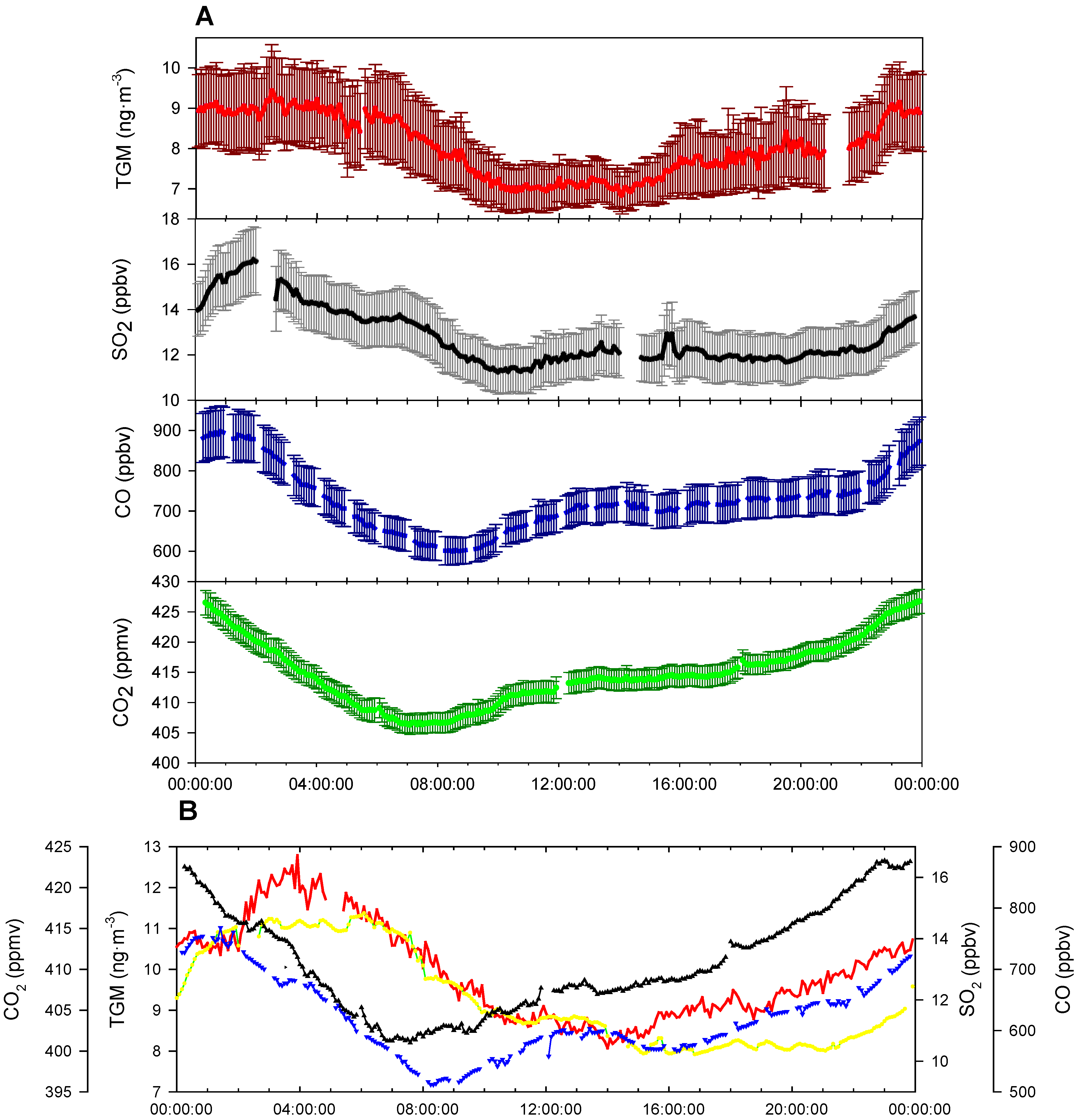
TGM shows a distinct diurnal pattern on a yearly averaged basis (
Figure 8A). On average, the TGM concentrations are mostly constant from 0:00 to 6:00 UT with a small spike in concentrations at 2:00 UT. From 6:00 to 10:00 UT there is a drop of ~2.0 ng∙m
−3 in TGM concentrations. Concentrations are rather constant from 10:00 to 13:00 UT. After 14:00, the concentrations in TGM begin to rise until 0:00, except for a small decline from 19:00 until 21:00 UT and a rapid rise between 22:00 and 23:00 UT. The nocturnal rise in TGM concentrations over 14:00–24:00 UT is indicative of the strong YRD emissions trapped under a low nocturnal boundary layer. The sources building under the nocturnal boundary layer are most likely of anthropogenic origin because legacy emission from soil and water are mediated by temperature and solar radiation [
30], both of which are low at night. A significant correlation was observed between the nine-month average diurnal cycles of TGM and SO
2 with r
2 = 0.70, indicating the possibility of a strong source for both TGM and SO
2 on the nine-month scale. The source for such co-emission is most likely coal combustion considering that >70% of SO
2 emissions in the YRD region were from industrial combustion and power generation using coal in 2010 [
31]. In contrast to the correlation of TGM and SO
2, the nine-month averaged diurnal cycles of TGM and other combustion products such as CO, NO
y, and CO
2 all have poor correlations with r
2 < 0.5, owing to a decline in concentrations of CO, NO
y, and CO
2 earlier in the morning and recovering sooner with near constant concentrations from 12:00 UT until 21:00 UT. This discrepancy indicates TGM has a different source profile than CO, CO
2, and NO
y and is likely co-emitted with SO
2. The line of best fit for the nine-month average diurnal cycles of TGM and SO
2 is:
The correlation slope translated to a molar ratio is 54.43 × 10−6 mol Hg∙mol−1 SO2. Normally trace gasses with such disparate lifetimes as TGM and SO2 (six months to one year and ~one day respectively) would not facilitate a comparison to direct emissions. In this case the nine-month average diurnal cycle allows for a unique focusing on processes with a fine temporal resolution, in this case emissions. Furthermore, the close proximity of major point sources of both pollutants allowed sampling before major transformation or aging of the air mass could occur. The slope value can be compared to emission inventories of pollutants in the Yangtze River Delta to investigate YRD source types.
The SO
2 emissions given in Fu
et al. [
31] provide an estimate of 3.35 × 10
10 mol total SO
2 emissions for the YRD region in 2010. Applying the molar ratio derived above, the total mercury emission from the YRD region would have been 366 tons or 1.8 × 10
6 mol. This is an upper end estimate assuming no transformation or loss of SO
2 before measurement. A lower end estimate can be established by assuming that on a daily time scale, the temporal scale the correlation is based on, 63.2% of SO
2 was lost due to its 0.693-day half-life. Using this assumption of maximum daily loss before measurement the minimum emission of mercury would be 138 tons of Hg per year. Streets
et al. [
8] estimated that in 1999 the total mercury emission from Jiangsu and Zhejiang provinces, plus Shanghai, making up the YRD, were 42.16 tons or 2.1 × 10
5 mol Hg, which is almost an order of magnitude lower than our upper end estimate and about three times smaller than our lower end estimate. Alternatively stated, emission inventories only account for as low as 12% to 31% of observed emissions. There are four possible reasons for this discrepancy: increased TGM emissions from 1999 to 2011, decreased sulfur dioxide emissions from 2010 to 2011, YRD emissions were much richer in mercury, and/or significant reemission of deposited mercury. The most likely scenario is a combination of mercury reemission as noted by Zhu
et al. [
9] and YRD emissions being enriched and, yet, underestimated in TGM compared to SO
2. Mercury emissions have most likely risen since 1999 due to continued and rapid economic development and increasing coal consumption. However, the existing emission inventories reported a meager increase in mercury emissions of 7.32% from 536 tons nationally in 1999 [
8] to 575 tons in 2010 [
4]. Additionally, emissions of sulfur dioxide appear to be declining according to Fu
et al. [
31] due to greater implementation of pollution controls. The opposing trends of increased TGM and decreased SO
2 emissions are not documented; therefore it is unknown if these trends constitute the order of magnitude required for the observed ratio of TGM to SO
2. Regarding the significance of legacy emissions, the rather strong reemission of deposited mercury, hypothesized by Zhu
et al. [
9] to contribute to enhance summertime TGM concentrations, was unlikely the majority of YRD emissions as would be required to make up the differential between observed emissions and inventory values. This is based on terrestrial reemissions constituting approximately 30% of annual global emissions [
4].
Figure 8.
(A) Nine-month average diurnal cycle of the sampling period of TGM (Red), SO2 (Black), CO (Blue), and CO2 (Green), with error bars representing 95% confidence interval. (B) Average diurnal cycle for summer season (June–August). The time axis is in UT, i.e., local time−eight hours.
Figure 8.
(A) Nine-month average diurnal cycle of the sampling period of TGM (Red), SO2 (Black), CO (Blue), and CO2 (Green), with error bars representing 95% confidence interval. (B) Average diurnal cycle for summer season (June–August). The time axis is in UT, i.e., local time−eight hours.
Comparing seasonal differences, the rapid TGM peak seen in the nine-month average cycle (
Figure 8A) occurs at approximately the same time, 22:00–23:00 UT near sunrise, in all seasons except summer. During the summer season, the increase was much delayed taking place at approximately 2:00 UT (
Figure 8B), albeit earlier sunrises. The rapid enhancement of TGM occurs at near 10:00 AM local time, the approximate time of flux enhancements over soil in Canada during August, observed by Boudala
et al. [
30]. The time series of TGM flux from soil from Boudala
et al. [
30] was similar in shape to the diurnal cycle observed in Nanjing rapidly peaking at 10:00 AM, leveling off, and then declining. Although, the decrease in TGM concentrations comes much earlier in Nanjing likely as a result of an increased ventilation coefficient that peaks in the afternoon [
25], and would not affect flux concentrations measured by a chamber as in Boudala
et al. [
30]. Legacy flux was unlikely the cause of the early morning peak throughout the rest of the year because the TGM peak was too early with respect to flux measurements and was concomitant with an SO
2 peak that has no photolytic mechanism in the troposphere [
32]. The summer enhancement in Nanjing in mid-morning was ~2.0 ng∙m
−3. Summer fluxes should be the greatest according to current understanding of mercury soil flux because the release of mercury appears to be a function of soil moisture, temperature [
33]. Soil moisture should be greatest during this period due to the summer monsoon. The temperature and solar radiation reached the annual maximum [
34]. It is highly likely that these three factors listed above interacted synergistically to increase soil evasion during the summer thereby contributing to the early 10:00 AM TGM peak observed in
Figure 8B. Furthermore, mixing layer heights should theoretically affect all trace gases equivalently and land-sea winds should not affect the site due to being >200 km inland. The short and rapid enhancement seen in TGM in summer is also not followed by SO
2 as would be expected of coal emissions, which rules out coal combustion as a source. This enhancement was conceivably the release of legacy mercury as TGM, which would constitute 16% of peak concentrations in the summer diurnal cycle, far less than the global estimate of ~30% of mercury emissions being from natural or legacy fluxes. Additionally, this 16% of emissions is not enough to explain the discrepancy between observed and emission inventory based TGM concentrations.
Going back to the nine-month diel cycle, the drop in TGM concentrations comes in the afternoon, delayed several hours compared to the rise of the boundary layer; therefore it seems unlikely that downward mixing of less polluted air aloft caused the afternoon drop in TGM concentrations, unlike CO and CO
2. It appears that the concentrations of SO
2 and TGM were strongly controlled by the ventilation co-efficient which reaches its daily maximum in the afternoon [
25] reasonably showing YRD emissions as the major source of TGM.
The emissions estimate above can be compared to the simulated mean mercury concentrations by Pan
et al. [
7] using the emissions inventory of Streets
et al. [
8], there is a clear underestimation of median mercury concentrations in Nanjing. The STEM-Hg∙model results estimate a median concentration of 1.6–1.7 ng∙m
−3 [
7], while Zhu
et al. [
9] found median mercury concentrations to be 6.2 ng∙m
−3 [
9]. Lin
et al. used CMAQ-Hg to investigate the mass balance of mercury in East Asia and found higher concentrations of atmospheric mercury, ~3–4 ng∙m
−3 [
11], than Pan
et al. [
7], using similar emissions inventories, but TGM was still underestimated in comparison with measurements in Zhu
et al. [
9]. This discrepancy highlights further the fact that YRD TGM emissions are much greater than documented in Streets
et al. for the region [
8]. It is noted in Pan
et al. [
7] that discrepancies between the modeled and observed concentrations were in part due to the inaccurate model representation of dispersion from point sources. However, this problem should not affect mercury concentrations in the lower quartile of measurements, 4.5 ng∙m
−3, which nearly triples the high estimate of 1.7 ng∙m
−3 from Pan
et al. The discrepancies between the emissions estimated in this study and models highlight the unique source profile in Nanjing and the possibility of large unaccounted for sources of TGM emissions in the YRD.
2.3. Peak Concentrations
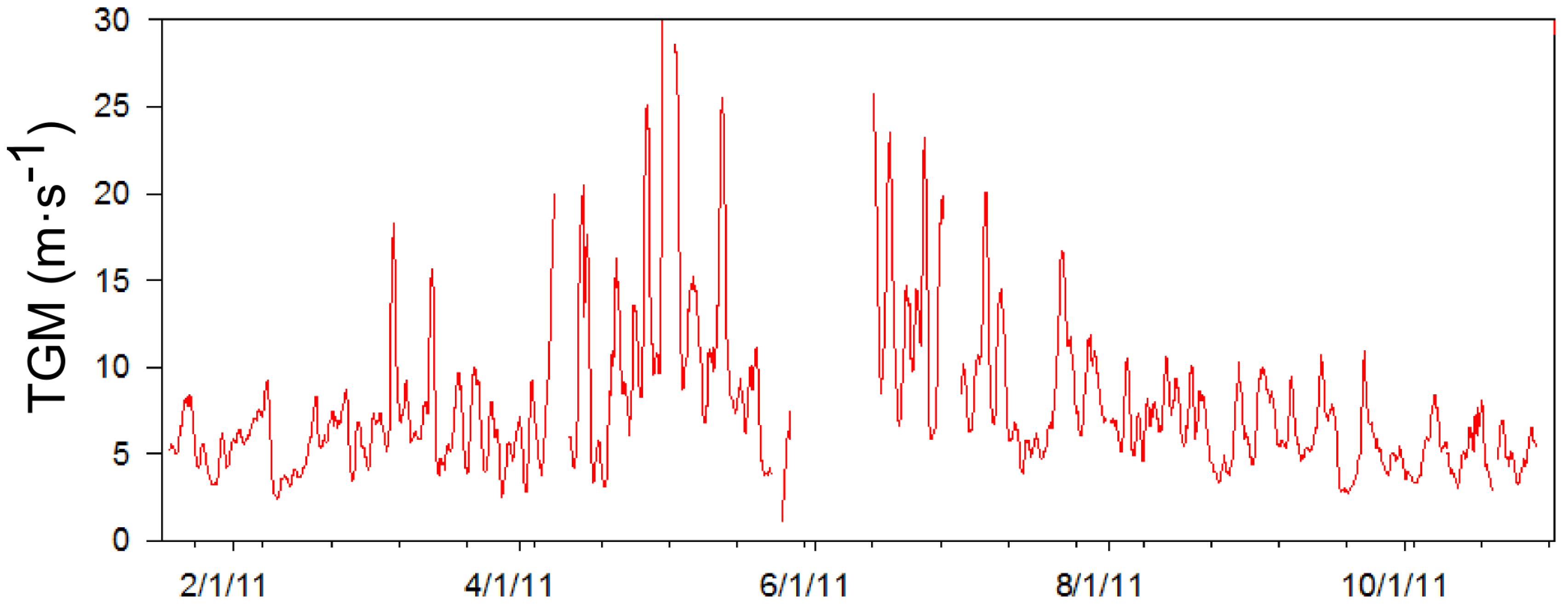
Throughout the measurement period TGM concentrations reached levels that were extraordinarily high compared to measurements made in North America and Europe [
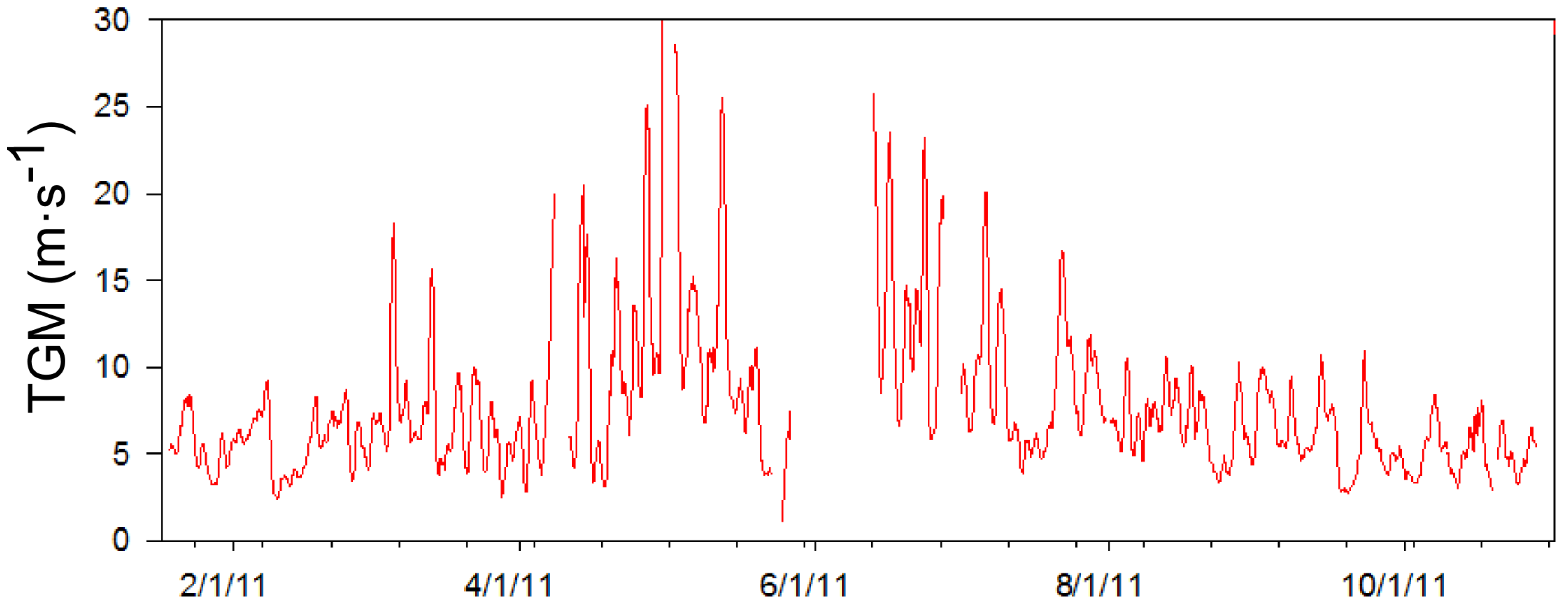
35]. In order to more clearly understand the sources and causes of these high concentrations, an episodic high concentration of TGM was defined for each season. One-day moving averages were used to smooth out diurnal variation, which was found to be the most frequent controlling interval based on a power spectrum analysis of the data (not shown). When the one-day moving average concentration of TGM (
Figure 9) exceeded the seasonal 90th percentile value it was considered an episode of high TGM concentrations. The seasons were defined as January and February for winter; March, April, and May for spring; June, July and August for summer; and September and October for fall. The episode concentration criterion was highest during the summer months at 17.1 ng∙m
−3 and lowest in winter at 8.3 ng∙m
−3. As a result, 20 high TGM episodes were identified in 2011 and are listed in
Table 1.
