High Contribution of Biomass Combustion to PM2.5 in the City Centre of Naples (Italy)

 , ,
, , 
Abstract
:1. Introduction
2. Methods
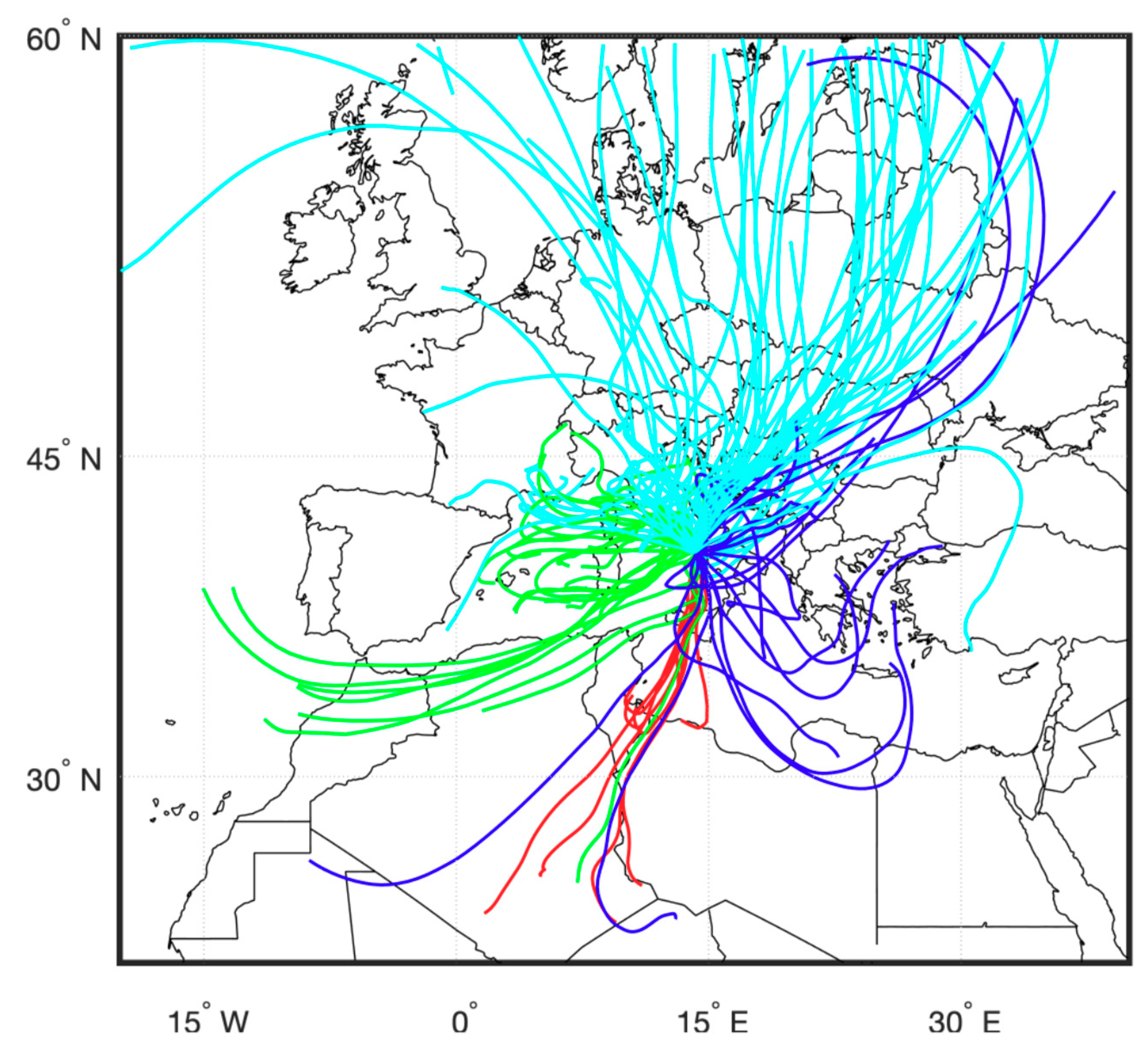
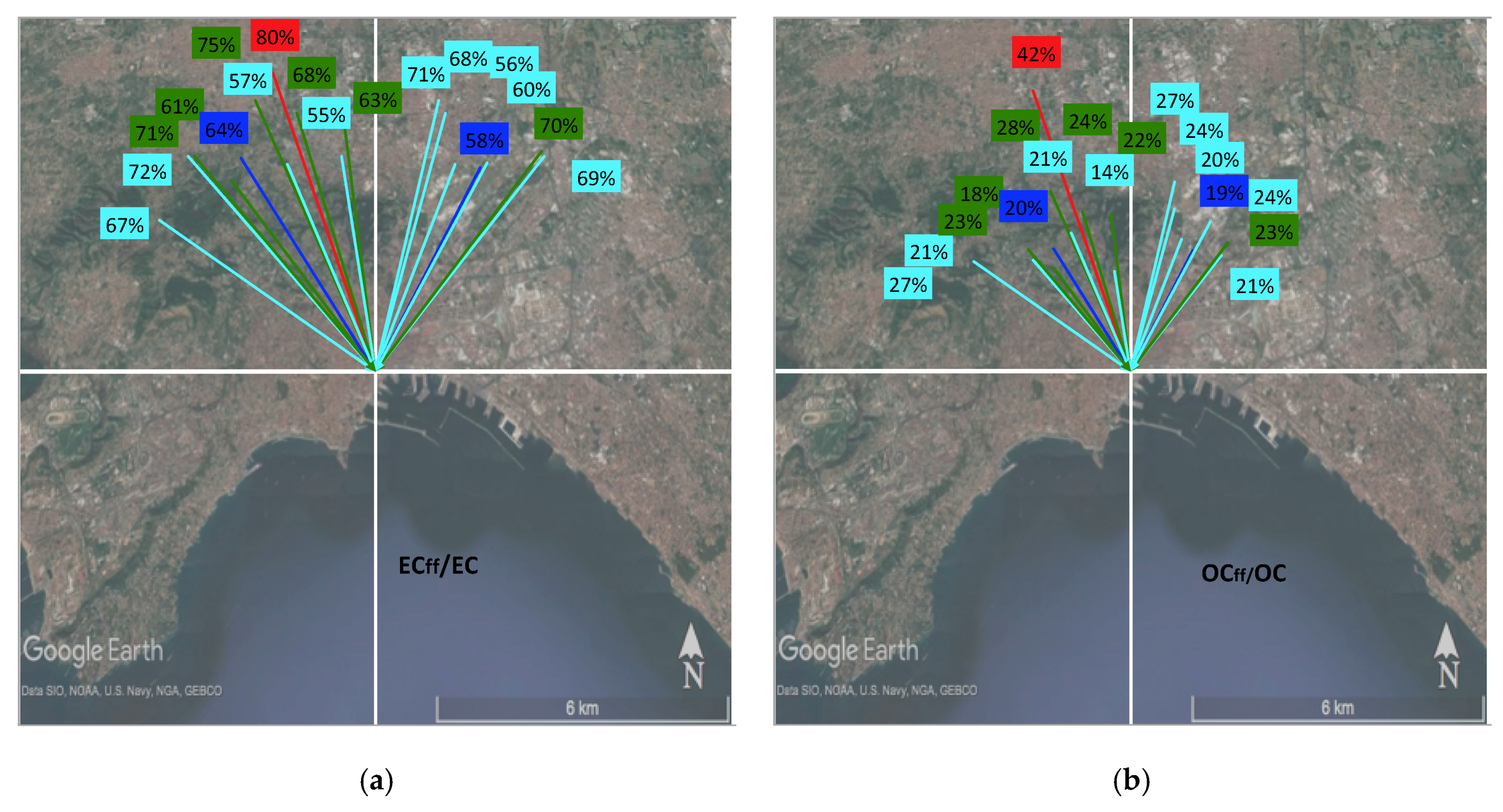
2.1. Sampling
- similar sample appearance, i.e., color (black, grey or sandy);
- similar average wind direction (within 6 degrees);
- similar precipitation condition (no precipitation or precipitations on both days);
- similar origins of the air masses, according to back-trajectories.
