Assessment of the Weather Research and Forecasting/Chemistry Model to Simulate Ozone Concentrations in March 2008 over Coastal Areas of the Sea of Japan

Abstract
:1. Introduction
2. Model Description
3. Simulation Method
3.1. Model Settings and Initialization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Horizontal grid (x, y) | 75, 70 |
| Grid spacing | 22 km |
| Meteorological time step | 180 s |
| Chemical time step | 180 s |
| Microphysics | WSM3-class simple ice scheme [56] |
| Advection scheme | 5th horizontal/3rd vertical [57,58] |
| Long wave radiation | RRTM [59] |
| Shortwave radiation | GODDARD [60,61] |
| Surface layer | Moni-Obukhov (Janjic Eta) [62] |
| Land surface model | NOAH [63] |
| Boundary layer | Mellor-Yamada-Janjic TKE [64,65] |
| Cumulus parameterization | Grell-Devenyi ensemble scheme [66] |
| Chemistry option | RADM2 [67] |
| Dry deposition | Wesley, 1989 [68] |
| Biogenic emissions | Guenther scheme [69,70] |
| Photolysis option | Madronich, 1987 [71] |
| Aerosol option | MADE/SORGAM [72] |
3.2. Data Acquisition and Pre-Processing of Emissions
| Inventory | Categories | Spatial resolution | Temporal resolution |
|---|---|---|---|
| POET [75,76] | Anthropogenic biomass burning (natural) | 1 × 1 | Annual (anthro.), monthly (biom. burn.), monthly (natural) |
| RETRO [77] | Anthropogenic biomass burning | 0.5 × 0.5 | Monthly |
| EDGAR [78 ,79,80] | Anthropogenic biomass burning | 1 × 1 | Annual |