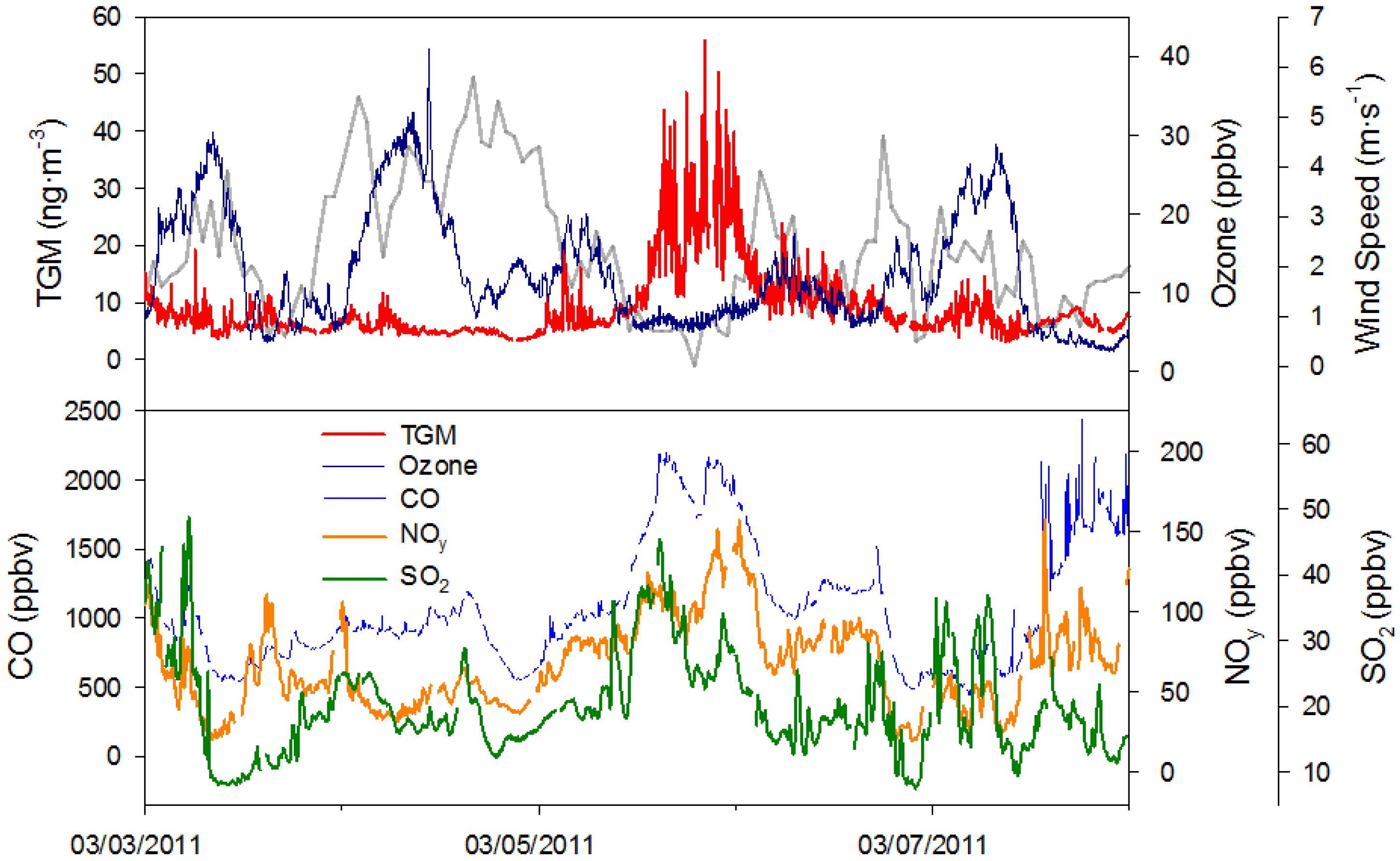
Once the episodes were determined they were plotted along with the time series of CO, SO
2, O
3, NO
y, wind speed, and direction. Using these four trace gases and wind, qualitative relationships between traces gases and peak TGM concentrations were determined. When TGM concentrations reached episode concentrations accompanied by spikes in urban tracers such as CO and SO
2 under weak wind conditions (wind speed <2 m∙s
−1), it was hypothesized that the source of higher TGM concentrations was the anthropogenic YRD emissions. Using this system, 50% of the episodes appeared due to stagnant conditions, including low O
3 episodes (
Table 1) and buildup of YRD sources in Nanjing. The largest controlling factor of TGM and high TGM concentrations was YRD emissions, while other controlling factors include the monsoon and transport from other source regions.
Figure 9.
The graph above shows the time series of the one-day moving average of mercury concentrations based on five-minute data.
Figure 9.
The graph above shows the time series of the one-day moving average of mercury concentrations based on five-minute data.
Table 1.
The table contains mercury episodes in chronological order. The date of the episode is the point of the peak. The concentration in column three is the highest achieved during a 24-hour moving average. The coincident peaks were qualitatively assigned from other trace gases. The other columns are the defining characteristics of the episode, the type or classification. The last three columns are the slope of the linear regression of the trace gas vs. mercury data from the 72 hours around the episode. Only slopes of correlations with an r2 > 0.2 and p-value < 0.001 were included.
Table 1.
The table contains mercury episodes in chronological order. The date of the episode is the point of the peak. The concentration in column three is the highest achieved during a 24-hour moving average. The coincident peaks were qualitatively assigned from other trace gases. The other columns are the defining characteristics of the episode, the type or classification. The last three columns are the slope of the linear regression of the trace gas vs. mercury data from the 72 hours around the episode. Only slopes of correlations with an r2 > 0.2 and p-value < 0.001 were included.
| # | Date | 24 Hr Moving avg Peak | Coincident Peaks | Characteristics | Classification | TGM-CO Slope | TGM-SO2 Slope | TGM-NOy Slope |
|---|
| 1 | 1/23 | 8.4 | CO, SO2 | Low winds | Local | 0.003 | N/A | 0.049 |
| 2 | 2/7 | 8.8 | CO, SO2, O3 | Low winds High O3 | Local | 0.003 | 0.074 | 0.063 |
| 3 | 2/17 | 8.3 | CO, SO2 | Low winds | Local | 0.004 | 0.141 | 0.051 |
| 4 | 2/24 | 8.7 | CO, SO2, O3 | Low winds High O3 | Local | 0.005 | 0.221 | 0.057 |
| 5 | 3/5 | 18.2 | CO, SO2 | Low Winds | Low O3 | 0.016 | 0.480 | 0.240 |
| 6 | 4/7 | 19.5 | CO, SO2, O3 | Low winds High O3 | Local | 0.017 | 0.382 | 0.242 |
| 7 | 4/13 | 20.5 | CO, Slight SO2 | Low winds | Local | N/A | N/A | 0.253 |
| 8 | 4/20 | 16.2 | CO, SO2 | Low winds & Sudden drop in O3 | Low O3 | 0.008 | 0.546 | 0.195 |
| 9 | 4/26 | 25.0 | None | High Winds & Other Gases low | Transport | N/A | N/A | N/A |
| 10 | 4/30 | 53.9 | None | High Winds & Other Gases low | Transport | N/A | N/A | N/A |
| 11 | 5/12 | 25.5 | SO2, O3 | Low CO, SO2 but elevated spiking O3 and moderate winds | Transport | N/A | N/A | N/A |
| 12 | 6/12 | 65.5 | CO, SO2 After TGM | Low Winds | Monsoon | 0.059 | N/A | N/A |
| 13 | 6/16 | 23.5 | CO, SO2 | Low winds | Monsoon | N/A | N/A | N/A |
| 14 | 6/23 | 23.1 | CO, SO2 | Low wind | Monsoon | 0.012 | 0.622 | N/A |
| 15 | 6/27 | 19.9 | CO, SO2, O3 | Trace gasses and Low winds | Monsoon | 0.011 | 0.914 | 0.377 |
| 16 | 7/6 | 20.1 | All | Low winds | Monsoon | N/A | 1.076 | 0.151 |
| 17 | 7/22 | 16.6 | All | Trace Gases peak and Low winds | Monsoon | N/A | 0.451 | N/A |
| 18 | 9/1 | 10.0 | All peak after | O3 Peaks and SO2 after low wind speeds | Local | N/A | N/A | N/A |
| 19 | 9/13 | 10.7 | None | CO drops O3 flat lines after peak SO2 drops Winds are moderate | Transport from Hangzhou | N/A | N/A | N/A |
| 20 | 9/22 | 10.9 | CO, O3 | Low winds, O3 peaks after CO peaks with TGM | Local | N/A | N/A | N/A |
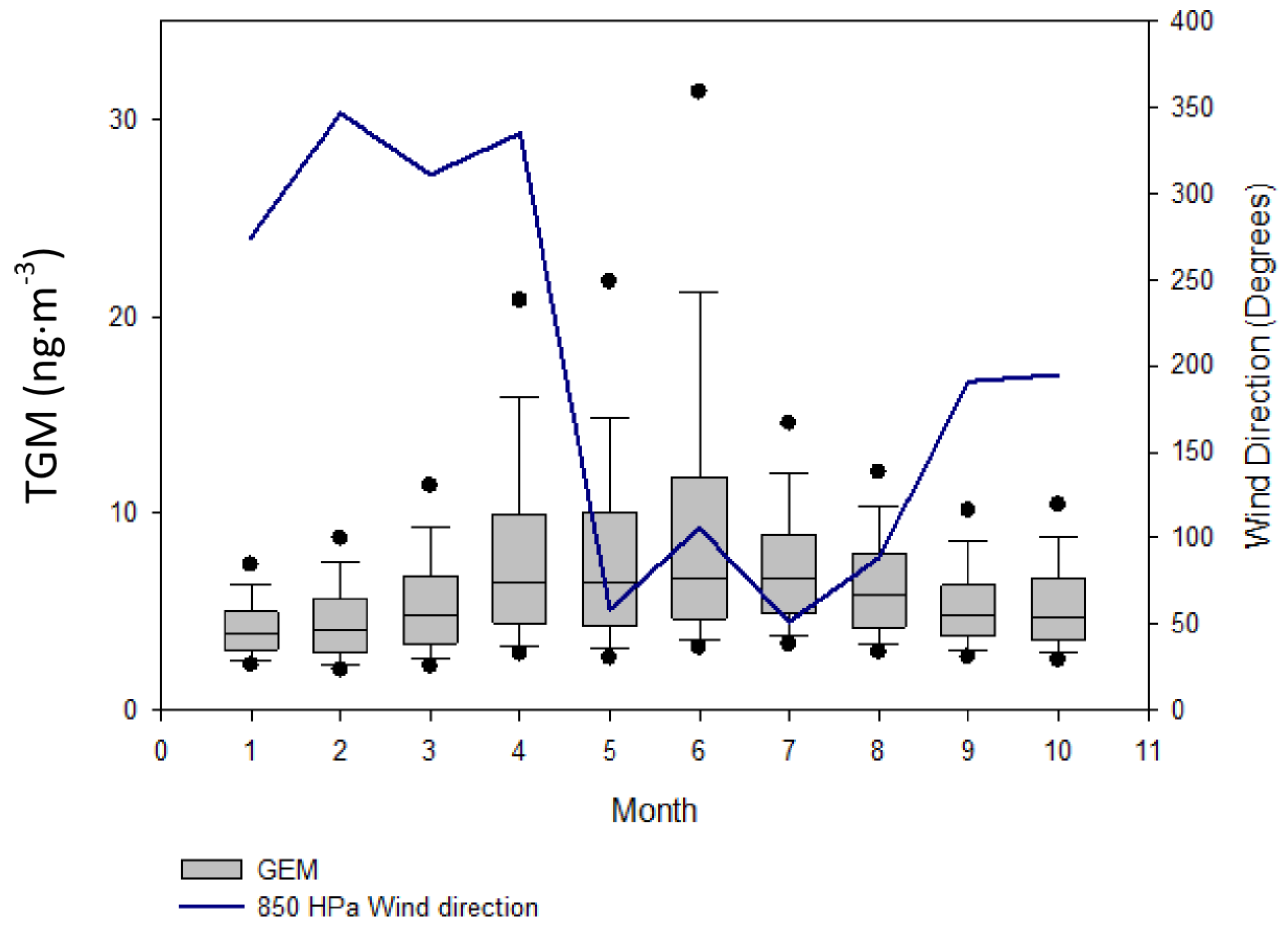
2.3.1. Monsoon Effects on TGM
The summer monsoon season in the YRD region comes with the shifting of prevailing flows at 850 hPa from the north, during most of the year, to south during mid-May [
16]. The winds at the surface level shift to east-southeast and at 500 hPa westerlies are observed. As the flows shift, rain bands move northward in early June signaling the beginning of the Meiyu season (
i.e., rainy season) in the YRD. The monsoon system sets up as a quasi-stationary front with an average eight day intervals of passage [
16]. These slow moving systems in the YRD in addition to larger summertime sources, such as biomass burning [
15], increased baseline due to higher revolatilization [
9], and changes to summer weather due to biomass burning [
18] may help explain the extraordinarily high concentrations of TGM during the mid-May–July monsoon season. The observed enhancement of TGM in Nanjing stands in contrast to other regions of China, such as the PRD, where the monsoon brought a decrease in atmospheric mercury concentrations over the season [
17].