2.2. Sample Processing and Mass Concentration Measurements
2.3. Radiocarbon Analysis
2.4. Source Apportionment
2.5. Uncertainty Assessment
3. Results and Discussions
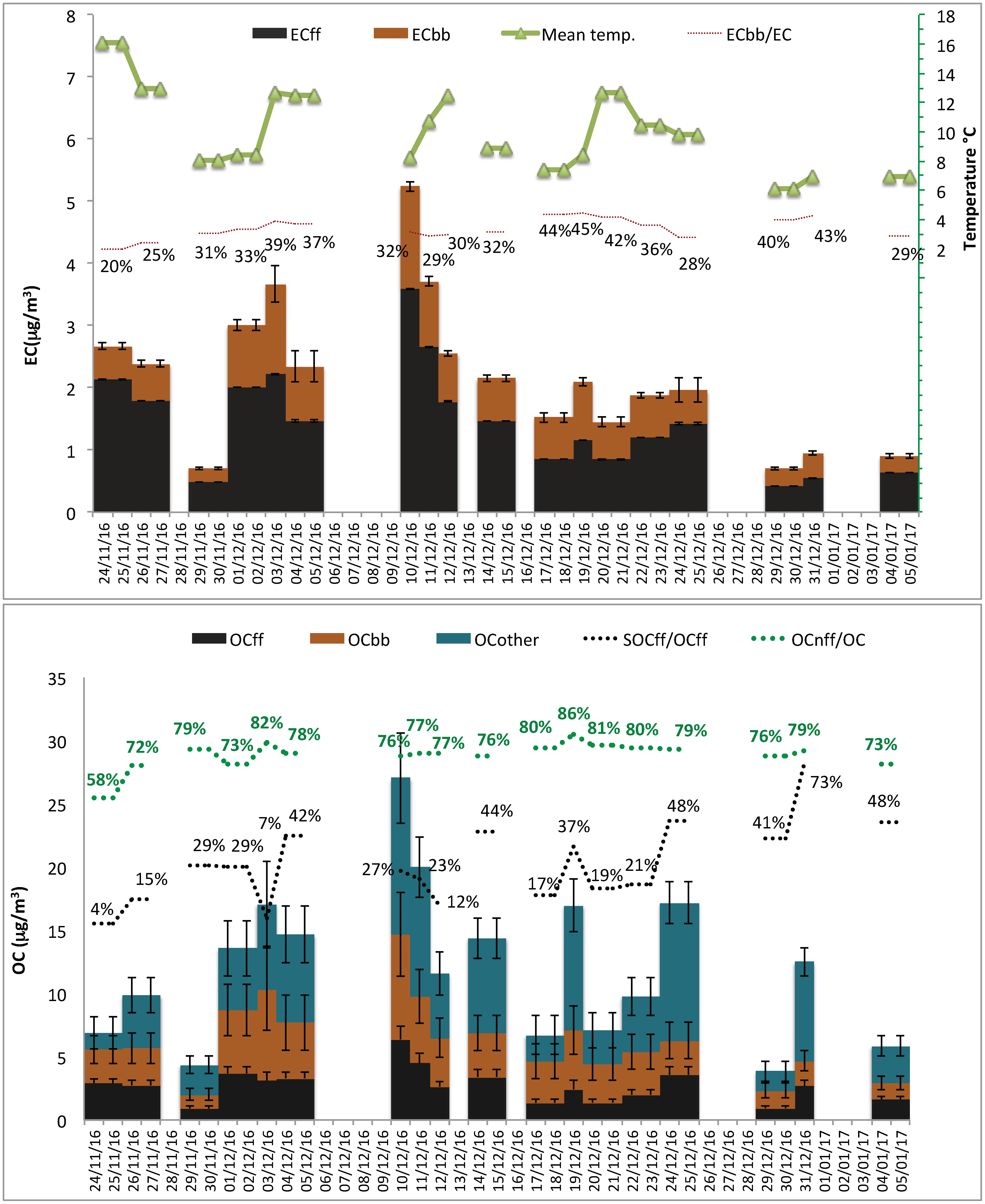
3.1. PM2.5 Concentration and Composition of Its Carbonaceous Fraction
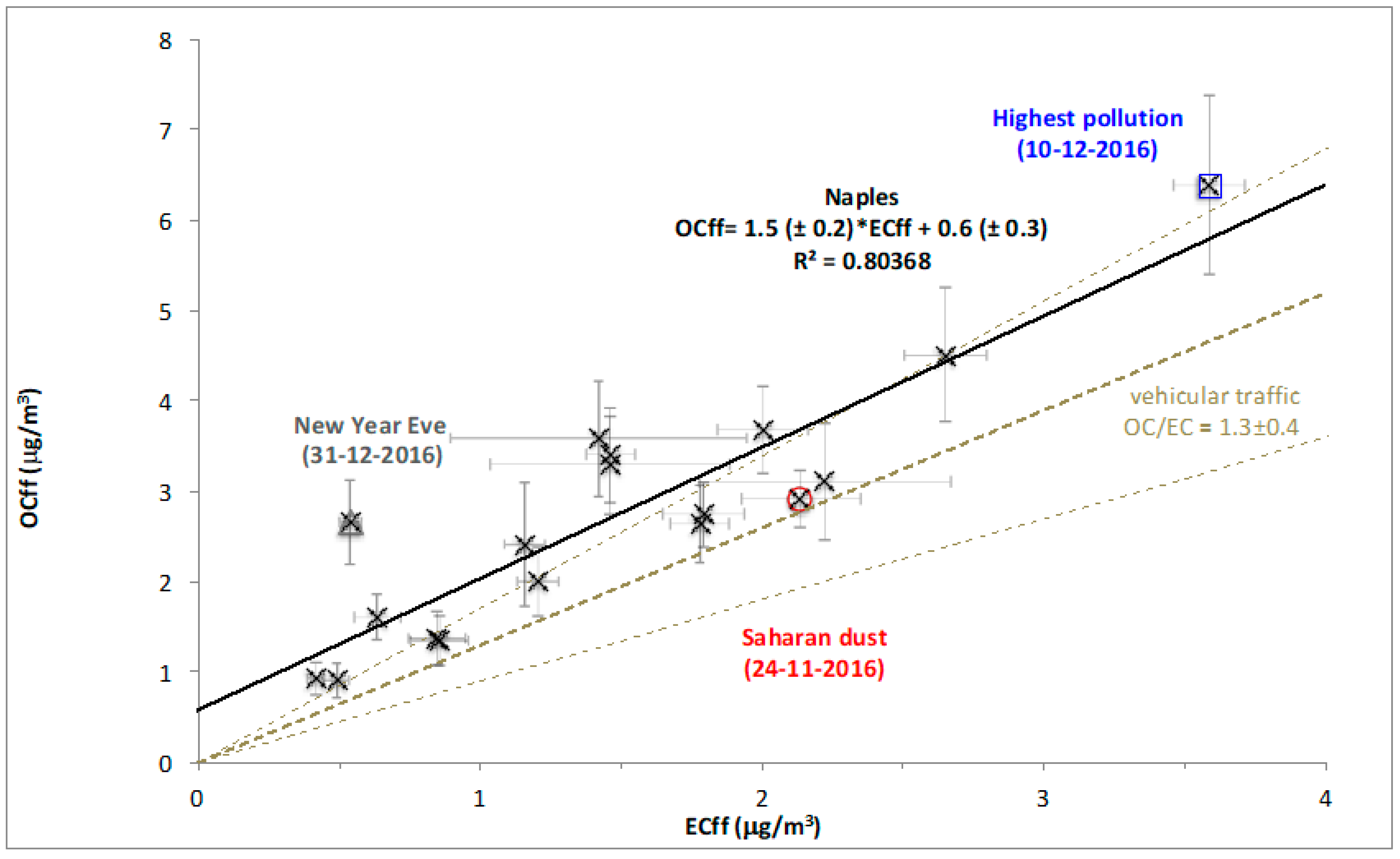
3.2. Fossil and Non-Fossil Contributions to EC and to OC
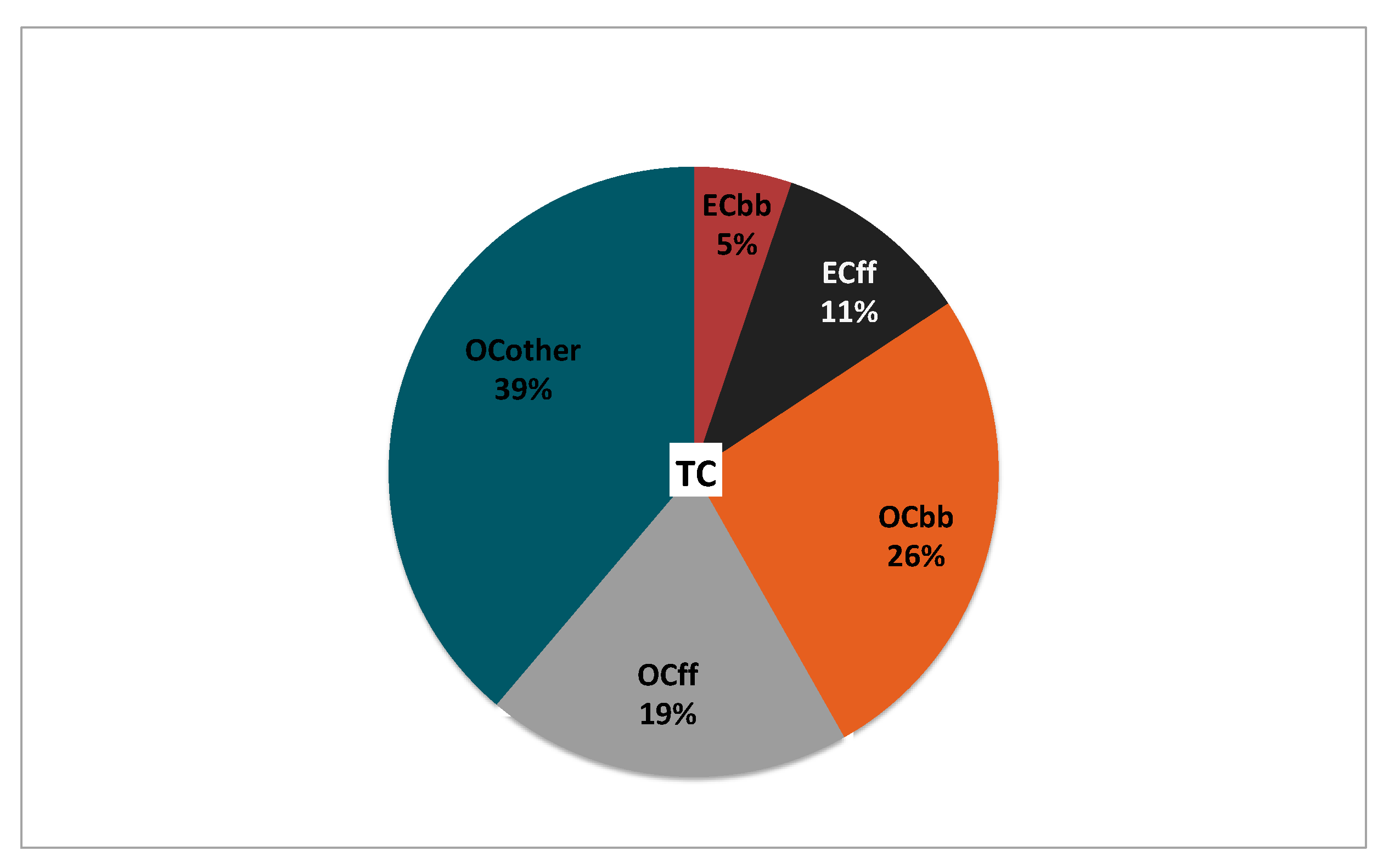
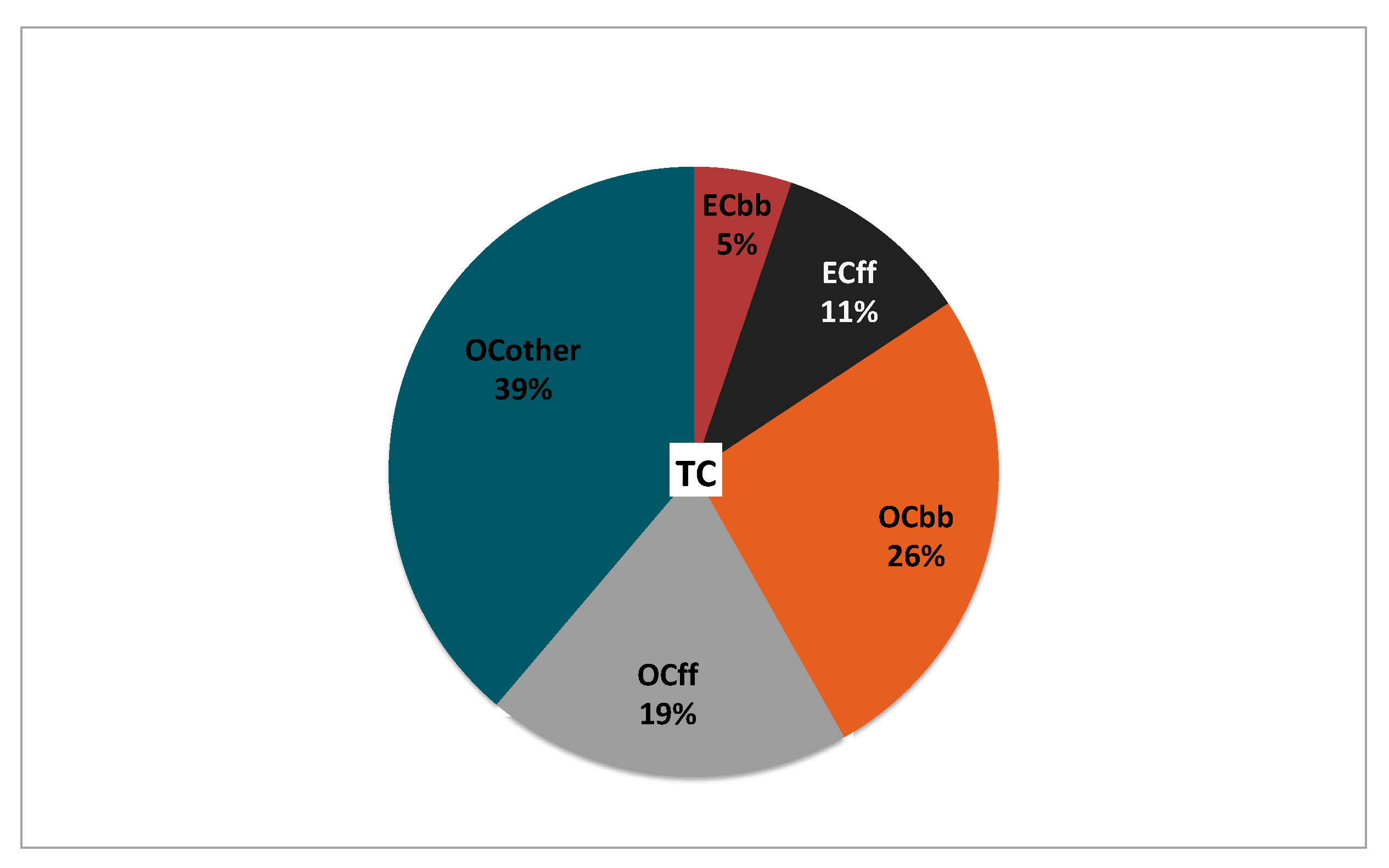
3.3. Source Apportionment of TC and Associated Uncertainties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuzzi, S.; Baltensperger, U.; Carslaw, K.; Decesari, S.; Denier van der Gon, H.; Facchini, M.C.; Fowler, D.; Koren, I.; Langford, B.; Lohmann, U.; et al. Particulate matter, air quality and climate: Lessons learned and future needs. Atmos. Chem. Phys. 2015, 15, 8217–8299. [Google Scholar] [CrossRef]
- Liang, C.S.; Duan, F.K.; He, K.-B.; Ma, Y.L. Review on recent progress in observations, source identifications and countermeasures of PM2.5. Environ. Int. 2016, 86, 150–170. [Google Scholar] [CrossRef] [PubMed]