| GFED v2 [81,82] | Biomass burning/wildfire emission | 1 × 1 | Monthly |
| CO2 [83] | Anthropogenic | 1 × 1 | Annual |
| GEIA v.1 [79] | Anthropogenic biomass burning naturally | 1 × 1 | Annual + monthly for NOx, SO2, and nat. VOC |
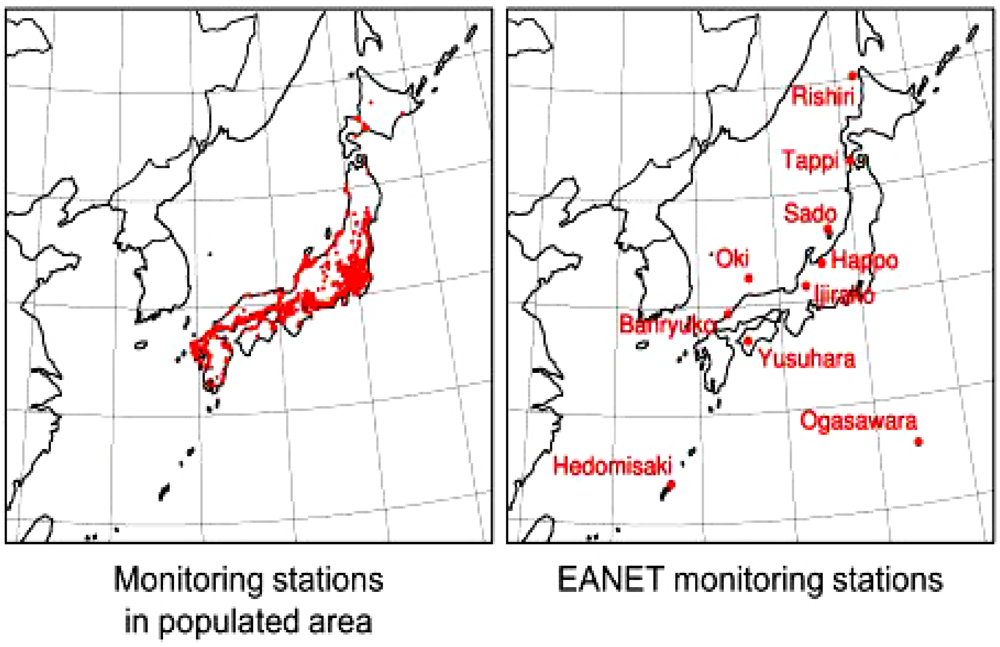
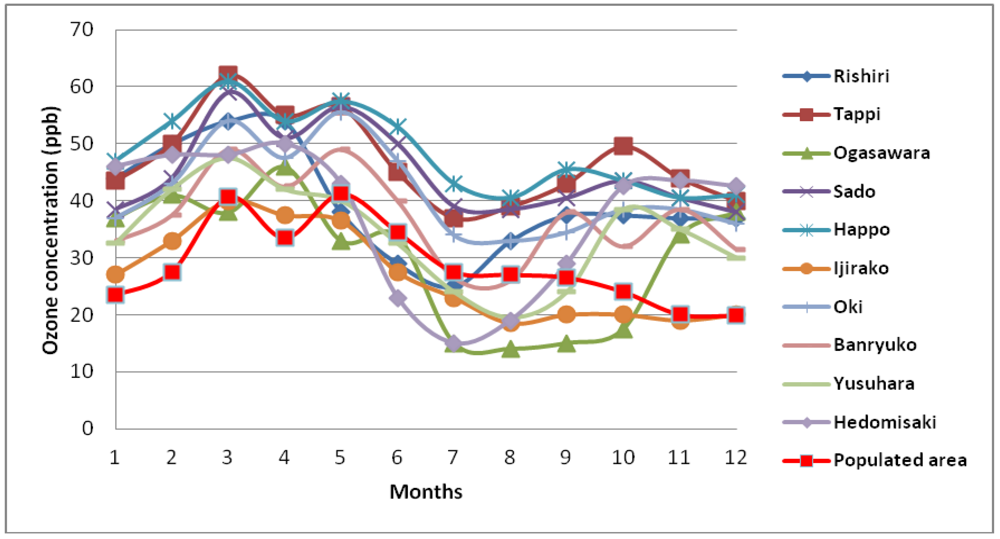
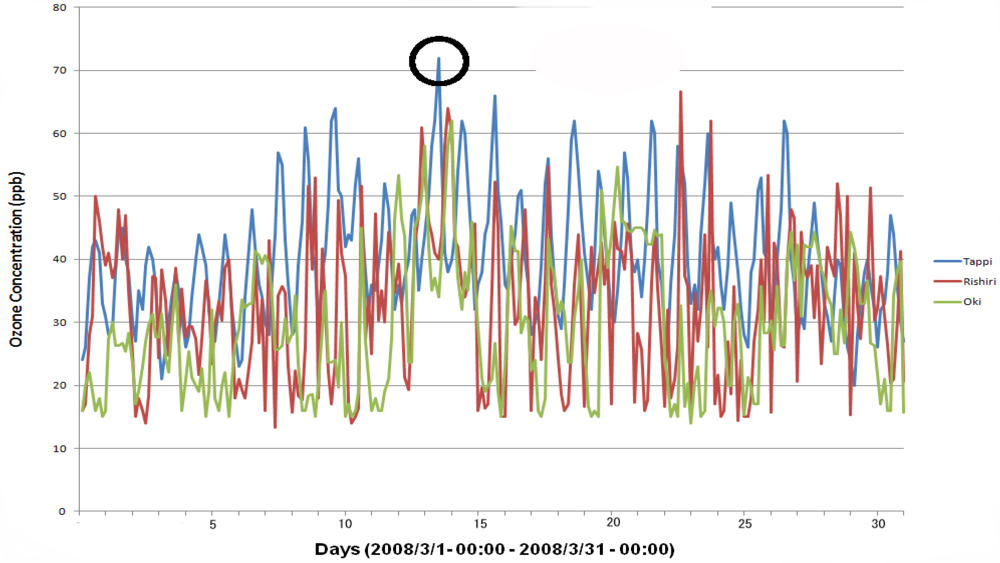
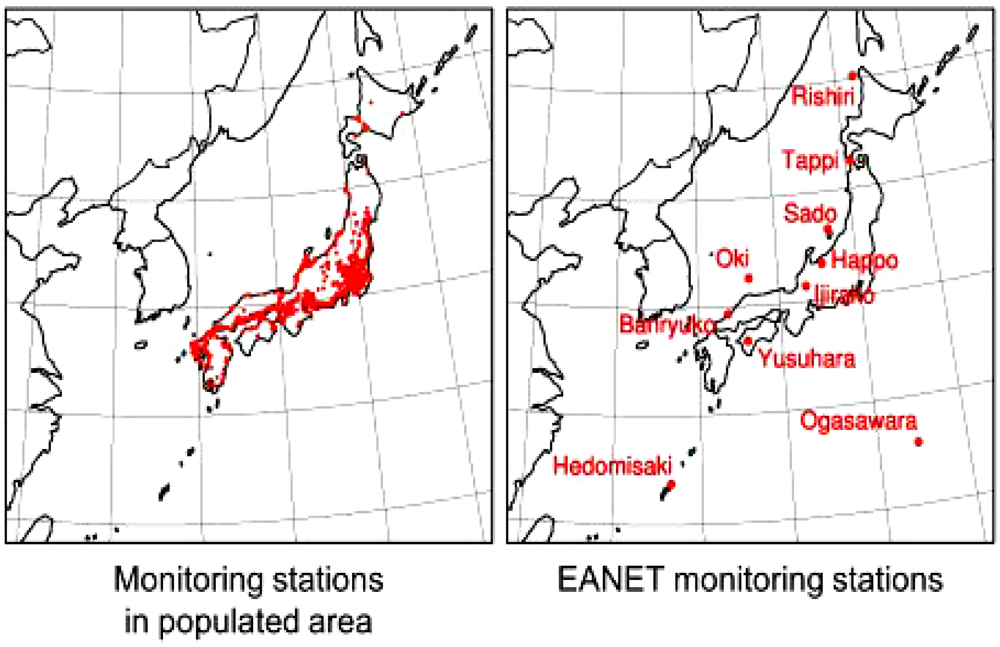
4. Observation of Ozone Concentrations over Japan
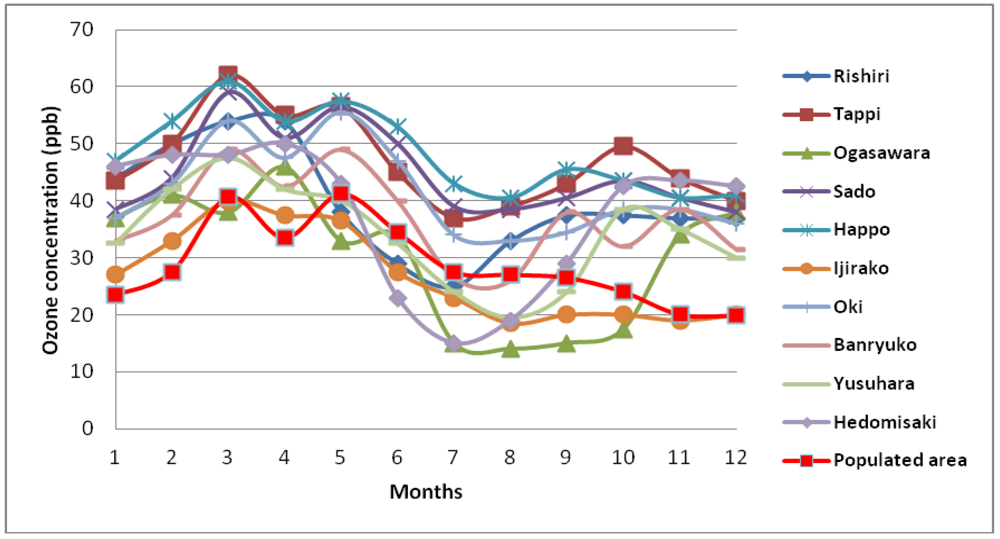
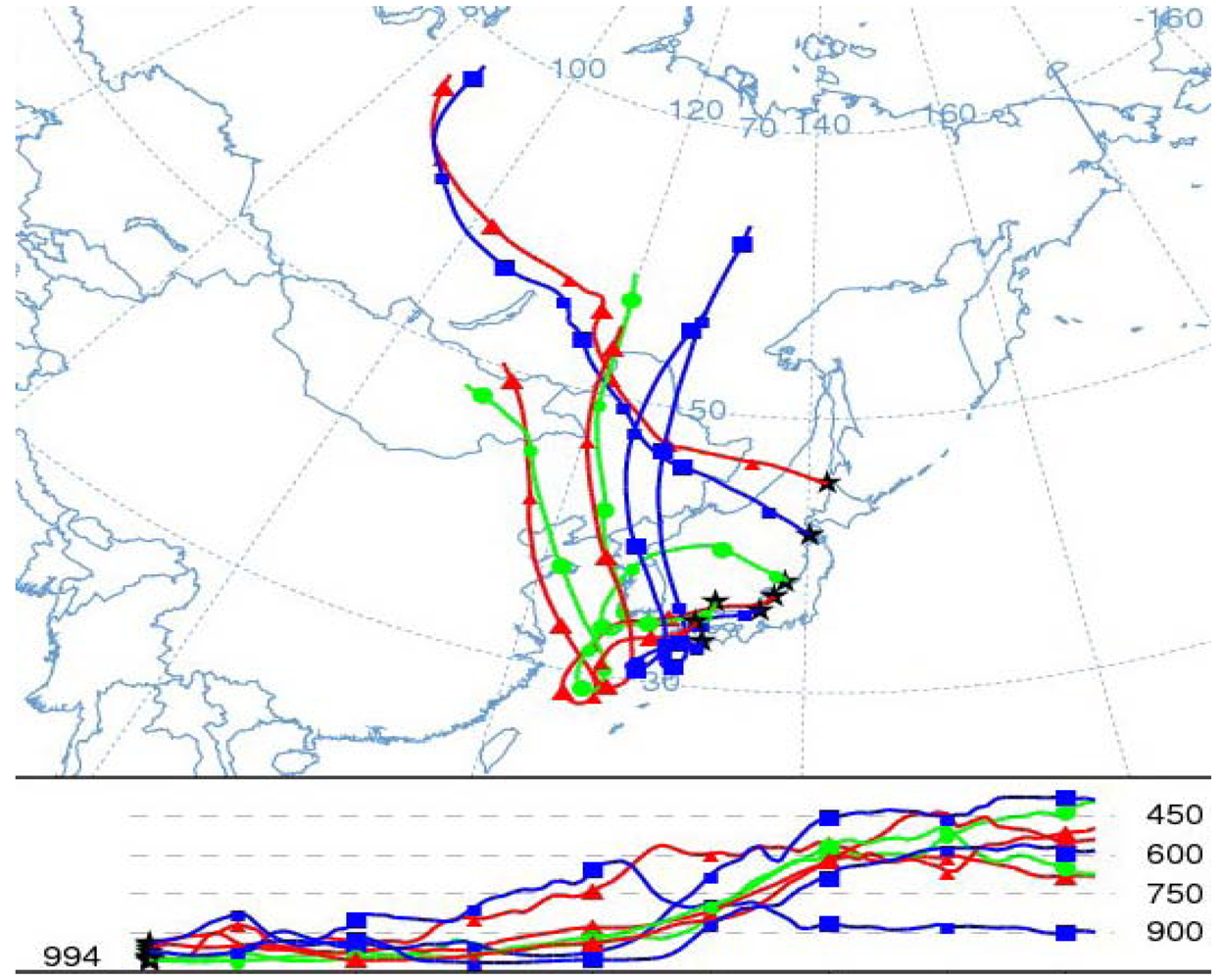
5. Characteristics of Ozone Episodes

6. Modeling Results and Discussion
6.1. Ozone Concentrations Based on WRF/Chem Simulations
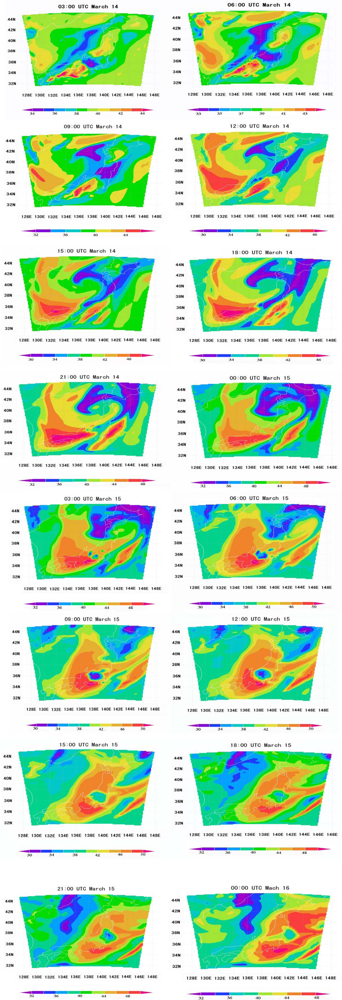
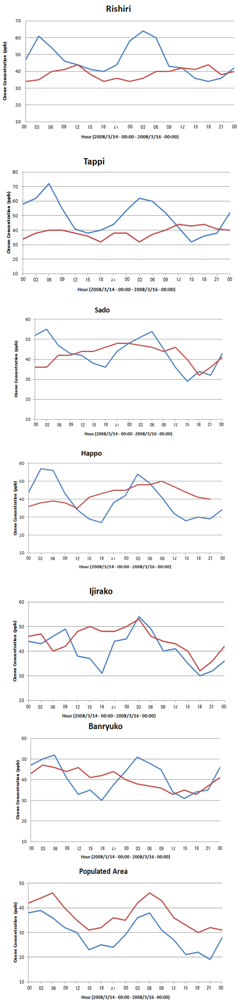
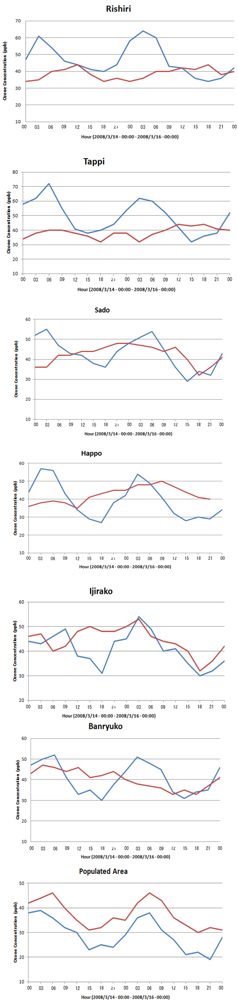