When the region was under the southwesterly flow at 850 hPa during∙mid-May to July 2011, TGM appears to spike. Spikes in TGM concentrations reaching high episode concentrations occurred on 12 June 2011, 16 June 2011, 23 June 2011, 27 June 2011, 6 July 2011, and 22 July 2011 corresponding to episodes numbered 12–17 in
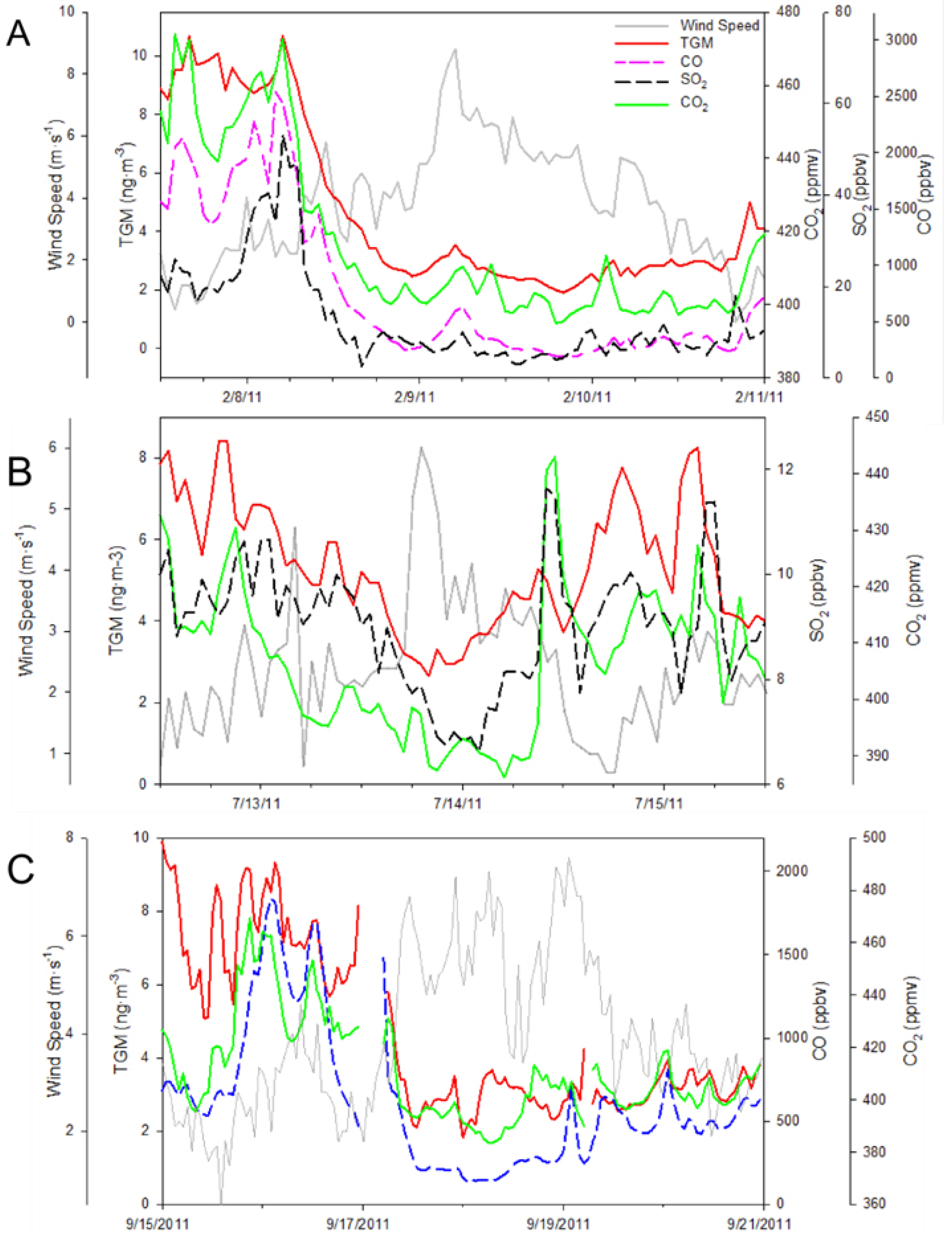
Table 1. In contrast, a drop in TGM concentrations most notably on background episode on 13 July 2011 discussed above was caused by strong flows from the southeast.
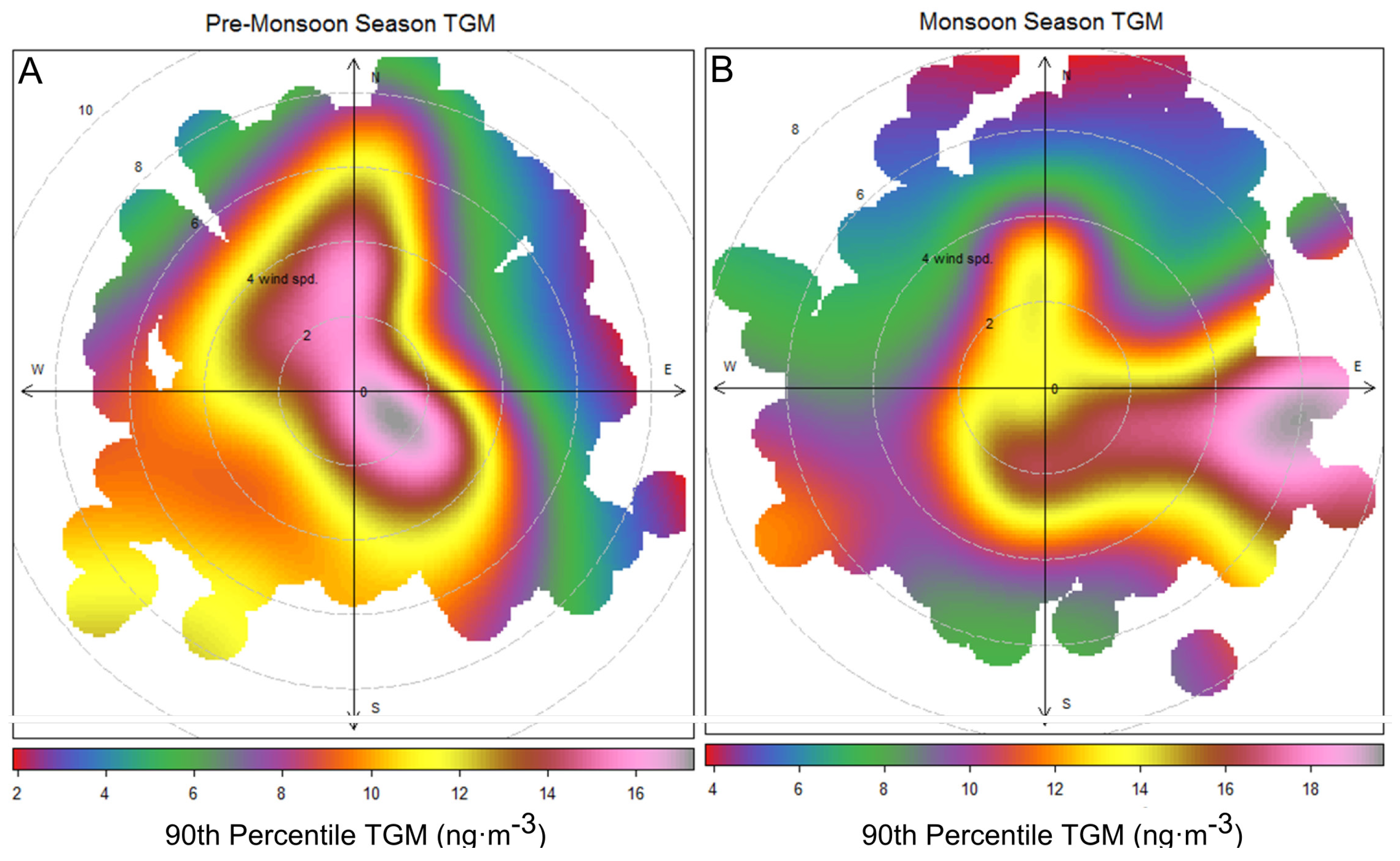
Figure 10.
TGM concentrations were binned by wind speed and direction. The 90th percentile was then plotted for (A) April–15 May, 2011 and (B) 16 May 2011–July 2011.
Figure 10.
TGM concentrations were binned by wind speed and direction. The 90th percentile was then plotted for (A) April–15 May, 2011 and (B) 16 May 2011–July 2011.
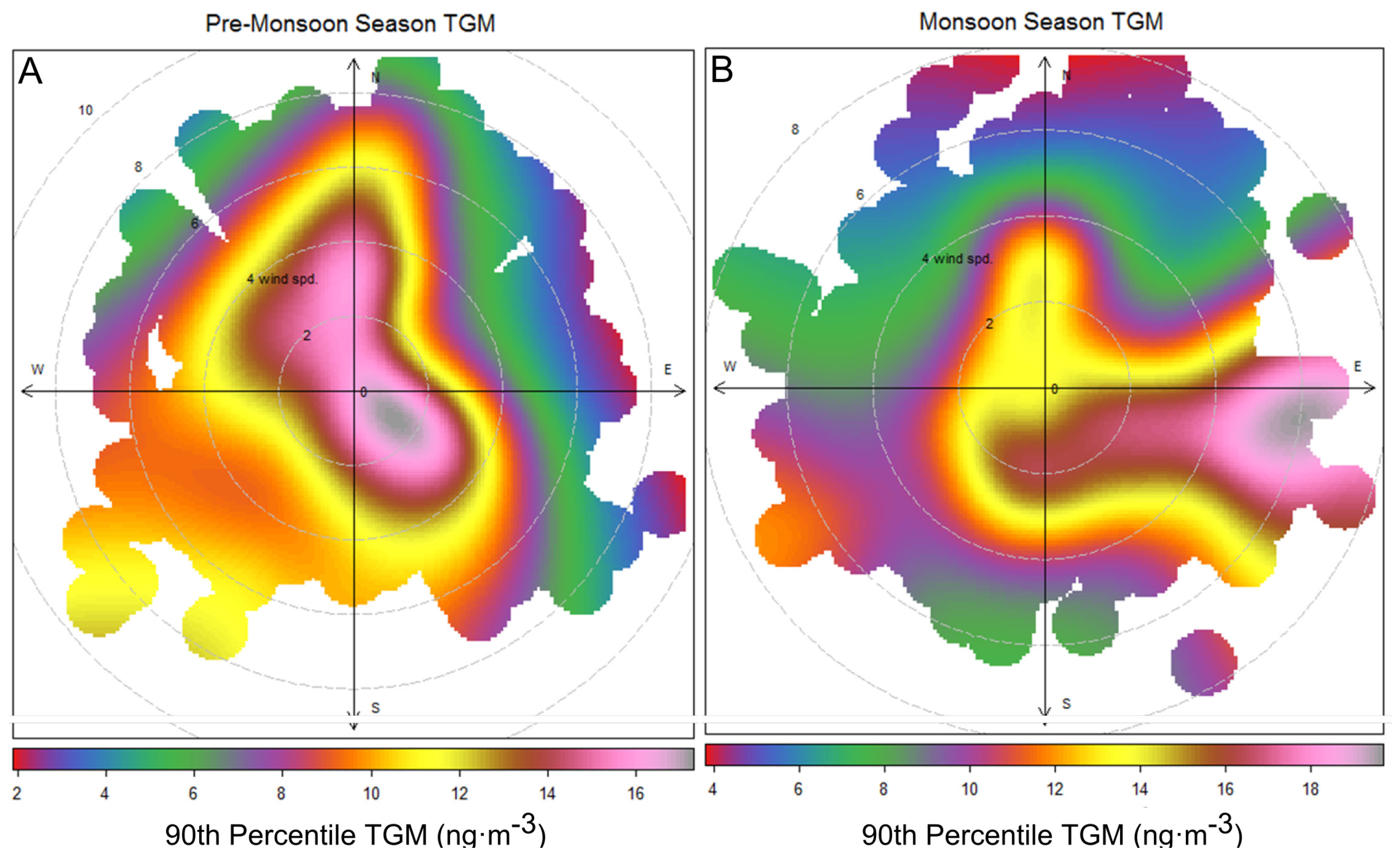
However, the arrival of monsoon season is not timed exactly with the increase in TGM values. The monthly median TGM value is elevated in April–May (
Figure 7) before the flow shift in mid-May. During April and the first half of May elevated TGM concentrations correspond with low wind speeds or northerly winds (
Figure 10A), and contrast this with the second half of May, June, and July where the highest TGM came from easterly directions (
Figure 10B). In June, the 10th, 75th, and 90th percentile values are much higher than in May indicating high episodic and baseline concentrations of TGM in June. The monsoon season may be only enhancing episodic concentrations of TGM during the summer while higher summer time temperatures and biomass burning raise median values in April through July [
9,
15].