- Putaud, J.P.; van Dingenen, R.; Alastuey, A.; Bauer, H.; Birmili, W.; Cyrys, J.; Flentje, H.; Fuzzi, S.; Gehrig, R.; Hansson, H.C.; et al. A European aerosol phenomenology—3: Physical and chemical characteristics of particulate matter from 60 rural, urban, and kerbside sites across Europe. Atmos. Environ. 2010, 44, 1308–1320. [Google Scholar] [CrossRef]
- Jacobson, M.C.; Hansson, H.-C.; Noone, K.J.; Charlson, R.J. Organic atmospheric aerosols: Review and state of the science. Rev. Geophys. 2000, 38, 267–294. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.; Alastuey, A.; Areskoug, H.; Ceburnis, D.; Čech, J.; Genberg, J.; Harrison, R.M.; Jaffrezo, J.L.; Kiss, G.; Laj, P.; et al. A European aerosol phenomenology-4: Harmonized concentrations of carbonaceous aerosol at 10 regional background sites across Europe. Atmos. Environ. 2016, 144, 133–145. [Google Scholar] [CrossRef]
- Petzold, A.; Ogren, J.A.; Fiebig, M.; Laj, P.; Li, S.M.; Baltensperger, U.; Holzer-Popp, T.; Kinne, S.; Pappalardo, G.; Sugimoto, N.; et al. Recommendations for reporting black carbon measurements. Atmos. Chem. Phys. 2013, 13, 8365–8379. [Google Scholar] [CrossRef]
- Turpin, B.J.; Huntzicker, J.J. Identification of secondary organic aerosol episodes and quantitation of primary and secondary organic aerosol concentrations during SCAQS. Atmos. Environ. 1995, 29, 3527–3544. [Google Scholar] [CrossRef]
- Pio, C.; Cerqueira, M.; Harrison, R.M.; Nunes, T.; Mirante, F.; Alves, C.; Oliveira, C.; Sanchez de la Campa, A.; Artíñano, B.; Matos, M. OC/EC ratio observations in Europe: Re-thinking the approach for apportionment between primary and secondary organic carbon. Atmos. Environ. 2011, 45, 6121–6132. [Google Scholar] [CrossRef]
- Wu, C.; Yu, J.Z. Determination of primary combustion source organic carbon-to-elemental carbon (OC/EC) ratio using ambient OC and EC measurements: Secondary OC-EC correlation minimization method. Atmos. Chem. Phys. 2016, 16, 5453–5465. [Google Scholar] [CrossRef]
- World Health Organization. Review of Evidence on Health Aspects of Air Pollution—REVIHAAP Project; World Health Organization: Copenhagen, Denmark, 2013. [Google Scholar]
- Landrigan, P.J.; Fuller, R.; Acosta, N.J.R.; Adeyi, O.; Arnold, R.; Basu, N.; Baldé, A.B.; Bertollini, R.; Bose-O’Reilly, S.; Boufford, J.I.; et al. The Lancet Commission on pollution and health. Lancet 2018, 391, 462–512. [Google Scholar] [CrossRef] [Green Version]
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.V.-M.; Kerminen, Y.; Kondo, H.; Liao, U.; Lohmann, P.; et al. Clouds and Aerosols. In Climate Change 2013—The Physical Science Basis; Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- World Health Organization. Ambient (Outdoor) Air Quality and Health. Available online: http://www.who.int/mediacenter/factsheets/fs313/en/ (accessed on 20 January 2018).