6.2. Comparison of WRF/Chem Simulation Results with Monitoring Data
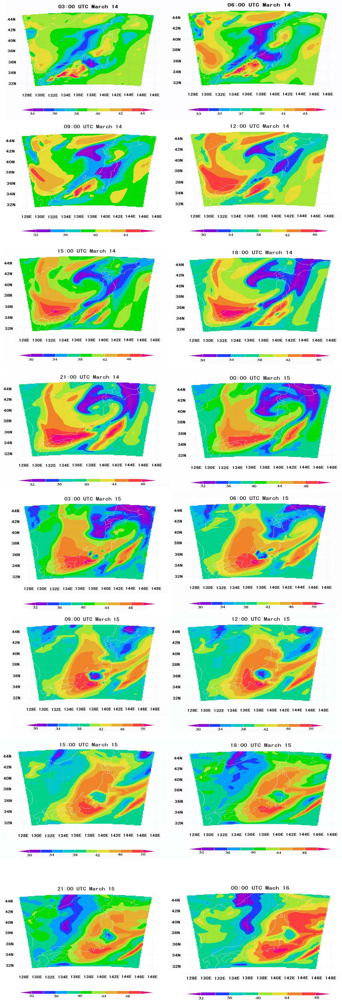
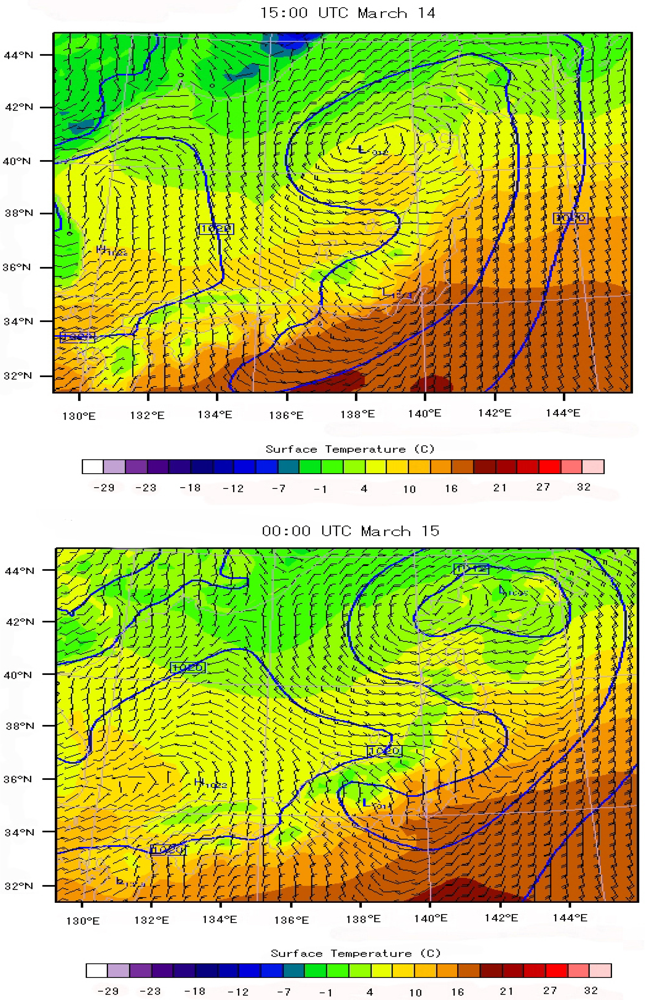
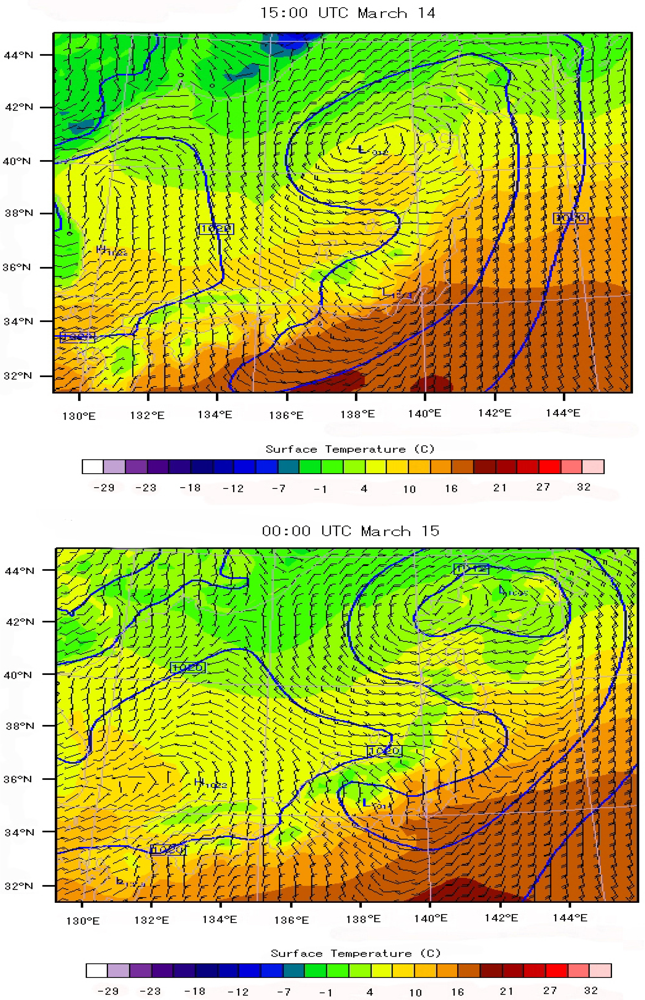
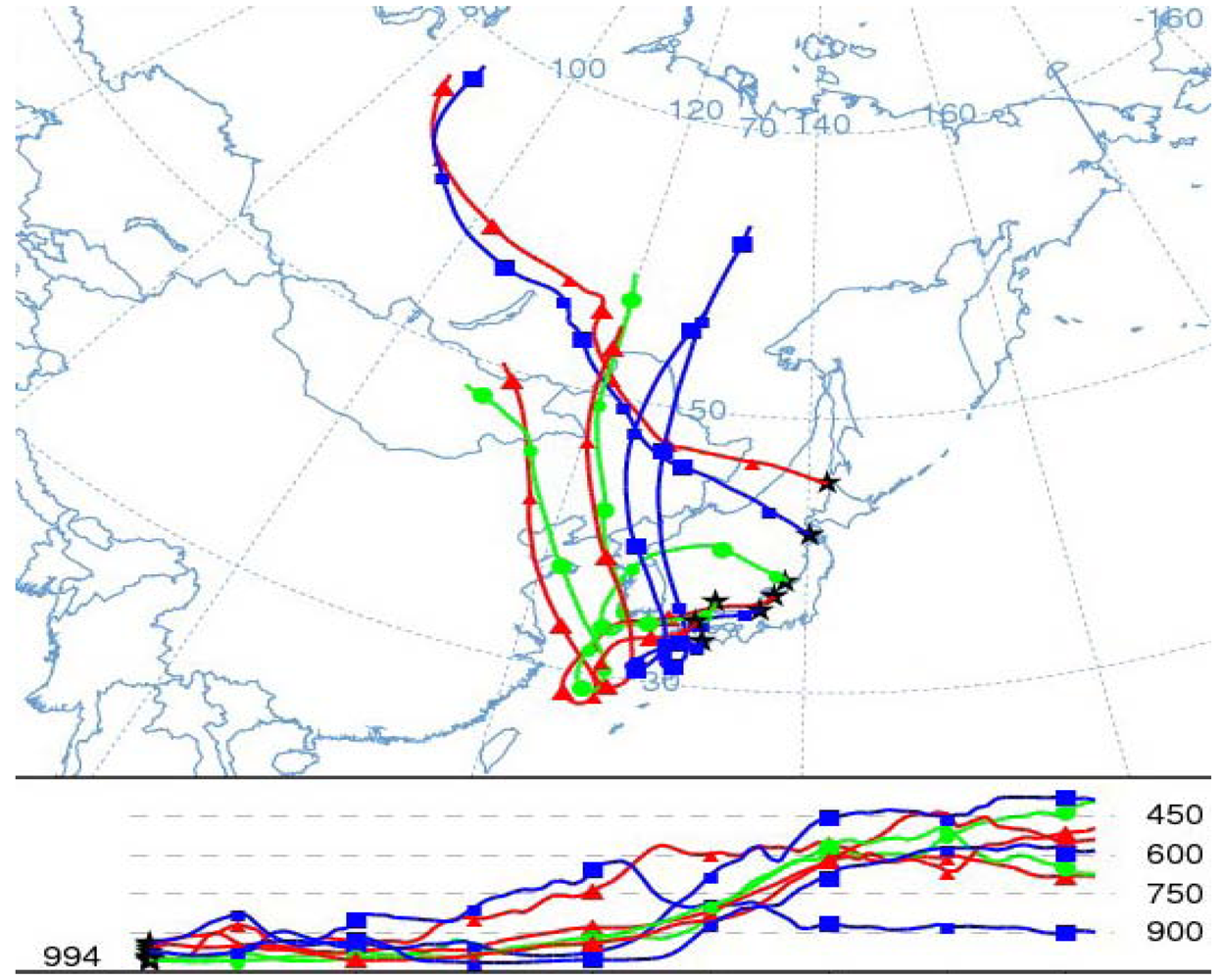
6.3. Local Meteorological Characteristics
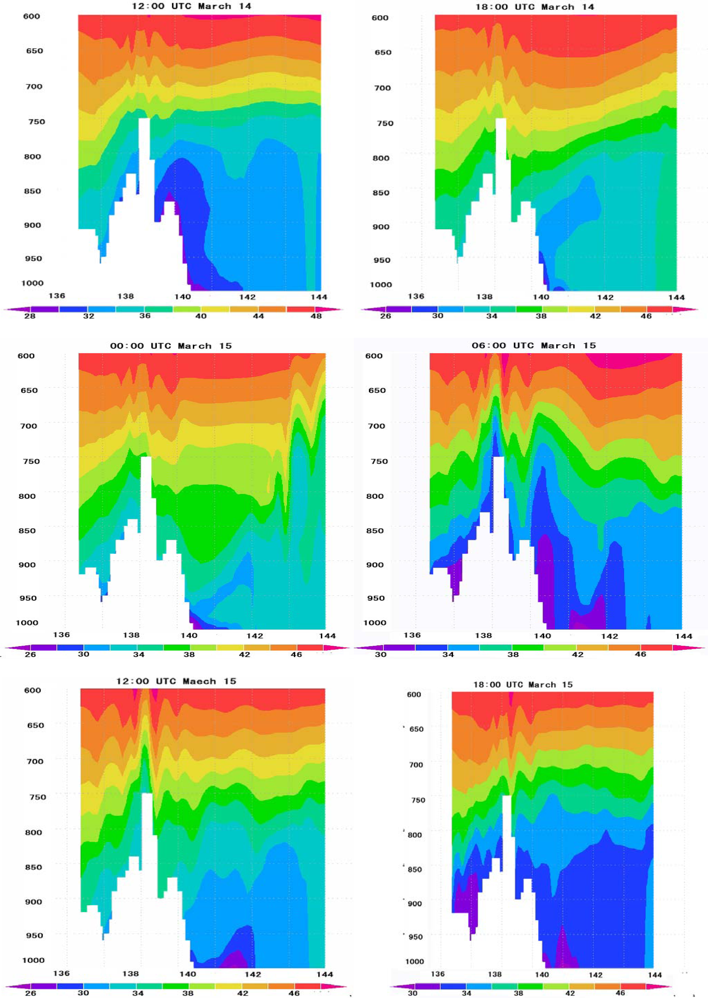
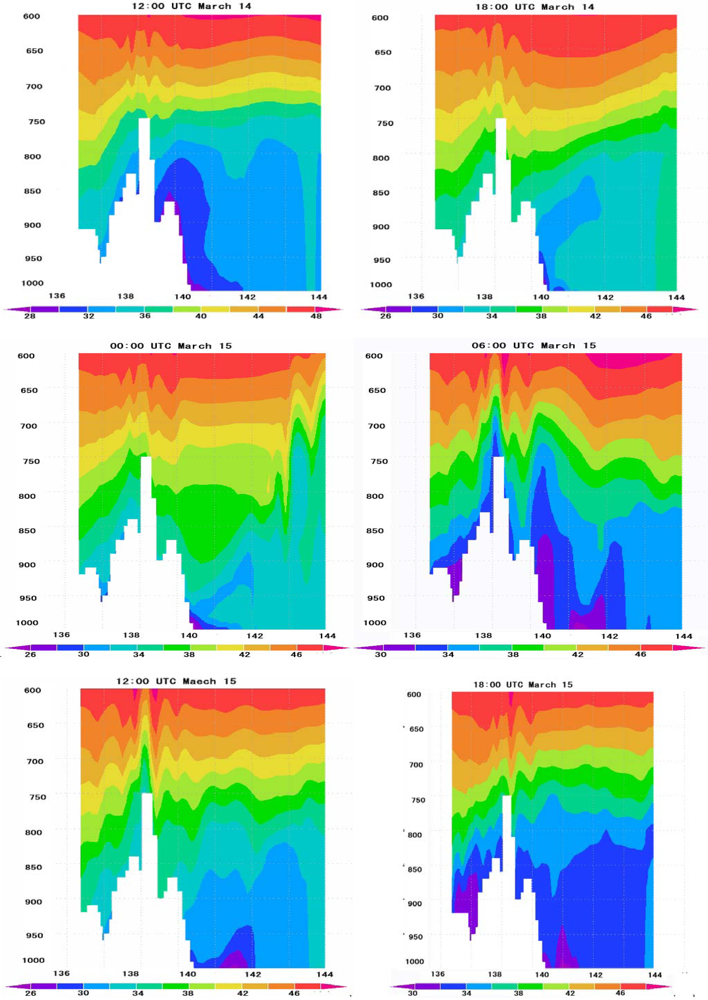
6.4. Vertical Distribution of Ozone Concentrations
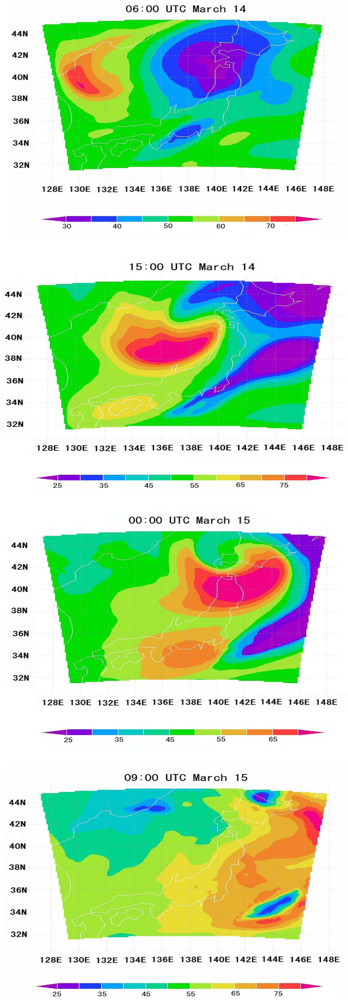
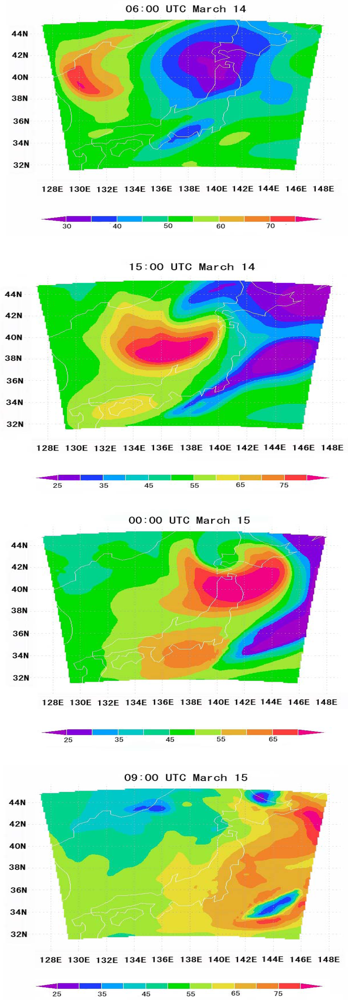
6.5. Tropospheric NOx Transportation
7. Conclusions
Acknowledgments
References
- Air Quality in the Mexico Mega City: An Integrated Assessment; Molina, L.; Molina, M. (Eds.) Massachusetts Institute of Technology: Cambridge ,MA, USA, 2002; p. 408.