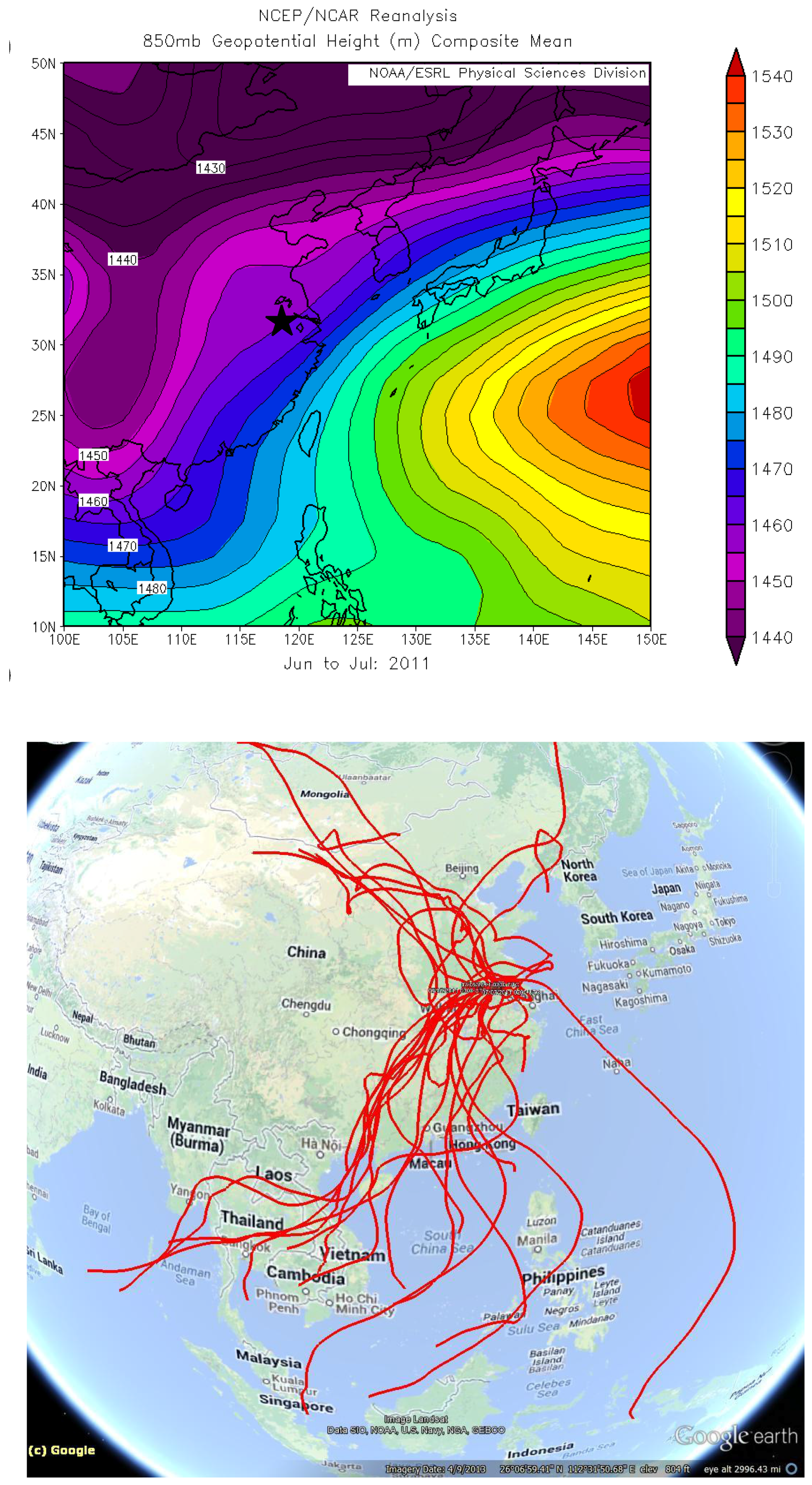
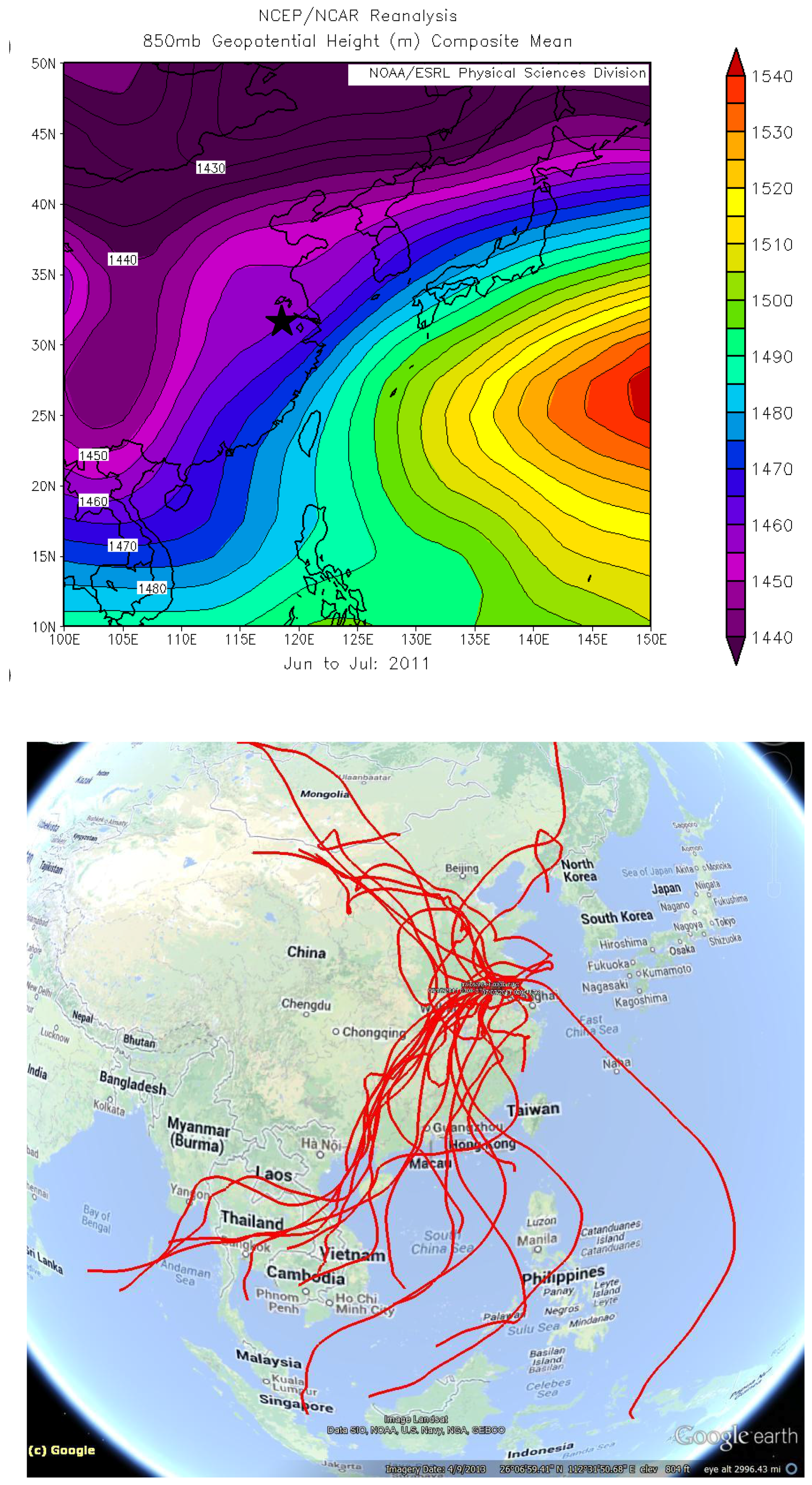
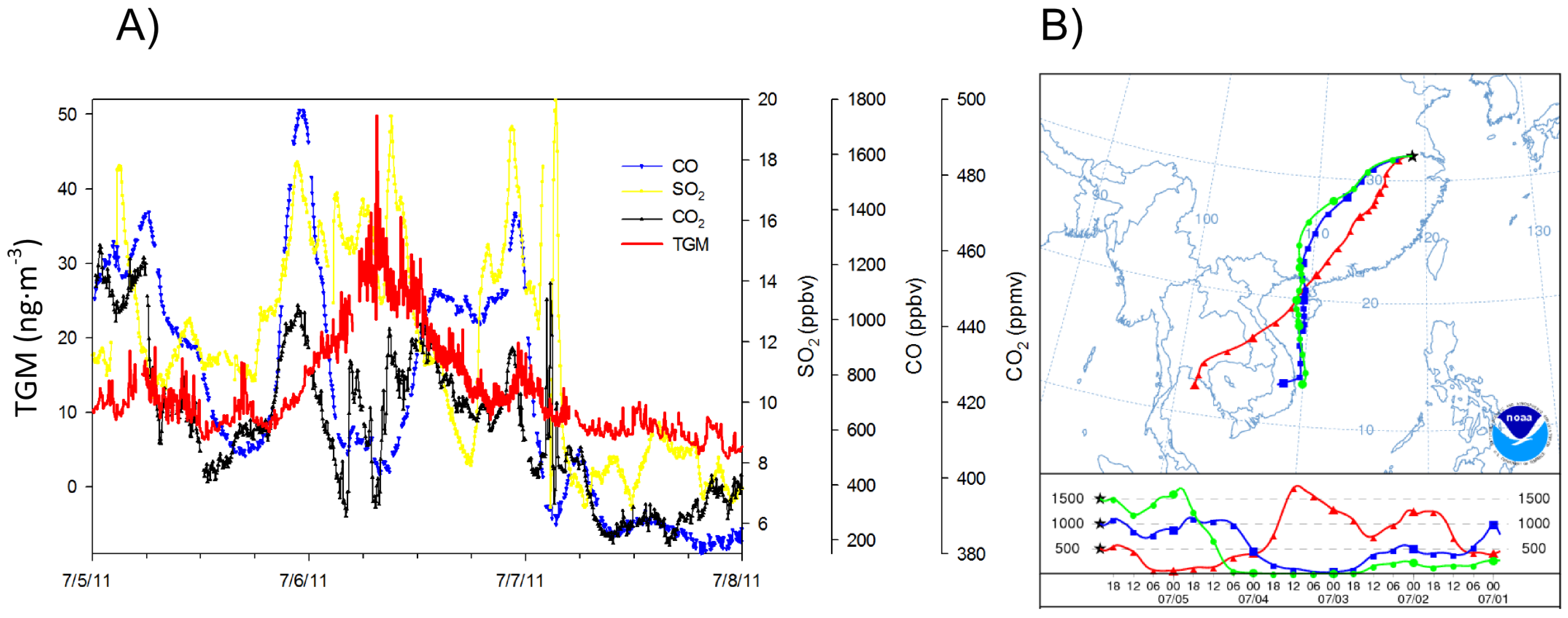
The extraordinary concentrations of TGM observed during June of 2011(
Figure 8,
Figure 9 and
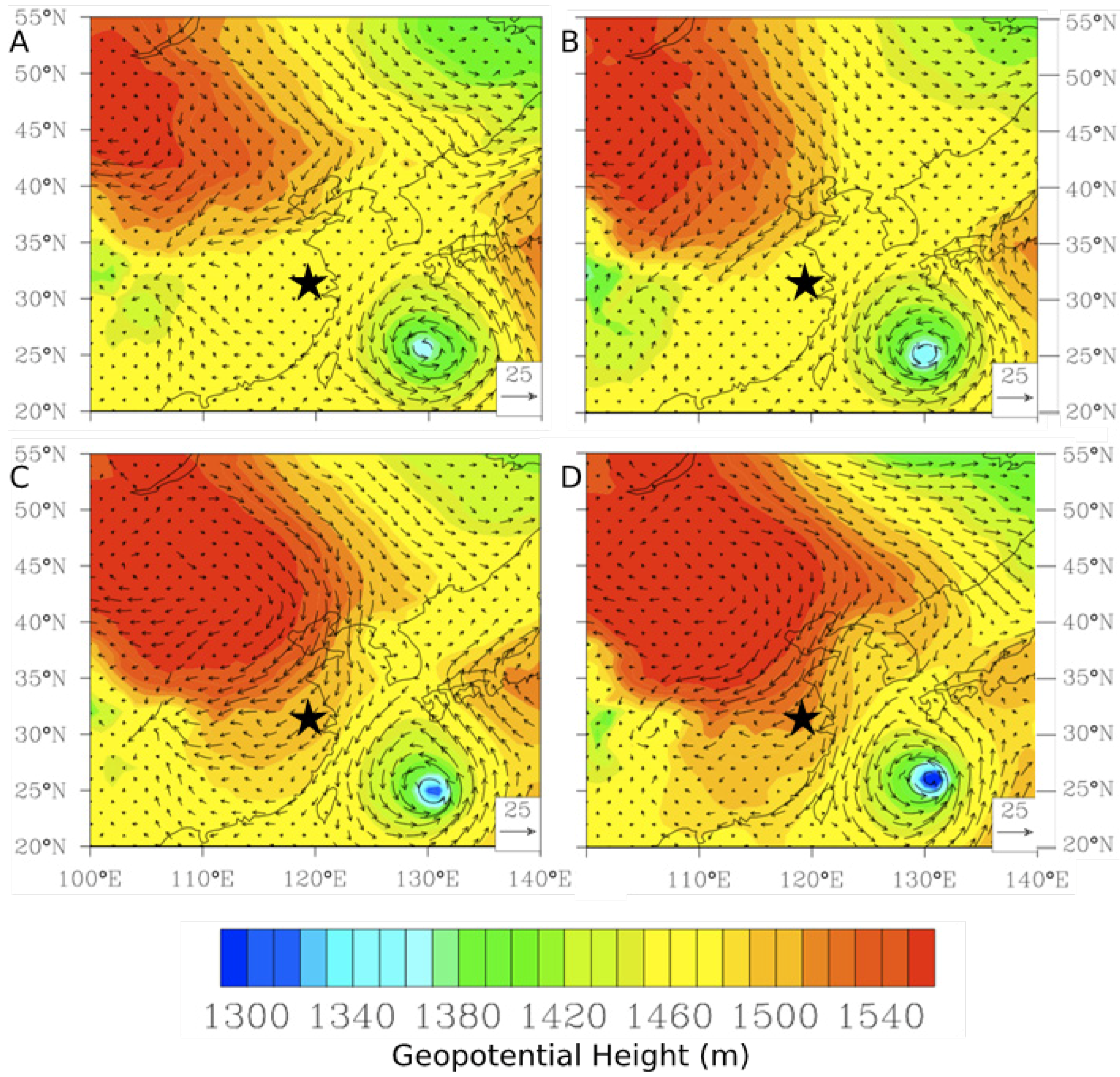
Figure 10B) were further investigated. The map of average geopotential height for 850 hPa shows prevalence of southwesterly flows in the YRD resulting from the low pressure system north of China and the subtropical high over the western Pacific during June and July of 2011 (
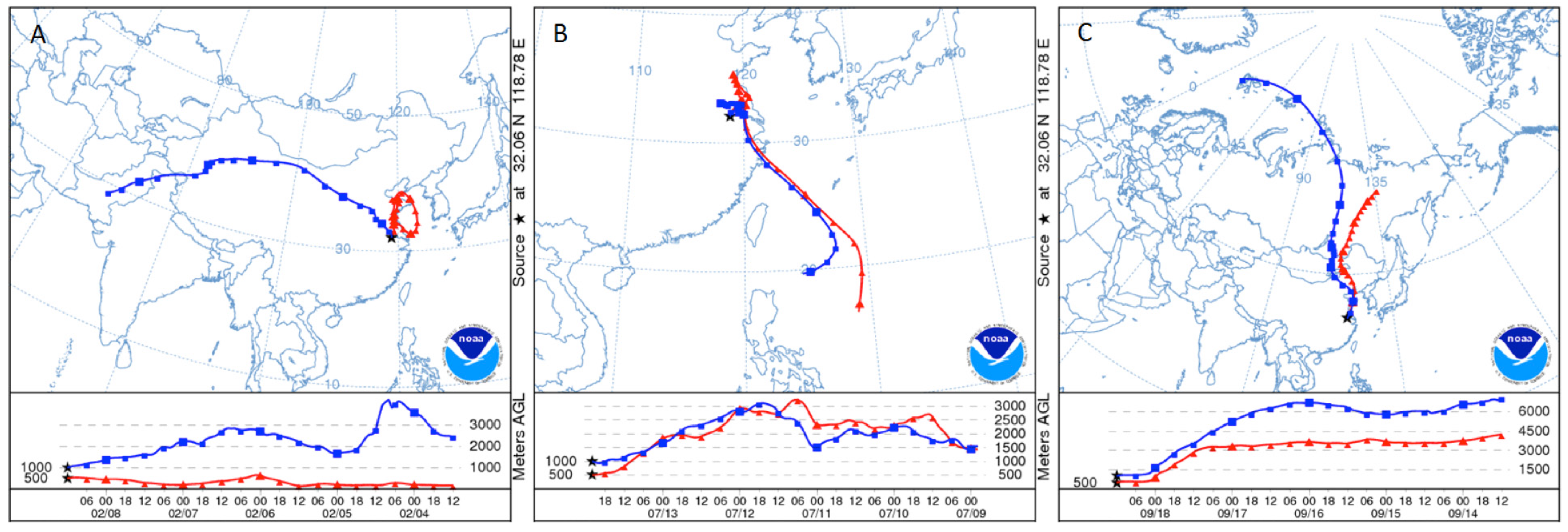
Figure 11A). To further investigate the effects of the monsoon, five-day backward trajectories for 7 June 2011, through 7 July 2011 starting at 1,500 m were plotted (
Figure 11B).
Figure 11B illustrates that 47% of the trajectories originated from southern China and the Indochina peninsula. Looking exclusively at episodically high day TGM concentrations there appear to be three ways in which TGM is enhanced in the region during the monsoon.
Figure 11.
Synoptic-scale flows during the monsoon season (A) Geopotential height for June and July, 2011, at the 850 hPa level (B) Five-day backward trajectories from Nanjing, China for 7 June 2011 through 7 July 2011 ending at 1500 m.
Figure 11.
Synoptic-scale flows during the monsoon season (A) Geopotential height for June and July, 2011, at the 850 hPa level (B) Five-day backward trajectories from Nanjing, China for 7 June 2011 through 7 July 2011 ending at 1500 m.