- Viana, M.; Kuhlbusch, T.A.J.; Querol, X.; Alastuey, A.; Harrison, R.M.; Hopke, P.K.; Winiwarter, W.; Vallius, M.; Szidat, S.; Prévôt, A.S.H.; et al. Source apportionment of particulate matter in Europe: A review of methods and results. J. Aerosol Sci. 2008, 39, 827–849. [Google Scholar] [CrossRef]
- Currie, L.A.; Klouda, G.A.; Continetti, R.E.; Kaplan, I.R.; Wong, W.W.; Dzubay, T.G.; Stevens, R.K. On the Origin of Carbonaceous Particles in American Cities: Results of Radiocarbon “Dating” and Chemical Characterization. Radiocarbon 1983, 25, 603–614. [Google Scholar] [CrossRef]
- Cachier, H.; Bremond, M.; Buat-Ménard, P. Determination of atmospheric soot carbon with a simple thermal method. Tellus B Chem. Phys. Meteorol. 1989, 41, 379–390. [Google Scholar] [CrossRef]
- Currie, L.A. Evolution and Multidisciplinary Frontiers of 14 C Aerosol Science. Radiocarbon 2000, 42, 115–126. [Google Scholar] [CrossRef]
- Szidat, S.; Jenk, T.M.; Gäggeler, H.; Synal, H.-A.; Fisseha, R.; Baltensperger, U.; Kalberer, M.; Samburova, V.; Reimann, S.; Kasper-Giebl, A.; et al. Radiocarbon (14C)-deduced biogenic and anthropogenic contributions to organic carbon (OC) of urban aerosols from Zürich, Switzerland. Atmos. Environ. 2004, 38, 4035–4044. [Google Scholar] [CrossRef]
- Ceburnis, D.; Garbaras, A.; Szidat, S.; Rinaldi, M.; Fahrni, S.; Perron, N.; Wacker, L.; Leinert, S.; Remeikis, V.; Facchini, M.C.; et al. Quantification of the carbonaceous matter origin in submicron marine aerosol by 13C and 14C isotope analysis. Atmos. Chem. Phys. 2011, 11, 8593–8606. [Google Scholar] [CrossRef] [Green Version]
- Heal, M.R. The application of carbon-14 analyses to the source apportionment of atmospheric carbonaceous particulate matter: A review. Anal. Bioanal. Chem. 2014, 406, 81–98. [Google Scholar] [CrossRef]
- Zotter, P.; Ciobanu, V.G.; Zhang, Y.L.; El-Haddad, I.; Macchia, M.; Daellenbach, K.R.; Salazar, G.A.; Huang, R.-J.; Wacker, L.; Hueglin, C.; et al. Radiocarbon analysis of elemental and organic carbon in Switzerland during winter-smog episodes from 2008 to 2012—Part 1: Source apportionment and spatial variability. Atmos. Chem. Phys. 2014, 14, 13551–13570. [Google Scholar] [CrossRef]
- Garbarienė, I.; Šapolaitė, J.; Garbaras, A.; Ežerinskis, Ž.; Pocevičius, M.; Krikščikas, L.; Plukis, A.; Remeikis, V. Origin Identification of Carbonaceous Aerosol Particles by Carbon Isotope Ratio Analysis. Aerosol Air Qual. Res. 2016, 16, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.; Tian, J.; Wang, X.; Wang, Q.; Han, Y.; Cao, J.; Long, X.; Chen, L.W.A.; Chow, J.C.; Watson, J.G.; et al. PM2.5 emissions and source profiles from open burning of crop residues. Atmos. Environ. 2017, 169, 229–237. [Google Scholar] [CrossRef]
- Ni, H.; Huang, R.J.; Cao, J.; Liu, W.; Zhang, T.; Wang, M.; Meijer, H.A.J.; Dusek, U. Source apportionment of carbonaceous aerosols in Xi’an, China: Insights from a full year of measurements of radiocarbon and the stable isotope 13C. Atmos. Chem. Phys. 2018, 18, 16363–16383. [Google Scholar] [CrossRef]
- Szidat, S.; Jenk, T.M.; Gäggeler, H.; Synal, H.-A.; Hajdas, I.; Bonani, G.; Saurer, M. THEODORE, a two-step heating system for the EC/OC determination of radiocarbon (14C) in the environment. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2004, 223, 829–836. [Google Scholar] [CrossRef]
- Calzolai, G.; Bernardoni, V.; Chiari, M.; Fedi, M.E.; Lucarelli, F.; Nava, S.; Riccobono, F.; Taccetti, F.; Valli, G.; Vecchi, R. The new sample preparation line for radiocarbon measurements on atmospheric aerosol at LABEC. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2011, 269, 203–208. [Google Scholar] [CrossRef]