- Paul, R.A.; Biller, W.F.; Mccurdy, T. National Estimates of Population Exposure to Ozone. In Presented at Air Pollution Control Association 80th Annual Meeting and Exhibition, New York, NY, USA, 21–26 June 1987.
- Tie, X.; Brasseur, G.; Zhao, C.; Granier, C.; Massie, S.; Qin, Y.; Wang, P.C.; Wang, G.L.; Yang, P.C. Chemical characterization of air pollution in eastern China and the eastern United States. Atmos. Environ. 2006, 40, 2607–2625. [Google Scholar]
- Geng, F.H.; Zhao, C.S.; Tang, X.; Lu, G.L.; Tie, X. Analysis of ozone and VOCs measured in Shanghai: A case study. Atmos. Environ. 2007, 41, 989–1001. [Google Scholar] [CrossRef]
- Deng, X.J.; Tie, X.; Wu, D.; Zhou, X.J.; Tan, H.B.; Li, F.; Jiang, C. Long-term trend of visibility and its characterizations in the Pearl River Delta Region (PRD), China. Atmos. Environ. 2008, 42, 1424–1435. [Google Scholar]
- Zhang, Q.; Zhao, C.; Tie, X.; Wei, Q.; Li, G.; Li, C. Characterizations of aerosols over the Beijing region: A case study of aircraft measurements. Atmos. Environ. 2006, 40, 4513–4527. [Google Scholar]
- Sillman, S. The use of NOx, H2O2, and HNO3 as indicators for ozone-NOx-hydrocarbon sensitivity in urban locations. J. Geophys. Res. 1995, 100, 14175–14188. [Google Scholar] [CrossRef]
- Kleinman, L.I.; Daum, P.H.; Imre, D.G.; Lee, J.H.; Lee, Y.N.; Nunnermacker, L.J.; Springston, S.R.; Weinstein-Lloyd, J.; Newman, L. Ozone production in the New York City urban plume. J. Geophys. Res. 2000, 105, 14495–14512. [Google Scholar]
- Lei, W.; De Foy, B.; Zavala, M.; Volkamer, R.; Molina, L.T. Characterizing ozone production in the Mexico City Metropolitan area, a case study using a chemical transport model. Atmos. Chem. Phys. 2007, 7, 1347–1366. [Google Scholar] [CrossRef]
- Zhang, R.; Lei, W.; Tie, X.; Hess, P. Industrial Emissions Cause Extreme Diurnal Urban Ozone Variability. In Proceedings of National Academy Science of the United States of America, Washington, DC, USA, 27 April 2004.
- Tie, X.; Madronich, S.; Li, G.H.; Ying, Z.M.; Zhang, R.; Garcia, A.; Lee-Taylor, J.; Liu, Y. Characterizations of chemical oxidants in Mexico City: A regional chemical/dynamical model (WRF/Chem) study. Atmos. Environ. 2007, 41, 1989–2008. [Google Scholar]
- Zavala, M.; Lei, W.; Molina, M.J.; Molina, L.T. Modeled and observed ozone sensitivity to mobile-source emissions in Mexico City. Atmos. Chem. Phys. 2009, 9, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Geng, F.H.; Qiang, Z.; Tie, X.; Huang, M.; Ma, X.; Deng, Z.; Quan, J.; Zhao, C. Aircraft measurements of O3, NOx, CO, VOCs, and SO2 in the Yangtze River Delta region. Atmos. Environ. 2009, 43, 584–593. [Google Scholar] [CrossRef]
- Stephens, S.; Madronich, S.; Wu, F.; Olson, J.B.; Ramos, R.; Retama, A.; Munoz, R. Weekly patterns of Mexico City’s surface concentrations of CO, NOx, PM10 and O3 during 1986-2007. Atmos. Chem. Phys. 2008, 8, 5313–5325. [Google Scholar] [CrossRef]
- Ying, Z.M.; Tie, X.; Li, G.H. Sensitivity of ozone concentrations to diurnal variations of surface emissions in Mexico City: A WRF/Chem modeling study. Atmos. Environ. 2009, 43, 851–859. [Google Scholar] [CrossRef]
- Kida, M. Countermeasures on Chemical Substances in Japan by Air Pollution Control Law. Available online: http://infofile.pcd.go.th/air/VOC_kida.pdf?CFID=1412050&CFTOKEN=60317530 (accessed on 10 August 2011).
- United Nations (UN) Statistics Division. Available online: http://unstats.un.org/unsd/methods/m49/m49regin.htm#asia (accessed on 1 April 2010).
- Finlayson-Pitts, B.J.; Pitts, J.N. References. In Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Seinfeld, J.H.; Pandis, S.N. References. In Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; John Wiley & Sons: New York, NY, USA, 1998. [Google Scholar]
- Thielmann, A.; Prevot, A.S.H.; Staehelin, J. Sensitivity of ozone production derived from field measurements in the Italian Po basin. J. Geophys. Res. 2002, 107. [Google Scholar] [CrossRef]
- Altshuller, A.P.; Lefohn, A.S. Background ozone in the planetary boundary layer over the United States. J. Air Waste Manag. Assoc. 1996, 46, 134–141. [Google Scholar] [CrossRef]
- Chameides, W.L.; Fehsenfeld, F.; Rodgers, M.O.; Cardelino, C.; Martinez, J.; Parrish, D.; Lonneman, W.; Lawson, D.R.; Rasmussen, R.A.; Zimmerman, P.; Greenberg, J.; Mlddleton, P.; Wang, T. Ozone precursor relationships in the ambient atmosphere. J. Geophys. Res. 1992, 97, 6037–6055. [Google Scholar]
- National Research Council (NRC), References. In Rethinking the Ozone Problem in Urban and Regional Air Pollution; National Academy Press: Washington, DC, USA, 1991.
- U.S. Environmental Protection Agency (U.S. EPA). Technology Transfer Network OAR Policy and Guidance. Available online: http://www.epa.gov/ttn/oarpg/t1/fr_notices/3866finalfrnotice.pdf (accessed on 25 March 2005).