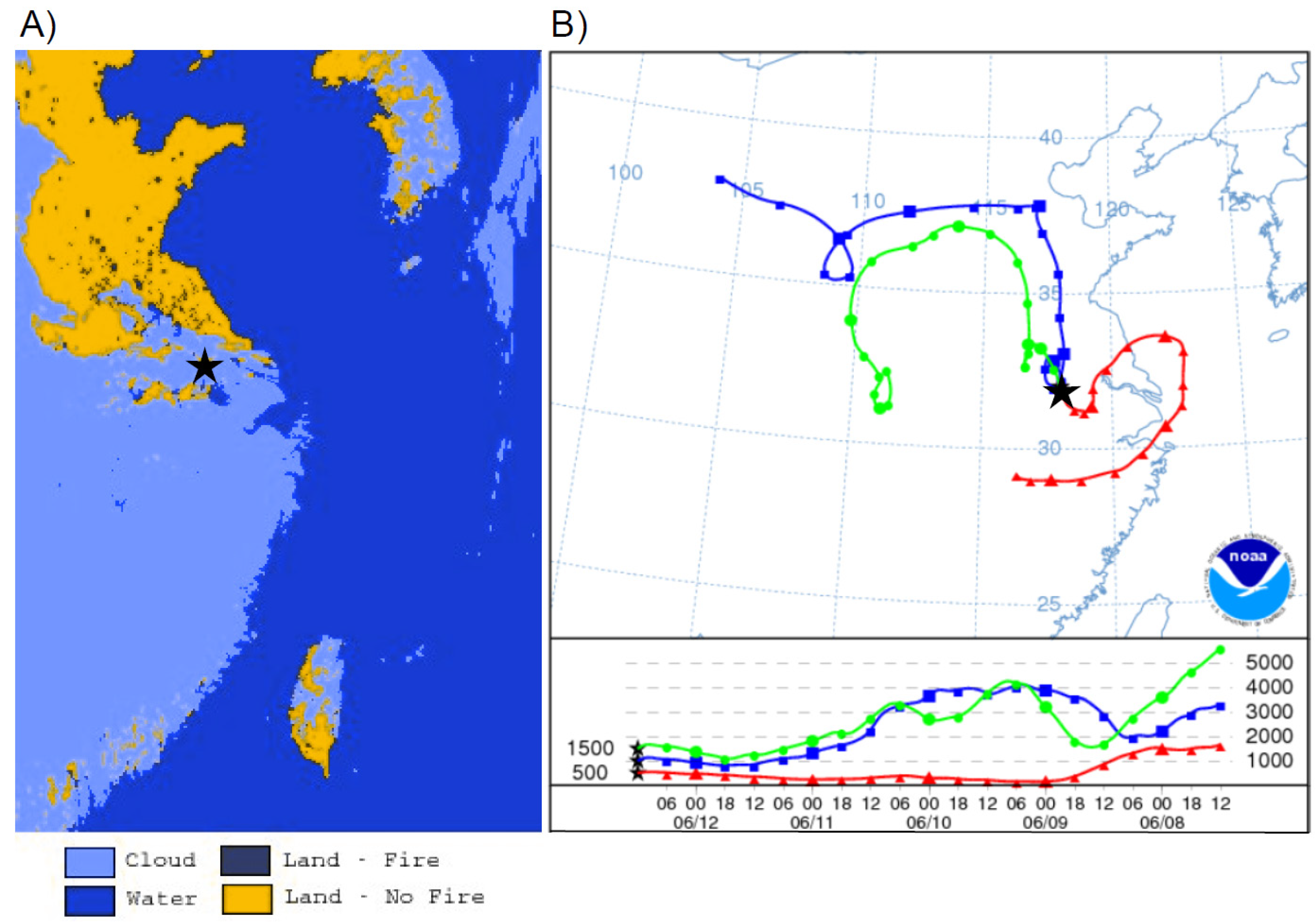
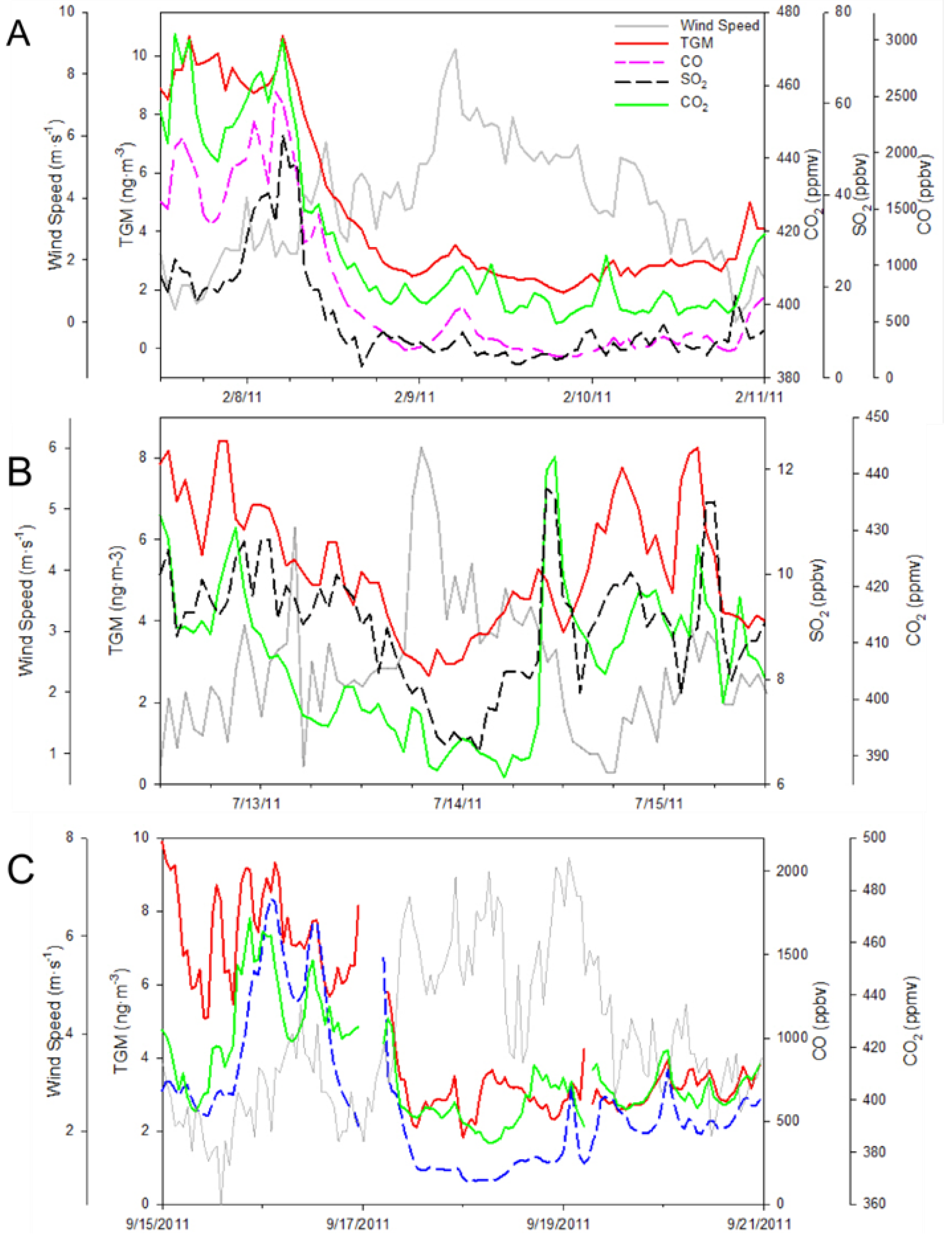
The first monsoon season episode happened on the 12 June episode with stagnant backward trajectories showing air masses building up within the YRD (
Figure 12B). Although there was a lack of correlation during this time period between TGM and SO
2 or NO
y, which was counterintuitive given the apparent urban nature of the episode, there was a strong correlation between TGM and CO with a slope of 0.059 ng∙m
−3 TGM ppbv
−1 CO. In addition, the Moderate Resolution Imaging Spectroradiometer (MODIS) fire map shown in
Figure 11A indicates a large number of agricultural fires, a source of CO and TGM, in the region possibly causing an inversion as noted by Ding
et al. [
18] that could build up TGM and CO concentrations in the YRD. This is especially interesting given the much higher CO and TGM correlation coefficient during this particular episode compared to other episodes in
Table 1.
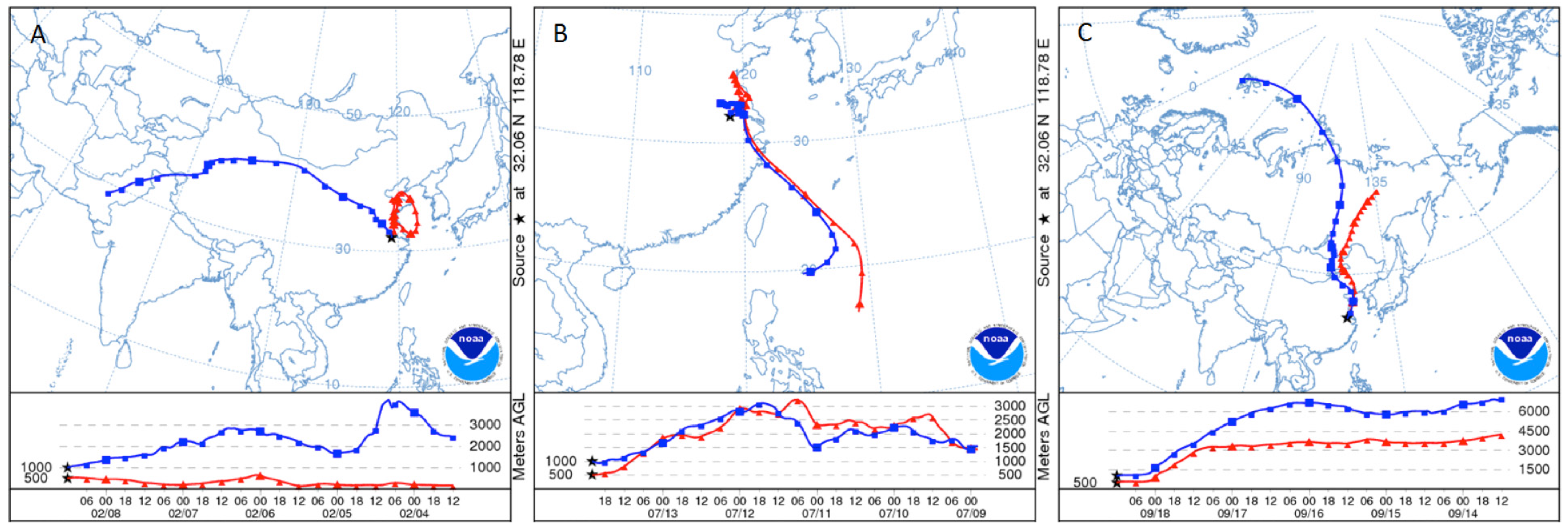
Figure 12.
On 12 June 2011 (
A) Fires from MODIS (defined in text) five-min Fire product V2 and (
B) a five-day backward trajectory from Nanjing starting at the 500, 1000, and 1500 m level at 12:00 UT. The star (
![Atmosphere 05 00124 i002]()
) indicates the location of Nanjing.
Figure 12.
On 12 June 2011 (
A) Fires from MODIS (defined in text) five-min Fire product V2 and (
B) a five-day backward trajectory from Nanjing starting at the 500, 1000, and 1500 m level at 12:00 UT. The star (
![Atmosphere 05 00124 i002]()
) indicates the location of Nanjing.
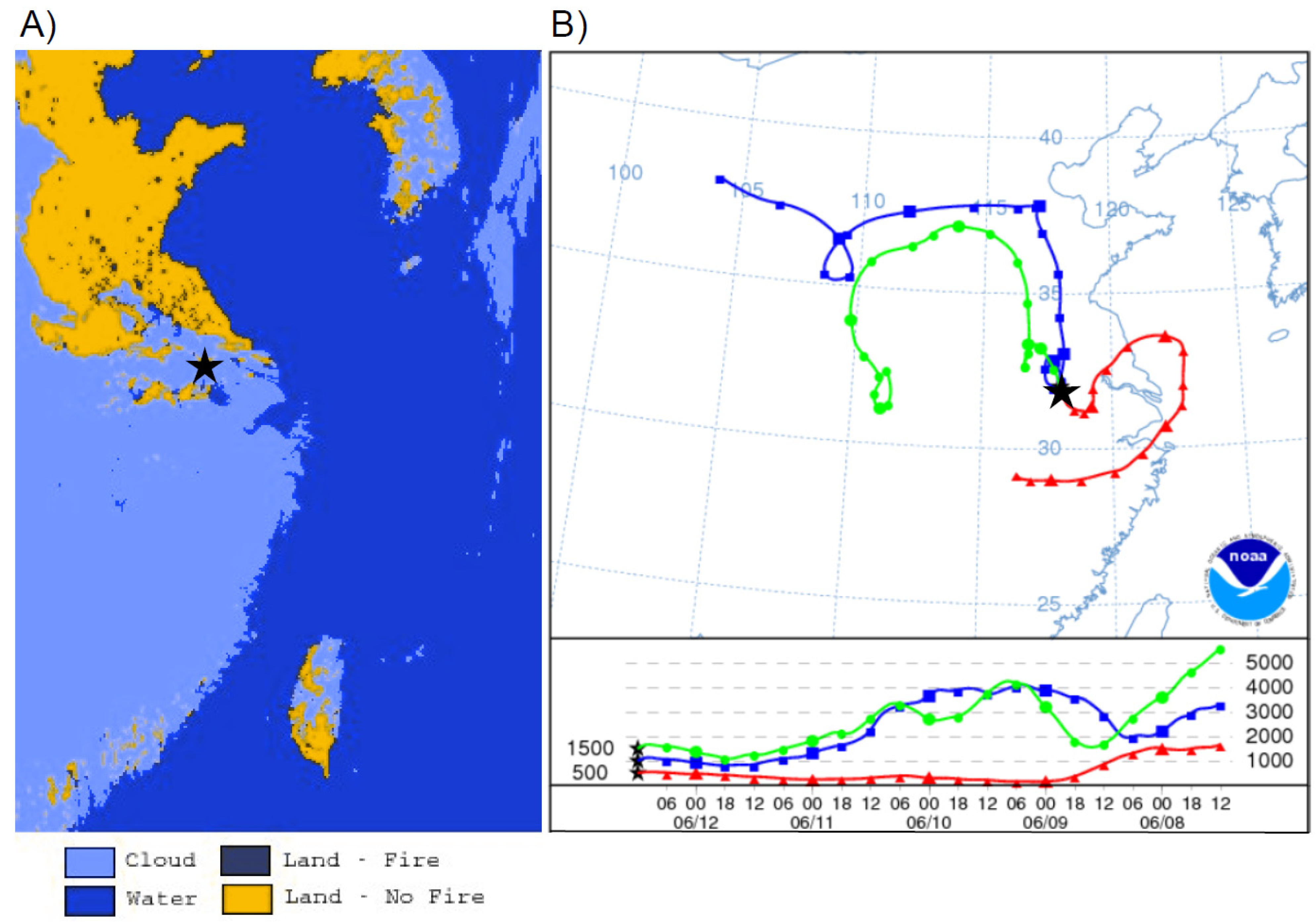
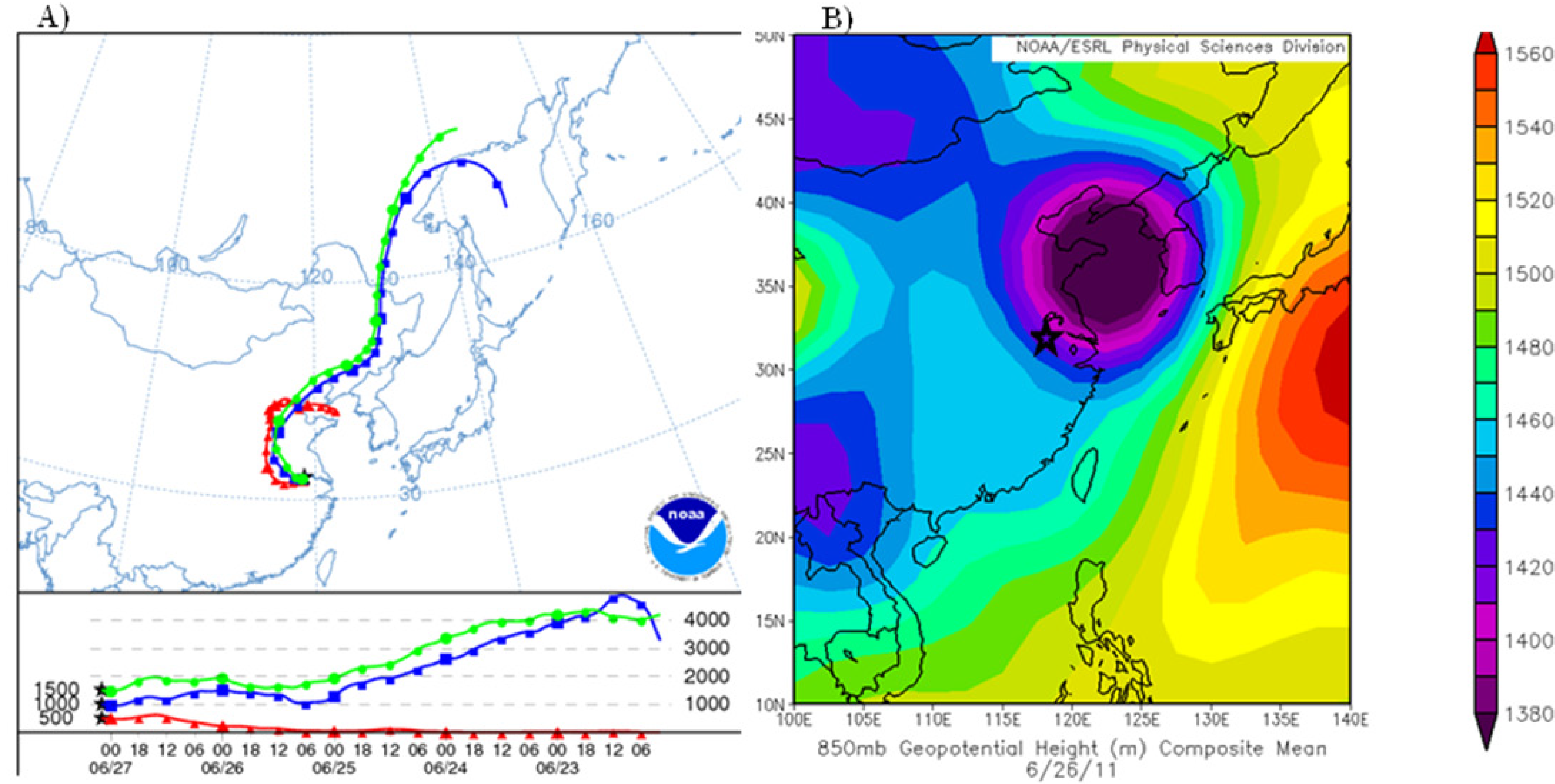
The second type was due to passing low-pressure systems causing a northerly circulation from the Beijing region, this occurred on 27 June. There was a low-pressure system on 26 June 2011 before concentrations of TGM began to rise (
Figure 13B). The accompanying HYSPLIT plot (
Figure 13A) illustrates air coming from the industrialized north of China ending at the time of the TGM peak 02:00 UT on 27 June. During the 27 June episode the correlations of TGM with CO, SO
2, and NO
y were good indicators of a strong urban source of TGM.
Figure 13.
(A) Five-day backwards trajectory from Nanjing ending on 27 June 2011, at 02:00 UT. (B) NCEP/NCAR reanalysis at 850 hPa showing low-pressure system that passed over Nanjing on 26 June 2011 and imported high mercury on 27 June 2011.
Figure 13.
(A) Five-day backwards trajectory from Nanjing ending on 27 June 2011, at 02:00 UT. (B) NCEP/NCAR reanalysis at 850 hPa showing low-pressure system that passed over Nanjing on 26 June 2011 and imported high mercury on 27 June 2011.
The third type of monsoon season episode is due to flows from the south southwest. The PRD was identified as the largest emissions region of TGM in the last published emissions inventory for China [
8] and therefore could have an effect on the mercury concentrations in Nanjing. Looking at the HYSPLIT model plot of 7 June through 7 July (
Figure 10B) it is clear that there is a common southwesterly flow during this period at the 1500 m level (
Figure 10A). Compared with the 850 hPa geopotential height map for May (not shown), it is clear that air more likely arrived from the southwest during the summer monsoon season. This seasonal enhancement is in stark contrast to the effect the monsoon had on mercury concentrations in the PRD region as noted by Chen
et al. [
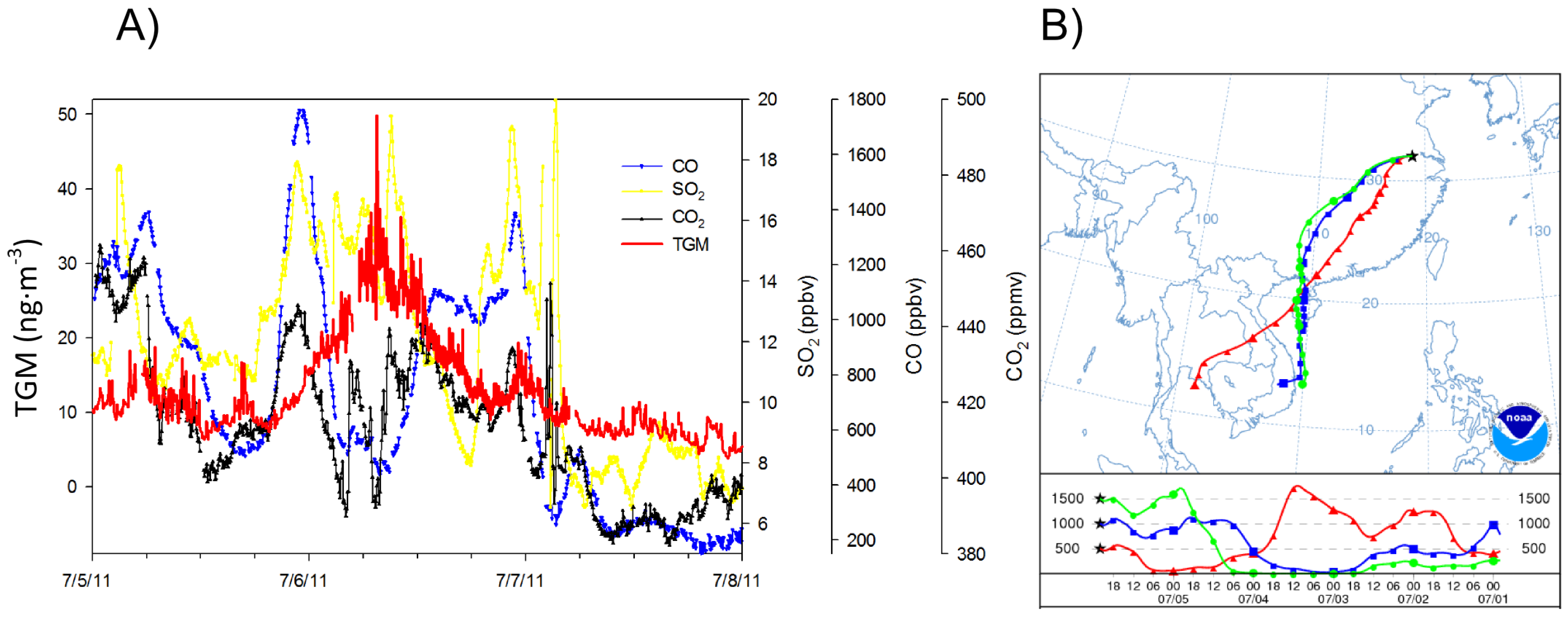
17], where mercury was diluted by relatively clean marine air masses entering the region via monsoonal southwesterly flows. A good example of an episode of high TGM concentrations transported from the southwest occurred on 6 July 2011. During this occurrence the wind speed was sustained at approximately 2 m∙s
−1 during the whole episode. Additionally there was a spike in CO and CO
2 (
Figure 14A) that preceded the TGM spike, most likely due to morning rush hour, as the peak occurred at approximately 7:00 a.m. local time while the HYSPLIT trajectory (
Figure 14B) was very similar to seasonal average. Moreover, wind speed and direction did not shift much, indicating consistent flows. All of these illustrate the possibility of transport from a strong source region southwest of the observation site.
The monsoon season in the YRD appears to have a very large impact on TGM concentrations. There are three separate ways in which TGM concentrations were enhanced these were: Biomass induced, northern transport, and southern transport. A fourth possible mechanism is increased legacy flux due to greater soil moisture discussed above. These factors in addition to the stronger summer sources noted in Lin
et al. [
11] contribute to a summertime concentration in the YRD that was anomalously large in comparison to Northern Hemispheric background.
Figure 14.
The high TGM episode on 6 July represented in (A) time series and (B) with the five-day backwards NOAA HYSPLIT plot at 500, 1,000, and 1,500 m.
Figure 14.
The high TGM episode on 6 July represented in (A) time series and (B) with the five-day backwards NOAA HYSPLIT plot at 500, 1,000, and 1,500 m.
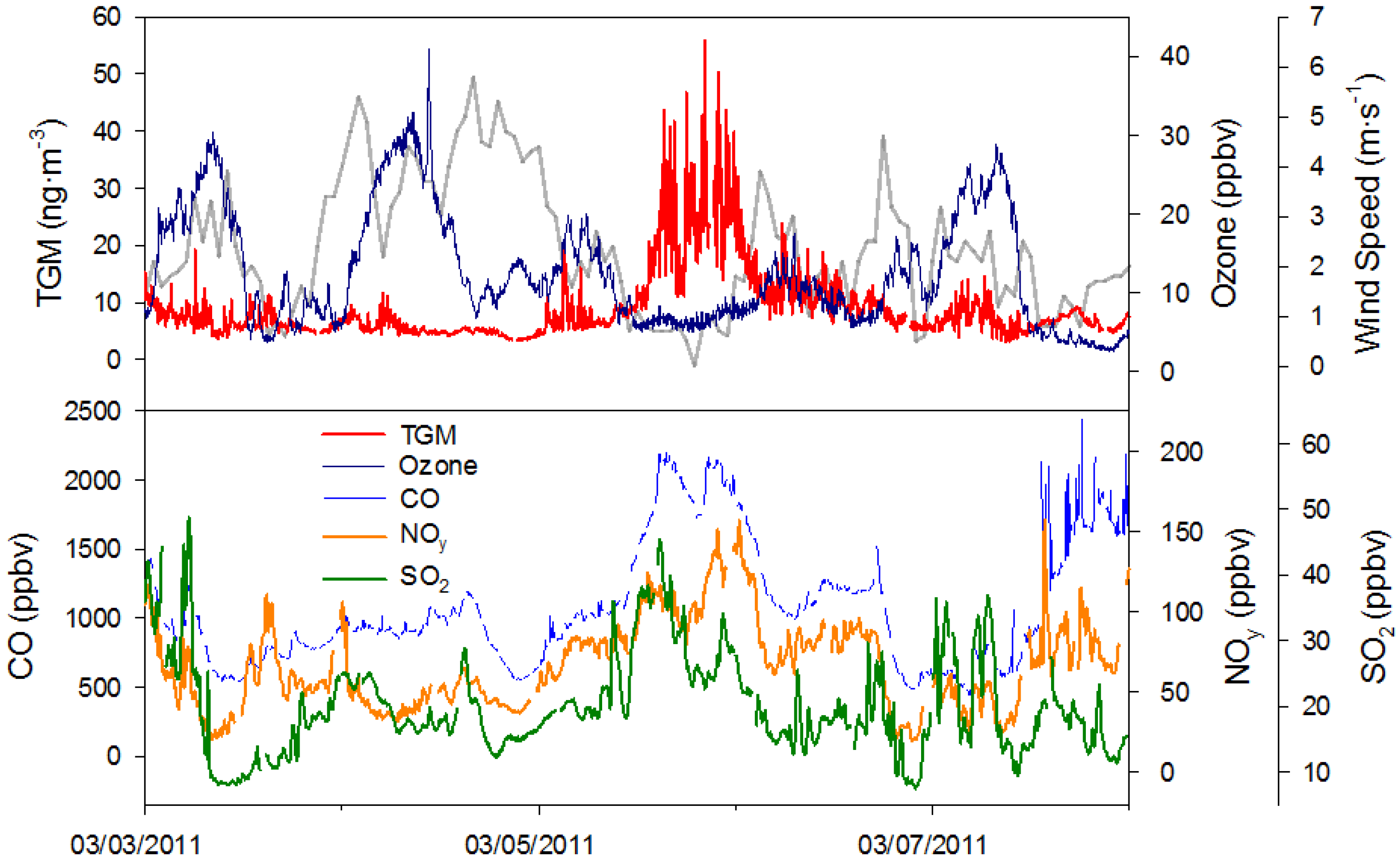
2.3.2. TGM Peaks during Low Ozone
There were two episodes on 5 March and 20 April, denoted as episodes #5 and #8 in
Table 1, where O
3 depletion was coincident with spikes in TGM concentrations over approximately a twelve hour period. On these occasions concentrations in SO
2, NO
y, and CO all became elevated in tandem with TGM concentrations, while O
3 concentrations remained under 20 ppbv. These episodes were also coincident with slow winds under 2 m∙s
−1 increasing buildup in the YRD, while Nanjing sat between fronts, similar to local episodes discussed below. The exceptional part about these episodes is the low O
3 that accompany them. The depressed concentrations of O
3 are not seen in other similar episodes.
Figure 15.
Time series of TGM, O
3, CO, NO
y, SO
2, and wind speed (gray) over the period 3 March 2011 through 7 March 2011. This represents episode #5 in
Table 1.
Figure 15.
Time series of TGM, O
3, CO, NO
y, SO
2, and wind speed (gray) over the period 3 March 2011 through 7 March 2011. This represents episode #5 in
Table 1.
The cause of low O
3 on 5 March 2011, appears to be titration by NO based on elevated concentrations of NO
y timed with the O
3 drop (
Figure 15). A time series of TGM, O
3, CO, NO
y, SO
2, and wind speed can be seen in
Figure 15. The low wind speeds (<2 m∙s
−1) during peak TGM concentrations coincide with increasing radiation flux to a maximum of 271 W∙m
−2 during the O
3 minimum on 5 March 2011. While O
3 rose the next day, 6 March 2011, the peak radiation flux was much lower at 65.6 W∙m
−2. On the previous day, 4 March 2011, radiation flux reached over 500 W∙m
−2 and the O
3 concentration was much higher near 30 ppbv.
In contrast to 5 March, low O3 on 21 April 2011 seems to be linked mostly to a very low radiation flux of 91.4 W∙m−2. This is coincident with low winds and high concentrations of urban tracers that indicate urban pollution. In addition, this area is under the influence of a low pressure system bringing in marine air and cloud cover.
Low O
3 only occurred in these two instances out of all of the high TGM episodes. It appears to be a coincident timing with decreased O
3 and a period of stagnation in the boundary layer and cloudy conditions, based on the radiation data. As both of these are a function of solar radiation, it seems unusual that this sort of stagnant, low O
3, high TGM episode only occurred on the two occasions listed above. The anomalous drop in O
3 may be due to mobile sources, which are known to be a strong source of NO. The other possible source of titrating nitrogen may be plumes of industrial and electrical boilers since these two sources account for over half of the NO
x emissions in the YRD [
31]. Furthermore, the linear correlation slope values between TGM and NO
y were unusually high at 0.24 ng∙m
−3 TGM ppbv
−1 NO
y on 5 March, 2011 and 0.20 ng∙m
−3 TGM ppbv
−1 NO
y on 20 April compared to 0.054 ng∙m
−3 TGM ppbv
−1 NO
y during the entire winter, indicating the increased importance of NO
y in the atmospheric composition and therefore as a titrant. The r
2 value for the correlation of NO
y and TGM was also high at 0.51 on 5 March 2011. Additionally, there was a linear correlation of TGM and SO
2 during both episodes showing a high slope value indicating a strong source of TGM with SO
2. Taken together these seem to indicate a possible plume from industrial or electrical boilers rather than mobile combustion as would normally be assumed in such a low O
3 environment. Overall, these episodes seem to speak to the importance of stagnant meteorology increasing TGM and other pollutants, such as NO
y and CO, while titrating or removing O
3 although YRD source density makes any conclusions about a single cause of low O
3 difficult.