- Skamarock, W.C.; Klemp, J.B.; Dudhia, J.; Gill, D.O.; Barker, D.M.; Wang, W.; Powers, J.G. A Description of the Advanced Research WRF Version 2; TN-468tSTR; NCAR Technical note. NCAR: Boulder, CO, USA, 2005; 88. [Google Scholar]
- Grell, G.A.; Peckham, S.E.; Schmitz, R.; McKeen, S.A.; Frost, G.; Skamarock, W.; Eder, B. Fully coupled “online” chemistry within the WRF model. Atmos. Environ. 2005, 39, 6957–6975. [Google Scholar]
- Byun, D.W.; Ching, J.K.S. Science algorithms of the EPA Models-3 Community Multiscale Air Quality Model (CMAQ) Modeling System; EPA/600/R-99/030. US Environmental Protection Agency, Office of Research and Development: Washington, DC, USA, 1999. [Google Scholar]
- Sistla, G.; Zhou, N.; Hao, W.; Ku, J.Y.; Rao, S.T.; Bornstein, R.; Freedman, F.; Thunis, P. Effects of uncertainties in meteorological inputs on urban airshed model predictions and ozone control strategies. Atmos. Environ. 1996, 30, 2011–2025. [Google Scholar] [CrossRef]
- Jimenez, P.; Jorba, O.; Parra, R.; Baldasano, J.M. Evaluation of MM5-EMICAT2000-CMAQ performance and sensitivity in complex terrain: High resolution application to the northeastern Iberian Peninsula. Atmos. Environ. 2006, 40, 5056–5072. [Google Scholar] [CrossRef]
- Mao, Q.; Gautney, L.L.; Cook, T.M.; Jacobs, M.E.; Smith, S.N.; Kelsoe, J.J. Numerical experiments on MM5-CMAQ sensitivity to various PBL schemes. Atmos. Environ. 2006, 30, 3092–3110. [Google Scholar]
- Zhang, Y.; Liu, P.; Pun, B.; Seigneur, C. A comprehensive performance evaluation of MM5-CMAQ for the summer 1999 southern oxidants study episode—Part I: Evaluation protocols, databases, and meteorological predictions. Atmos. Environ. 2006, 40, 4825–4838. [Google Scholar] [CrossRef]
- Otte, T.L. The impact of nudging in the meteorological model for retrospective air quality simulations. Part I: Evaluation against national observation networks. J. Appl. Meteorol. Climat. 2008, 47, 1853–1867. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, X.M.; Howell, G.W.; Sills, E.; Fast, J.D.; Gustafson, W.I.; Zaveri, R.A.; Grell, G.A.; Peckham, S.E.; McKeen, S.A. Modeling Atmospheric Aerosols in WRF/Chem. In Proceedings of 6th/15th MM5 Users’ Workshop, Boulder, CO, USA, 27-30 June 2005.
- Fast, J.D.; Gustafson, W.I.; Easter, R.C.; Zaveri, R.A.; Barnard, J.C.; Chapman, E.G.; Grell, G.A.; Peckham, S.E. Evolution of ozone, particulates, and aerosol direct radiative forcing in the vicinity of Houston using a fully coupled meteorology chemistry aerosol model. J. Geophys. Res. Atmos. 2006, 111. [Google Scholar] [CrossRef]
- Misenis, C.; Hu, X.M.; Krishnan, S.; Zhang, Y.; Fast, J.D. Sensitivity of WRF/Chem Predictions to Meteorological Schemes. In Proceedings of the 86th Annual AMS Annual Meeting, 14th Joint Conference on the Applications of Air Pollution Meteorology with the Air and Waste Management Associated, Atlanta, GA, USA, 27 January-3 February; 2006. [Google Scholar]
- Jiang, F.; Wang, T.J.; Wang, T.T.; Xie, M.; Zhao, H. Numerical modeling of a continuous photochemical pollution episode in Hong Kong using WRF-CHEM. Atmos. Environ. 2008, 42, 8717–8727. [Google Scholar] [CrossRef]
- De Foy, B.; Fast, J.D.; Paech, S.J.; Phillips, D.; Walters, J.T.; Coulter, R.L.; Martin, T.J.; Pekour, M.S.; Shaw, W.J.; Kastendeuch, P.P.; Marley, N.A.; Retama, A.; Molina, L.T. Basin scale wind transports during the MILAGRO field campaign and comparison to climatology using cluster analysis. Atmos. Chem. Phys. 2008, 8, 1209–1224. [Google Scholar]
- Tie, X.; Madronich, S.; Li, G.; Ying, Z.; Weinheimer, A.; Apel, E.; Campos, T. Simulation of Mexico City plumes during the MIRAGE-Mex field campaign using the WRF-Chem model. Atmos. Chem. Phys. 2009, 9, 4621–4638. [Google Scholar] [CrossRef]
- Zhang, Y.; Dubey, M.K.; Olsen, S.C.; Zheng, J.; Zhang, R. Comparisons of WRF/Chem simulations in Mexico City with ground-based RAMA measurements during the 2006-MILAGRO. Atmos. Chem. Phys. 2009, 9, 3777–3798. [Google Scholar] [CrossRef]
- Chatani, S.; Sudo, K. Influences of the variation in inflow to East Asia on surface ozone over Japan during 1996-2005. Atmos. Chem. Phys. 2011, 11, 8745–8758. [Google Scholar] [CrossRef]
- Masanori, N.; Masayuki, T.; Hajime, A.; Masahisa, N.; Tomohiro, N.; Tetsu, S.; Yuzo, M. Evaluation of vertical ozone profiles simulated by WRF/Chem using lidar-observed data. SOLA 2007, 3, 133–136. [Google Scholar] [CrossRef]
- Kurokawa, J.; Ohara, T.; Uno, I.; Hayasaki, M.; Tanimoto, H. Influence of meteorological variability on interannual variations of the springtime boundary layer ozone over Japan during 1981-2005. Atmos. Chem. Phys. 2009, 9, 6287–6304. [Google Scholar] [CrossRef]
- Kurokawa, J.; Ohara, T.; Uno, I.; Hayasaki, M. Analysis of episodic pollution of photochemical ozone during 8-9 May 2007 over Japan using the nesting RAMS/CMAQ modeling system. J. Japan Soc. Atmos. Environ. 2008, 43, 209–224. [Google Scholar]
- Hayasaki, M.; Ohara, T.; Kurokawa, J.; Uno, I.; Shimizu, A. Episodic pollution of photochemical ozone during 8-9 May 2007 over Japan: Observational data analysis. J. Japan Soc. Atmos. Environ. 2008, 43, 225–237. (in Japanese).. [Google Scholar]
- Saikawa, E.; Kurokawa, J.; Takigawa, M.; Mauzerall, D. L.; Horowitz, L. W.; Ohara, T. The impact of China’s vehicle emissions on regional air quality in 2000 and 2020: A scenario analysis. Atmos. Chem. Phys. Discuss. 2011, 11, 13141–13192. [Google Scholar] [CrossRef]
- Wesley, M.L. Parameterization of surface resistance to gaseous dry deposition in regional numerical models. Atmos. Environ. 1989, 16, 1293–1304. [Google Scholar]
- Madronich, S.; Flocke, S. References. In The Handbook of Environmental Chemistry; Springer-Verlag: Heidelberg, Berlin, Germany, 1999; pp. 1–26. [Google Scholar]
- Tie, X.; Madronich, S.; Walters, S.; Zhang, R.; Rasch, P.; Collins, W. Effect of clouds on photolysis and oxidants in the troposphere. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
- Chang, J.S.; Middleton, P.B.; Stockwell, W.R.; Walcek, C.J.; Pleim, J.E.; Lansford, H.H.; Madronich, S.; Binkowski, F.S.; Seaman, N.L.; Stauffer, D.R. The Regional Acid Deposition Model and Engineering Model; Report No. 4; NAPAP SOS/T: Washington, DC, USA, 1989. [Google Scholar]
- Schell, B.; Ackermann, I.J.; Hass, H.; Binkowski, F.S.; Ebel, A. Modeling the formation of secondary organic aerosol within a comprehensive air quality model system. J. Geophys. Res. 2001, 106, 28275–28293. [Google Scholar]
- S Korea Bid to Solve Sea Dispute. BBC News. 2007. Available online: http://news.bbc.co.uk/2/hi/asia-pacific/6240051.stm (accessed on 8 January 2007).