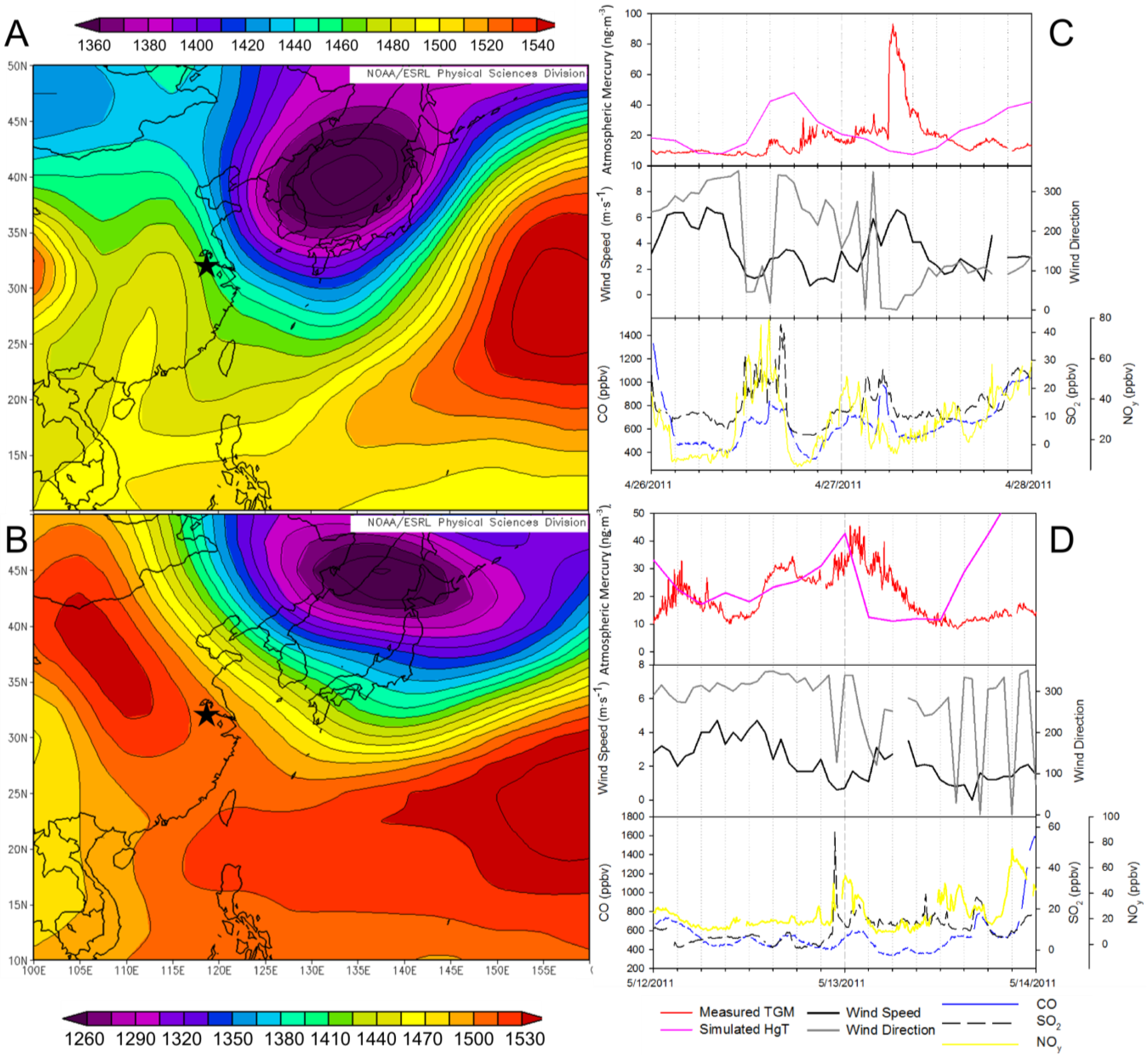
2.3.3. Transport of TGM from Non-Urban Sources
Three episodes on 26 April, 13 May, and 13 September 2011, appear to be due to transport. The spring episodes occurred with no correlated trace gases and during periods of high winds in contrast to most localized episodes coincident with wind speeds less than 2 m∙s
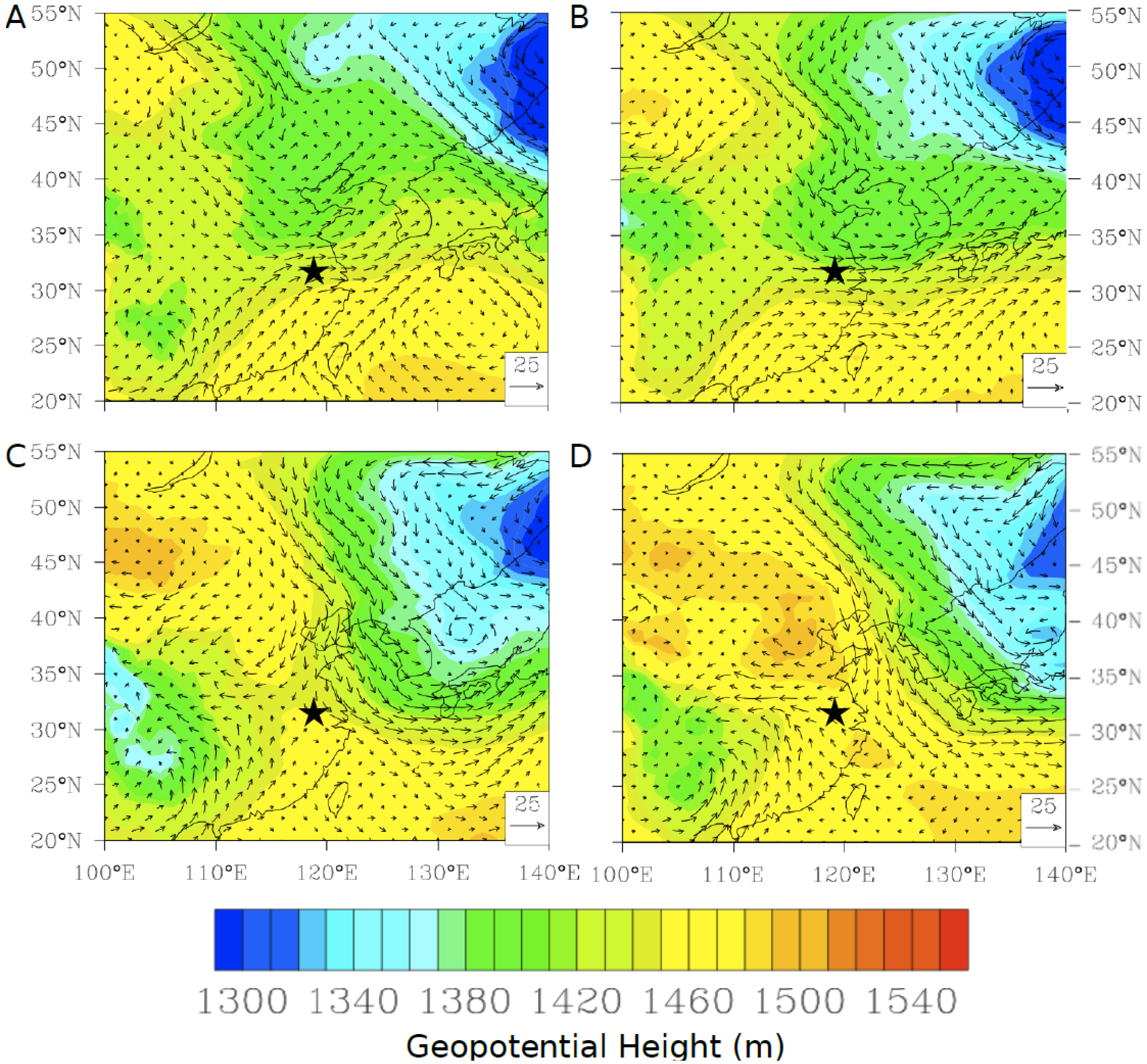
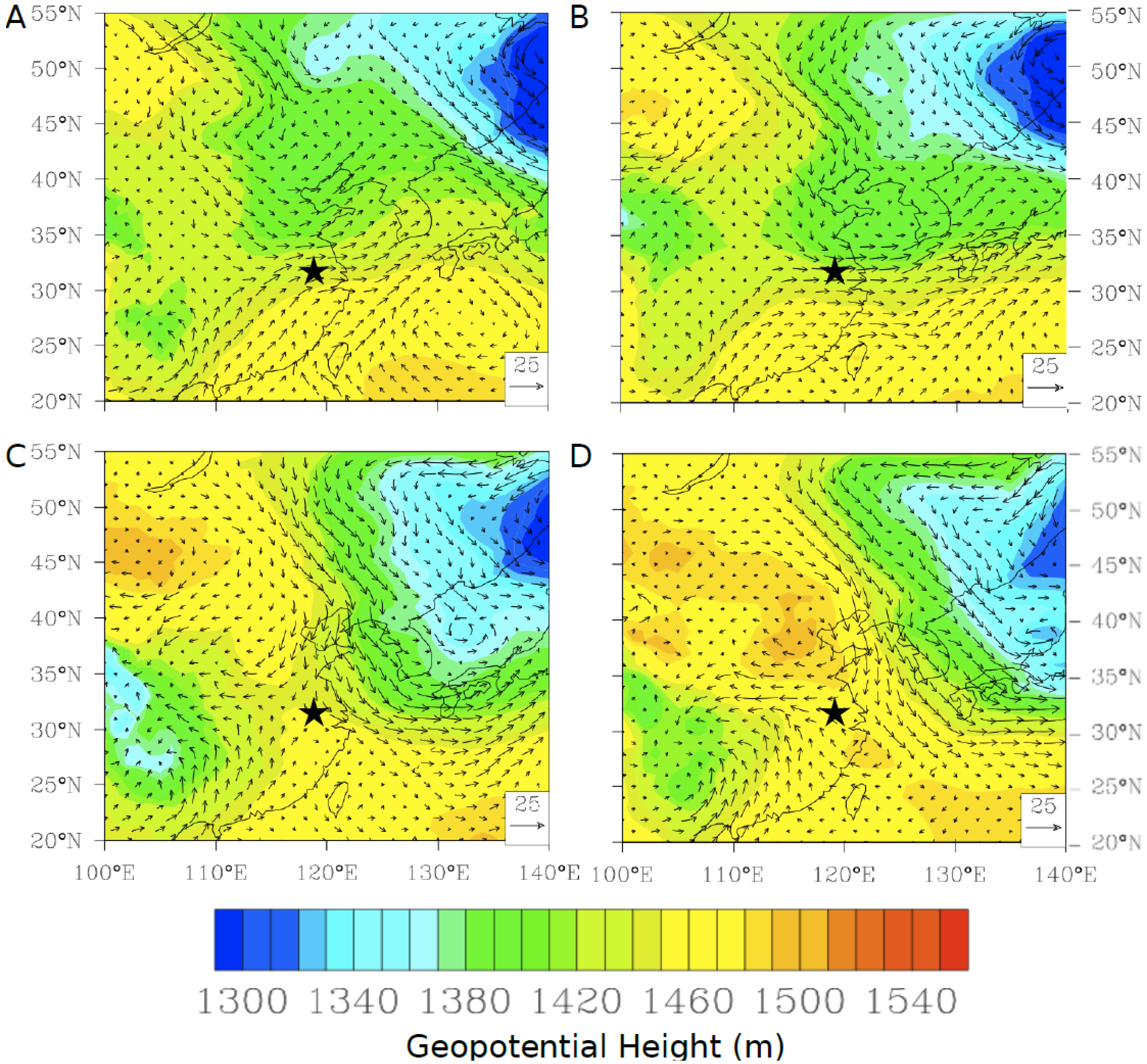
−1. Further investigation of the geopotential height at 850 mbar height using daily NCEP/NCAR reanalysis data shows that in both cases during the spring there was a low pressure system passing north of Nanjing over the Korean peninsula and into the Sea of Japan (
Figure 16A,B). The strength of the gradient flow varies from episode to episode. On 27 April 2011, Nanjing was immediately under the influence of strong gradient flows formed by a high-pressure ridge west of Nanjing and a strong low pressure system to the northeast (
Figure 16A). In comparison, on 13 May 2011, Nanjing was sitting outside of strong gradient flows as shown in
Figure 16B. These meteorological systems appear to transport polluted air masses rich in mercury from the heavily industrialized north of China into Nanjing, although the lack of correlation with any other trace gas (
Figure 16C,D) indicates otherwise. Possible explanations for the lack of correlation are industrial sources that intentionally use elemental mercury such as compact florescent light manufacturing, artisanal small scale gold mining, wet deposition or removal of more reactive gases, e.g., SO
2 and NO
y, and/or transport from rural areas.
Compact florescent light and battery manufacturing are major contributors to mercury emissions in the PRD region of China [
36]. These industrial emissions appear to be due to accidental release of mercury used in the final product, not combustion and so therefore are not coincident with combustion tracers such as CO, SO
2, and NO
y. However, such industrial sites are expected to be collocated with urban emissions. The fact that while mercury concentrations spiked, urban tracers were flat, indicates a non-urban source of emissions.
Wet deposition of the more reactive gases during transport is unlikely since NCEP/NCAR reanalysis data for all three occasions shows no precipitation in and around Nanjing or north of the location. Additionally, if the mercury were of urban origin and the other trace gases were removed by a process that acted on reactive gases, then CO should remain intact as a tracer of urban emissions. In the case of these episodes it is unlikely that the mercury is of urban origin because of lack of high concentrations of CO, a relatively long lived and insoluble gas.
Rural sources of mercury in China generally include biomass burning and artisanal mining [
4,
15]. Biomass burning of fields and crop residue is a source of mercury in China, especially in Shandong, north of Nanjing, and the Jiangsu provinces [
8,
15]. Biomass burning as an inefficient combustion source is generally considered to emit CO which does not spike during the three episodes considered here. In addition to the lack of correlation with CO, episodes in the spring occurred before summer field burning began [
18], ruling out biomass burning as the source of these episodes. Artisanal mining can produce mercury emissions, ~45% of the released mercury is atmospheric, through the use of mercury amalgamation to recover gold from ore and is the single largest source of atmospheric mercury emissions in China, making up ~29% [
4]. Although mercury amalgam processes are illegal in China, they are still in use [
14], and, thus, many of these artisanal mining sources have not been included in emission inventories. Artisanal mining is greatest in Hebei and Shandong both due north of Nanjing [
14]. In
Figure 16, for both episodes there is a common theme of air masses passing over these provinces before reaching Nanjing. Additionally there are north to northwesterly flows (
Figure 16C,D). Furthermore, 27 April reached higher TGM concentrations and had a stronger gradient and therefore winds. In comparison, TGM concentrations on 13 May did not reach as extraordinary concentrations, while the pressure gradient was not located directly over Nanjing and consequently wind speeds facilitating transport to Nanjing were not as fast. The difference in TGM concentrations corresponds to the strength of transport phenomena; therefore transport from the Shandong and Hebei are likely candidates for the source of the high TGM concentrations during these episodes. Artisanal mining would explain why TGM concentrations spiked while combustion gases concentrations stayed low and wind speeds rose (
Figure 16C,D).
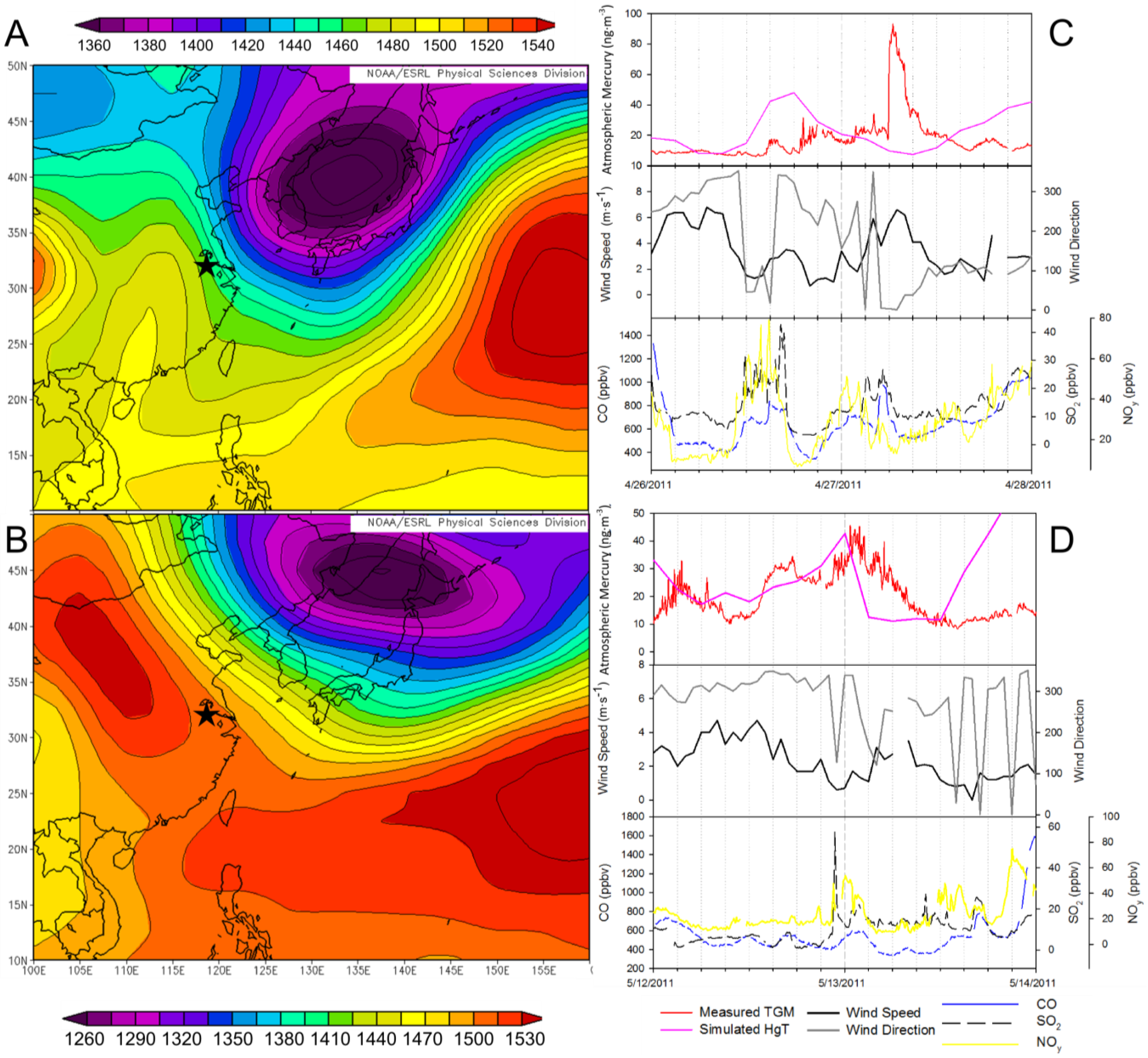
To determine if TGM concentrations were a function of known emission sources the concentration of TGM were simulated using Lagrangian particle dispersion modeling (LPDM). Simulated mercury concentrations do not peak simultaneously with the observed TGM concentrations (
Figure 16C,D). In the 27 April 2011instance the observed peak comes arrives between the two simulated peaks and was much greater in magnitude. On 13 May 2011the simulated peak comes before the actual TGM peak but was approximately equal in magnitude. On 13 May 2011 the simulated mercury peak declines to the baseline within 3 hours, whereas the observed decrease lasted 12 hours. The simulations employed gridded emissions in the AMAP/UNEP for the year 2010. The fact that the TGM concentrations are not captured in the model indicates missing∙mercury sources in emission inventories, such as artisanal mining sources in and around China. Such evidence seems to support undocumented artisanal mines in the provinces north of Nanjing releasing TGM that was transported on a regional scale to Nanjing.
Figure 16.
The geopotential height at 850 hPa over East Asia from NCEP/NCAR reanalysis for (
A) 27 April 2011 and (
B) 13 May 2011. A time series of measured TGM (red), Flexpart simulated TGM (magenta), wind speed (black), wind direction (grey), CO (blue), NO
y (yellow), and SO
2 (black dash) for (
C) 26 and 27 April 2011, (
D) 12 and 13 May 2011. The star (
![Atmosphere 05 00124 i002]()
) indicates the location of Nanjing.