- The Ministry of Foreign Affairs and Trade, Republic of Korea (MFAT). Report on the Progress in Consultations on the Naming of the Sea Area between the Korean Peninsula and the Japanese Archipelago. In Proceedings of Ninth United Nations Conference on the Standardization of Geographical Names, 21-30 August 2007; E/CONF.98/CRP.81.. United Nations Economic and Social Council: New York, NY, USA.
- David, W.A.; Ashby, N.; Hodge, C.C. The Science of Timekeeping. Hewlett-Packard; Application Note No. 1289. Allan’s Time Interval Metrology Enterprise: Fountain Green, UT, USA, 1997. [Google Scholar]
- Hanado, Y.; Imamura, K.; Kotake, N.; Nakagawa, F.; Shimizu, Y.; Tabuchi, R.; Takahashi, Y.; Hosokawa, M.; Morikawa, T. The new generation system of Japan standard time at NICT. Int. J. Nav. Observ. 2008. [Google Scholar] [CrossRef]
- Binkowski, F.S.; Shankar, U. The regional particulate matter model, 1. Mode description and preliminary results. J. Geophys. Res. 1995, 100, 26191–26209. [Google Scholar] [CrossRef]
- Hong, S.Y.; Dudhia, J.; Chen, S.H. A revised approach to ice microphysical processes for the bulk parameterization of clouds and precipitation. J. Amer. Met. Soc. 2004, 132, 103–120. [Google Scholar]
- Ahmad, N.; Linderman, J. A godunov-type finite volume scheme for meso- and micro-scale flows in three dimensions. Pure Appl. Geophys. 2008, 165, 1929–1939. [Google Scholar] [CrossRef]
- Baldauf, M.; Skamarock, W.C. An Improved Third Order Vertical Advection Scheme for the Runge-Kutta Dynamical Core. In Proceedings of8th International SRNWP-Workshop on Non-Hydrostatic Modelling, Bad Orb, Germany, 26-28 October 2009.
- Mlawer, E.J.; Taubman, S.J.; Brown, P.D.; Iacono, M.J.; Clough, S.A. Radiative transfer for inhomogeneous atmosphere: RRTM, a validated correlated-k model for the long wave. J. Geophys. Res. 1997, 102, 16663–16682. [Google Scholar]
- Shin, H.H.; Hong, S.-Y.; Dudhia, J.; Kim, Y.-J. Orography-induced gravity wave drag parameterization in the global WRF: Implementation and sensitivity to shortwave radiation schemes. Adv. Meteorol. 2010, 2010. [Google Scholar] [CrossRef]
- Dudhia, J. Numerical study of convection observed during the winter monsoon experiment using a mesoscale two-dimensional model. J. Atmos. Sci. 1989, 46, 3077–3107. [Google Scholar] [CrossRef]
- Monin, A.S.; Obukhov, A.M. Basic laws of turbulent mixing in the surface layer of the atmosphere. Contrib. Geophys. Inst. Slov. Acad. Sci. 1954, 151, 163–187. (in Russian).. [Google Scholar]
- Ek, M.B.; Mitchell, K.E.; Lin, Y.; Rogers, E.; Grunmann, P.; Koren, V.; Gayno, G.; Tarpley, J.D. Implementation of Noah land surface model advances in the National Centers for Environmental Prediction operational mesoscale Eta model. J. Geophys. Res. 2003, 108, 8851–8867. [Google Scholar]
- Janjic, Z.I. The step-mountain coordinate: Physical package. Mon. Wea. Rev. 1990, 118, 1429–1443. [Google Scholar] [CrossRef]
- Janjic, Z.I. The step-mountain ETA coordinate model: Further development of the convection, viscous sub-layer, and turbulent closure schemes. Mon. Wea. Rev. 1994, 122, 927–945. [Google Scholar] [CrossRef]
- Grell, G.A.; Devenyi, D. A generalized approach to parameterizing convection combining ensemble and data assimilation techniques. Geophys. Res. Lett. 2002, 29. [Google Scholar] [CrossRef]
- Stockwell, W.R.; Middleton, P.; Chang, J.S. The second generation regional acid deposition model chemical mechanism for regional air quality modeling. J. Geophys. Res. 1990, 95, 16343–16367. [Google Scholar] [CrossRef]
- Wesley, M.L. Parameterization of surface resistance to gaseous dry deposition in regional numerical models. Atmos. Environ. 1989, 16, 1293–1304. [Google Scholar]
- Guenther, A.B.; Zimmerman, P.R.; Harley, P.C.; Monson, R.K.; Fall, R. Isoprene and monoterpene emission rate variability: Model evaluations and sensitivity analyses. J. Geophys. Res. Atmos. 1993, 98, 12609–12617. [Google Scholar]
- Guenther, A.; Zimmerman, P.; Wildermuth, M. Natural volatile organic compound emission rate estimates for US woodland landscapes. Atmos. Environ. 1994, 28, 1197–1210. [Google Scholar] [CrossRef]
- Madronich, S. Photodissociation in the atmosphere 1. Actinic flux and the effects of ground reflections and clouds. J. Geophys. Res. 1987, 92, 9740–9752. [Google Scholar] [CrossRef]
- Schell, B.; Ackermann, I.J.; Hass, H.; Binkowski, F.S.; Ebel, A. Modeling the formation of secondary organic aerosol within a comprehensive air quality model system. J. Geophys. Res. 2001, 106, 28275–28293. [Google Scholar]
- Freitas, S.R.; Longo, K.; Silva Dias, M.; Silva Dias, P.; Chatfield, R.; Prins, E.; Artaxo, P.; Grell, Y.G.; Recuero, F. Monitoring the transport of biomass burning emissions in South America. Environ. Fluid Mech. 2005, 5, 135–167. [Google Scholar] [CrossRef]
- Longo, K.; Freitas, S. R.; Setzer, A.; Prins, E.; Artaxo, P.; Andreae, M. The coupled aerosol and tracer transport model to the Brazilian developments on the regional atmospheric modeling system (CATT-BRAMS). Part 2: Model sensitivity to the biomass burning inventories. Atmos. Chem. Phys. Discuss. 2007, 8571–8595.
- Granier, C.; Niemeir, U.; Muller, J. F.; Olivier, J.; Peters, J.; Richter, A.; Nuss, H.; Burrows, J. Variation of the Atmospheric Composition over the 1990-2000 Period; EU project EVK2-1999-00011, Report No. 6. POET: Paris, France, 2003. [Google Scholar]
- Olivier, J.; Peters, J.; Granier, C.; Petron, G.; Muller, J.F.; Wallens, S. Present and Future Surface Emissions of Atmospheric Compounds; EU project EVK2-1999-00011, POET report No. 2. POET POET: Paris, France, 2003. [Google Scholar]
- Schultz, M. RETRO team. RETRO Emission Trends and Variability. Presented at 1st ACCENT Symposium, Urbino, Italy, 12–16 September 2005; 2005. Available online: http://retro.enes.org/index.shtml (accessed on 8 April 2008).
- Olivier, J.G.J.; Bouwman, A.F.; Van der Maas, C.W.M.; Berdowski, J.J.M.; Veldt, C.; Bloos, J.P.J.; Visschedijk, A.J.H.; Zandveld, P.Y.J.; Haverlag, J.L. Description Of EDGAR Version 2.0: A Set of Global Emission Inventories of Greenhouse Gases and Ozone-Depleting Substances for All Anthropogenic and Mos-Natural Sources on a Per Country Basis and On a 1 × 1 Grid; RIVM report No. 771060 002. RIVM: Bilthoven, The Netherlands, 1996. Available online: http://www.mnp.nl/edgar/introduction (accessed on 5 October 2010).
- Olivier, J.G.J.; Bloos, J.P.J.; Berdowski, J.J.M.; Visschedijk, A.J.H.; Bouwman, A.F. A 1990 global emission inventory of anthropogenic sources of carbon monoxide on 1 × 1 developed in the framework of EDGAR/GEIA. Chemosphere: Global Change Sci. 1999, 1, 1–17. [Google Scholar] [CrossRef]
- Olivier, J.G.J.; Berdowski, J.J.M.; Peters, J.A.H.W.; Bakker, J.; Visschedijk, A.J.H.; Bloos, J.-P.J. Applications of EDGAR. Including a Description of EDGAR 3.0: Reference Database with Trend Data for 1970-1995; Bilthoven; RIVM report no. 773301 001/ NOP report no. 410200 051. RIVM: Bilthoven, The Netherlands, 2001. [Google Scholar]
- Van der Werf, G.R.; Randerson, J.T.; Collatz, G.J.; Giglio, L.; Kasibhatla, P.S.; Avelino, A.; Olsen, S.C.; Kasischke, E.S. Continental-scale partitioning of fire emissions during the 1997-2001 El Nino/La Nina period. Science. 2004, 303. Available online: http://daac.ornl.gov/VEGETATION/guides/global_fire_emissions_v2.1.html. (accessed on 22 June 2010).
- Van der Werf, G.R.; Randerson, J.T.; Giglio, L.; Collatz, G.J.; Kasibhatla, P.S. Interannual variability in global biomass burning emission from 1997 to 2004. Atmos. Chem. Phys. 2006, 6, 3423–3441. [Google Scholar] [CrossRef]
- Andres, R.J.; Fielding, D.J.; Marland, G.; Boden, T.A.; Kumar, N. Carbon dioxide emissions from fossil-fuel use 1751-1950. Tellus 1999, 51, 759–765. [Google Scholar]
- Saiki, M. Annual Report on Atmospheric and Marine Environment Monitoring: Observation Results for 2008; No. 10. Global Environment and Marine Department,Japan Meteorological Agency (JMA): Tokyo, Japan, 2010. [Google Scholar]
- Segami, A. Annual Report on Atmospheric and Marine Environment Monitoring: Observation result for 2009; No. 11. Global Environment and Marine Department,Japan Meteorological Agency (JMA): Tokyo, Japan, 2011. [Google Scholar]
- National Institute for Environmental Studies. Observation Data Download for Pollutant Concentration in the Atmospheric Environment. Available online: http://www.nies.go.jp/igreen/td_down.html (accessed on 20 July 2010) (in Japanese).
- Ministry of the Environment (MOE). The Status of Atmosphere Pollution. Available online: http://www.env.go.jp/air/osen/index.html (accessed on 20 August 2010) (in Japanese).
- Simpson, D.; Olendrzynacuteski, K.; Semb, A.; Storen, E.; Unger, S. Photochemical Oxidant Modeling In Europe: Multi-Annual Modeling and Source Receptor Relationships; EMEP MSC-W Report 3/97. Norwegian Meteorological Institute: Oslo, Norway, 1997. [Google Scholar]
- Giorgi, F.; Meleux, F. Modelling the regional effects of climate change on air quality. C. R. Geosci. 2007, 339, 721–733. [Google Scholar] [CrossRef]
- Hirofumi, A. Air Pollution Control Policy in Japan for Mitigating Sulphur Emission. In Presentation of the International Conference on Transboundary Air Pollution in North-East Asia, Tokyo, Japan, 17–19 December 2008.
- Val, B. Cyclones and Anticyclones in the Mid-Latitudes. Available online: www.aos.wisc.edu/~aos101vb/val_cyclones.ppt (accessed on 2 January 2010).
- National Weather Service. Available online: http://www.srh.weather.gov/srh/jetstream/index.htm (accessed on 24 October 2006).
- Vautard, R.; Honore, C.; Beekmanna, M.; Rouil, L. Simulation of ozone during the August 2003 heat wave and emission control scenarios. Atmos. Environ. 2005, 39, 2957–2967. [Google Scholar] [CrossRef]
- Seaman, N.L. Meteorological modeling for air-quality assessments. Atmos. Environ. 2000, 34, 2231–2259. [Google Scholar] [CrossRef]
- Logan, J.A. Ozone in rural areas of the United States. J. Geophys. Res. 1989, 94, 8511–8532. [Google Scholar] [CrossRef]
- Parrish, D.D.; Buhr, M.P.; Trainer, M.; Norton, R.B.; Shimshock, J.P.; Fehsenfeld, C.F.; Anlauf, K.G.; Bottenheim, J.W.; Tang, Y.Z.; Wiebe, H.A.; Roberts, J.M.; Tanner, R.L.; Newman, L.; Bowersox, V.C.; Olszyna, K.J.; Bailey, E.M.; Rodgers, M.O.; Wang, T.; Berresheim, H.; Roychowdhury, U.K.; Demerjiani, K.L. The total oxidized nitrogen levels and the partitioning between the individual species at six rural sites in eastern North America. J. Geophys. Res. 1993, 98, 2927–2939. [Google Scholar]
- Trainer, M.; Williams, E.J.; Parrish, D.D.; Buhr, M.P.; Allwine, E.J.; Westberg, H.H.; Fehsenfeld, F.C.; Liu, S.C. Models and observations of the impact of natural hydrocarbons on rural ozone. Nature 1987, 329, 705–707. [Google Scholar] [CrossRef]
- Warneck, P. Chemistry of the Natural Atmosphere; Academic Press Inc.: London, UK, 1988. [Google Scholar]
- National Research Council (NRC), References. In Rethinking the Ozone Problem in Urban and Regional Air Pollution; National Academy Press: Washington, DC, USA, 1992.
- Lin, X.; Trainer, M.; Liu, S.C. On the nonlinearity of the tropospheric ozone production. J. Geophys. Res. 1988, 93, 15879–15888. [Google Scholar]
- Levy, H.; Moxim, W.J.; Klonecki, A.A.; Kasibhatla, P.S. Simulated tropospheric NOx: Its evaluation, global distribution and individual source contributions. J. Geophys. Res. 1999, 104, 26279–26306. [Google Scholar]
- Johnson, C.; Henshaw, J.; McInnes, G. Impact of aircraft and surface emissions of nitrogen oxides on tropospheric ozone and global warming. Nature 1992, 355, 69–71. [Google Scholar]
- Stoddard, J.L.; Jeffries, D.S.; Lukeville, A.; Clair, T.A.; Dillon, P.J.; Driscoll, C.T.; Forsius, M.; Johannessen, M.; Kahl, J.S.; Kellogget, J.H.; et al. Regional trends in aquatic recovery from acidification in North America and Europe. Nature 1999, 401, 575–578. [Google Scholar]
- Stoddard, J.L.; Jeffries, D.S.; Lükewille, A.; Clair, T.A.; Dillon, P.J.; Driscoll, C.T.; Forsius, M.; Johannessen, M.; Kahl, J.S.; Kellogg, J.H.; et al. Sources and chemistry of NOx in the upper troposphere over the United States. Geophys. Res. Lett. 1998, 25, 1705–1708. [Google Scholar]
- Stohl, A.; Eckhardt, S.; Forster, C.; James, P.; Spichtinger, N. On the pathways and timescales of intercontinental air pollution transport. J. Geophys. Res. 2002, 107. [Google Scholar] [CrossRef]
- Murphy, D.; Fahey, D.; Profitt, M.; Liu, S.; Chan, K.; Eubank, C.; Kawa, S.; Kelly, K. Reactive nitrogen and its correlation with ozone in the lower stratosphere and upper troposphere. J. Geophys. Res. 1993, 98, 8751–8773. [Google Scholar]
- Von Kuhlmann, R.; Lawrence, M.G.; Crutzen, P.J.; Rasch, P.J. A model for studies of tropospheric ozone and nonmethane hydrocarbons: Model description and ozone results. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al Razi, K.M.H.; Hiroshi, M. Assessment of the Weather Research and Forecasting/Chemistry Model to Simulate Ozone Concentrations in March 2008 over Coastal Areas of the Sea of Japan. Atmosphere 2012, 3, 288-319. https://doi.org/10.3390/atmos3030288
Al Razi KMH, Hiroshi M. Assessment of the Weather Research and Forecasting/Chemistry Model to Simulate Ozone Concentrations in March 2008 over Coastal Areas of the Sea of Japan. Atmosphere. 2012; 3(3):288-319. https://doi.org/10.3390/atmos3030288
Chicago/Turabian StyleAl Razi, Khandakar Md. Habib, and Moritomi Hiroshi. 2012. "Assessment of the Weather Research and Forecasting/Chemistry Model to Simulate Ozone Concentrations in March 2008 over Coastal Areas of the Sea of Japan" Atmosphere 3, no. 3: 288-319. https://doi.org/10.3390/atmos3030288