Figure 16.
The geopotential height at 850 hPa over East Asia from NCEP/NCAR reanalysis for (
A) 27 April 2011 and (
B) 13 May 2011. A time series of measured TGM (red), Flexpart simulated TGM (magenta), wind speed (black), wind direction (grey), CO (blue), NO
y (yellow), and SO
2 (black dash) for (
C) 26 and 27 April 2011, (
D) 12 and 13 May 2011. The star (
![Atmosphere 05 00124 i002]()
) indicates the location of Nanjing.
2.3.4. Localized Emissions
This grouping of episodes includes 50% of episodes and generally happened during periods of low wind speed (<2 m∙s
−1). There is a diversity of slope values of correlations between TGM and other tracers (
Table 1). The values for localized episodes with an r
2 > 0.2 between CO and TGM ranged from 0.003 to 0.017 ng∙m
−3 TGM ppbv
−1 CO. For SO
2 the slope values reported above the criteria listed previously range from 0.074 ng∙m
−3 TGM ppbv
−1 SO
2 to 0.382 ng∙m
−3 TGM ppbv
−1 SO
2, while TGM-NO
y slope values ranged from 0.049 ng∙m
−3 TGM ppbv
−1 NO
y to 0.253 ng∙m
−3 TGM ppbv
−1 NO
y. These different slope values give an idea of the diversity and density of mercury sources in the Nanjing region and the difficulty of identifying specific sources when the emission correlations span such a large range.
Table 2 illustrates slope values for GEM or TGM to CO correlations found in other locations such as Mt Bachelor Oregon [
20], Beijing China [
10], New Hampshire [
37], Southern England [
38]. The linear relationship between Hg and SO
2 was also found in Southern England [
38] and Canada [
39]. The levels correlations in
Table 1 range from similar to values in
Table 2 to an order of magnitude higher for all values. This indicates in many cases emissions that are generally enriched with high gas phase mercury compared to other locations. In order to more clearly understand the conditions that cause these peak episodes, one episode, 24 February 2011, shown in
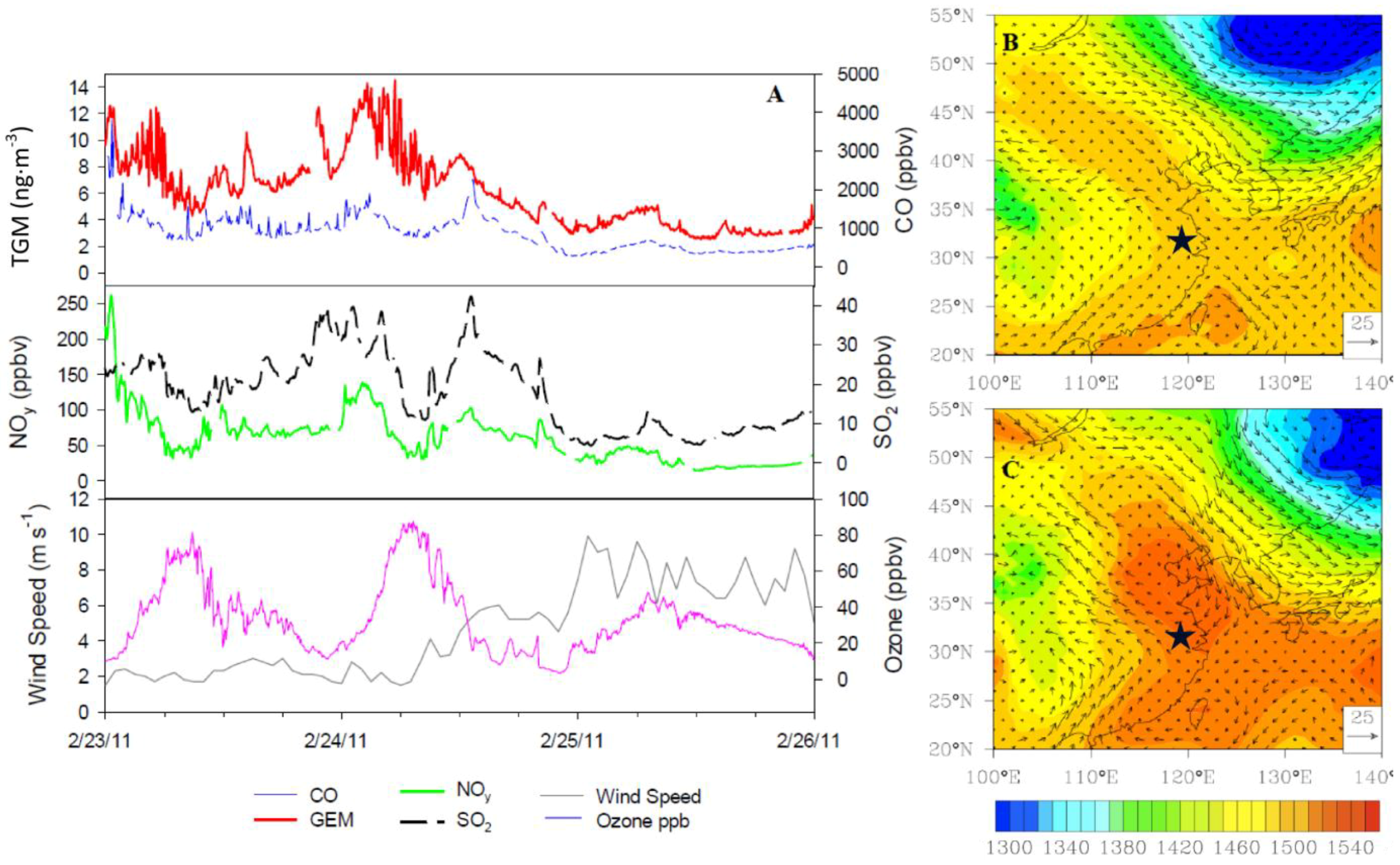
Figure 17, illustrates the progression of a typical locally driven, non-monsoon peak in mercury concentrations.
Table 2.
Slope values of Total Gaseous Mercury or Gaseous Elemental Mercury (TGM/GEM) correlations from other studies and years all in the format ng∙m−3 ppbv−1.
Table 2.
Slope values of Total Gaseous Mercury or Gaseous Elemental Mercury (TGM/GEM) correlations from other studies and years all in the format ng∙m−3 ppbv−1.
| Location & Year | Hg/CO | Reference |
|---|
| Mt. Bachelor, 2004 | 0.005 | [20] |
| Beijing, 2008/09 | 0.0015–0.00324 | [10] |
| New Hampshire, 2004–07 | 5.4 × 10−4 to 0.0019 | [37] |
| Hg/SO2 | |
| S. England, 1995 | 2.8 × 10−4–0.00156 | [38] |
| Canada, 2005–2008 | 0.003–0.01 | [39] |
| Hg/NOx | |
| Canada, 2005–2008 | 0.002–0.024 | [39] |
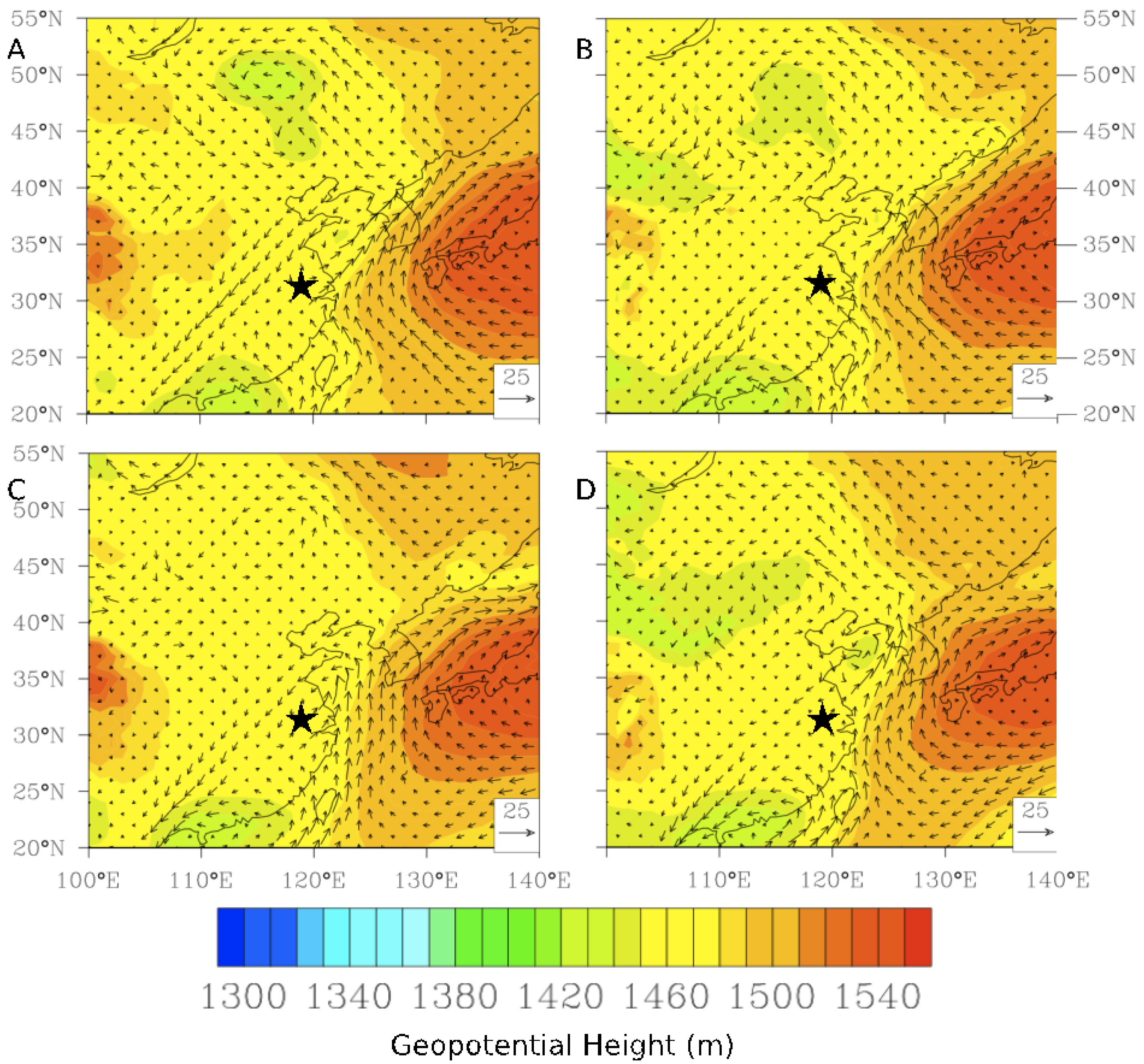
The accumulation of high TGM concentrations in this region started approximately five days earlier on 17 February 2011 (not shown). The TGM concentrations built up in nightly peaks and daytime lows over the course of the preceding week until reaching peak concentrations at approximately 2:30 UT on 24 February 2011. Illustrated in
Figure 17, the trace gases CO, SO
2, and NO
y behave similarly to TGM, while O
3 acts differently and peaks in the afternoon as expected. Wind speeds remain low around 2 m∙s
−1 allowing emissions to build up locally until approximately 6:00 UT when wind speeds rose and all pollutant concentrations fell due to ventilation. Furthermore there is a stationary front observed at the 850 hPa level on the 24 February 2011causing the stagnant conditions to evolve to increased winds, which finally ventilated the region (
Figure 17B,C).
The episodes have differing behavior in terms of correlation of TGM with different trace gases (
Table 1). Generally if one trace gas was correlated with TGM, then all trace gases were. The defining characteristic of these episodes was the low wind speeds indicative of YRD sources. Although the sources were within the YRD, fingerprinting individual point sources of pollution proved intractable due to the density and diversity of source types within the YRD.
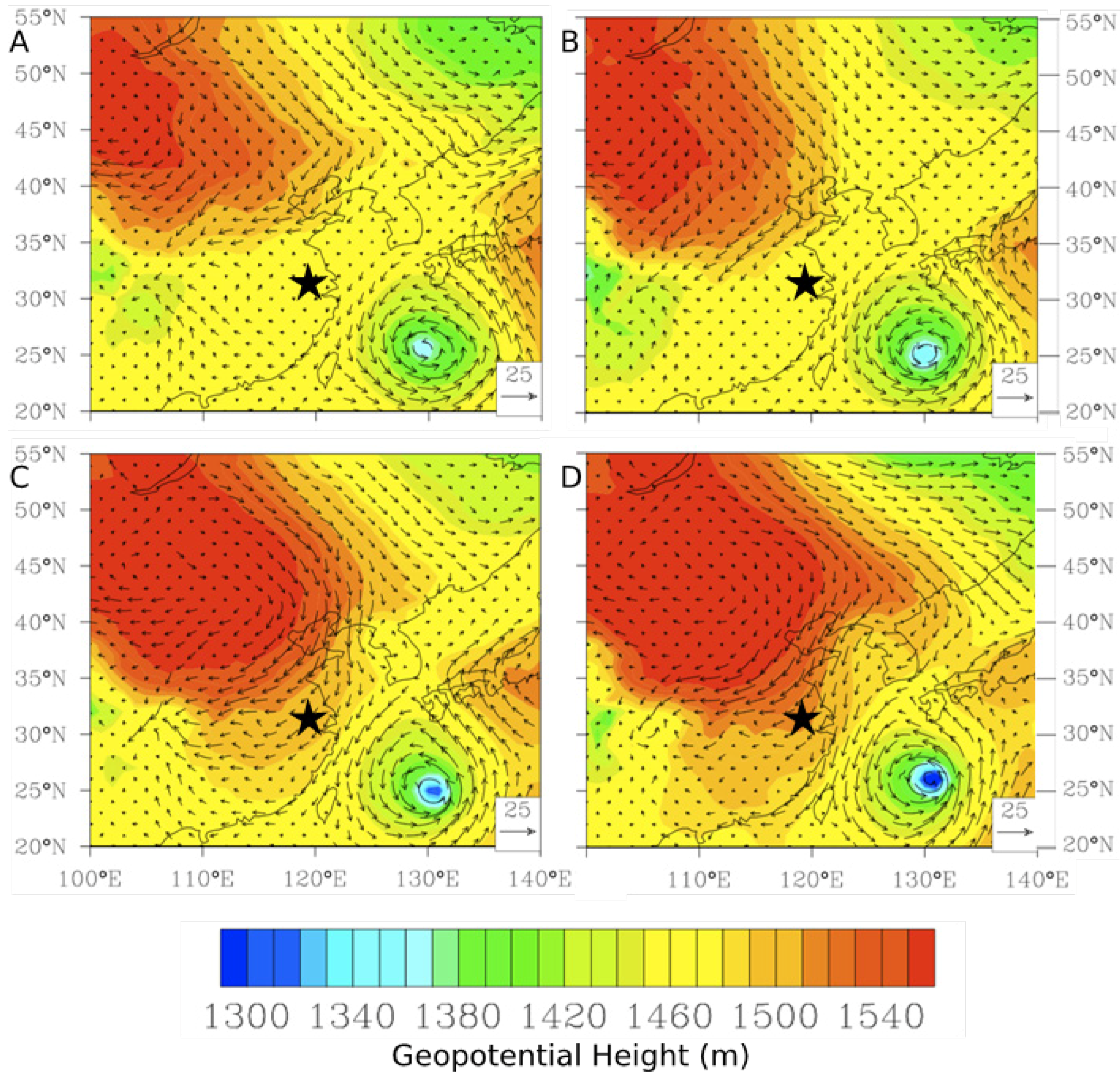
Figure 17.
Evolution of an episode of high TGM on 24 February 2011 (
A) Time series of five-min average concentrations of trace gases and hourly wind speed. (
B) Geopotential height at the 850 hPa level along with wind vectors at 6:00 UT on 24 February 2011. (
C) Geopotential height at the 850 hPa level along with wind vectors at 0:00 UT on 25 February 2011. The star (
![Atmosphere 05 00124 i002]()
) indicates the location of Nanjing.
Figure 17.
Evolution of an episode of high TGM on 24 February 2011 (
A) Time series of five-min average concentrations of trace gases and hourly wind speed. (
B) Geopotential height at the 850 hPa level along with wind vectors at 6:00 UT on 24 February 2011. (
C) Geopotential height at the 850 hPa level along with wind vectors at 0:00 UT on 25 February 2011. The star (
![Atmosphere 05 00124 i002]()
) indicates the location of Nanjing.

 ,
, 
 ) indicates the location of Nanjing.
) indicates the location of Nanjing.

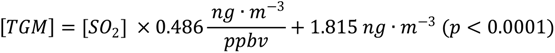

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}