- Dusek, U.; Monaco, M.; Prokopiou, M.; Gongriep, F.; Hitzenberger, R.; Meijer, H.A.J.; Röckmann, T. Evaluation of a two-step thermal method for separating organic and elemental carbon for radiocarbon analysis. Atmos. Meas. Tech. 2014, 7, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Briggs, N.L.; Long, C.M. Critical review of black carbon and elemental carbon source apportionment in Europe and the United States. Atmos. Environ. 2016, 144, 409–427. [Google Scholar] [CrossRef]
- Gelencsér, A.; May, B.; Simpson, D.; Sánchez-Ochoa, A.; Kasper-Giebl, A.; Puxbaum, H.; Caseiro, A.; Pio, C.A.C.; Legrand, M. Source apportionment of PM2.5 organic aerosol over Europe: Primary/secondary, natural/anthropogenic, and fossil/biogenic origin. J. Geophys. Res. 2007, 112, D23S04. [Google Scholar] [CrossRef]
- Szidat, S.; Jenk, T.M.; Synal, H.-A.; Kalberer, M.; Wacker, L.; Hajdas, I.; Kasper-Giebl, A.; Baltensperger, U. Contributions of fossil fuel, biomass-burning, and biogenic emissions to carbonaceous aerosols in Zurich as traced by 14C. J. Geophys. Res. Atmos. 2006, 111, 111. [Google Scholar] [CrossRef]
- Szidat, S.; Ruff, M.; Perron, N.; Wacker, L.; Synal, H.-A.; Hallquist, M.; Shannigrahi, A.S.; Yttri, K.E.; Dye, C.; Simpson, D. Fossil and non-fossil sources of organic carbon (OC) and elemental carbon (EC) in Göteborg, Sweden. Atmos. Chem. Phys. 2009, 9, 1521–1535. [Google Scholar] [CrossRef]
- Heal, M.R.; Naysmith, P.; Cook, G.T.; Xu, S.; Duran, T.R.; Harrison, R.M. Application of 14C analyses to source apportionment of carbonaceous PM2.5 in the UK. Atmos. Environ. 2011, 45, 2341–2348. [Google Scholar] [CrossRef] [Green Version]
- Minguillón, M.C.; Perron, N.; Querol, X.; Szidat, S.; Fahrni, S.M.; Alastuey, A.; Jimenez, J.L.; Mohr, C.; Ortega, A.M.; Day, D.A.; et al. Fossil versus contemporary sources of fine elemental and organic carbonaceous particulate matter during the DAURE campaign in Northeast Spain. Atmos. Chem. Phys. 2011, 11, 12067–12084. [Google Scholar] [CrossRef] [Green Version]
- Yttri, K.E.; Simpson, D.; Stenstr, K.; Puxbaum, H.; Svendby, T. Source apportionment of the carbonaceous aerosol in Norway—quantitative estimates based on 14 C, thermal-optical and organic tracer analysis. Atmos. Chem. Phys. 2011, 11, 9375–9394. [Google Scholar] [CrossRef]
- Keuken, M.P.; Moerman, M.; Voogt, M.; Blom, M.; Weijers, E.P.; Röckmann, T.; Dusek, U. Source contributions to PM2.5 and PM10 at an urban background and a street location. Atmos. Environ. 2013, 71, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.L.; Zotter, P.; Perron, N.; Prévôt, A.S.H.; Wacker, L.; Szidat, S. Fractions in Fine and Coarse Particles by Radiocarbon Measurement. Radiocarbon 2013, 55, 1510–1520. [Google Scholar] [CrossRef]
- Salma, I.; Németh, Z.; Weidinger, T.; Maenhaut, W.; Claeys, M.; Molnár, M.; Major, I.; Ajtai, T.; Utry, N.; Bozóki, Z. Source apportionment of carbonaceous chemical species to fossil fuel combustion, biomass burning and biogenic emissions by a coupled radiocarbon-levoglucosan marker method. Atmos. Chem. Phys. 2017, 17, 13767–13781. [Google Scholar] [CrossRef]
- Dusek, U.; Brink, H.M.T.; Meijer, H.A.J.; Kos, G.; Mrozek, D.; Röckmann, T.; Holzinger, R.; Weijers, E.P.; ten Brink, H.M.; Meijer, H.A.J.; et al. The contribution of fossil sources to the organic aerosol in the Netherlands. Atmos. Environ. 2013, 74, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Dusek, U.; Hitzenberger, R.; Kasper-Giebl, A.; Kistler, M.; Meijer, H.A.J.; Szidat, S.; Wacker, L.; Holzinger, R.; Röckmann, T. Sources and formation mechanisms of carbonaceous aerosol at a regional background site in the Netherlands: Insights from a year-long radiocarbon study. Atmos. Chem. Phys. 2017, 17, 3233–3251. [Google Scholar] [CrossRef]
- Glasius, M.; Hansen, A.M.K.; Claeys, M.; Henzing, J.S.; Jedynska, A.D.; Kasper-Giebl, A.; Kistler, M.; Kristensen, K.; Martinsson, J.; Maenhaut, W.; et al. Composition and sources of carbonaceous aerosols in Northern Europe during winter. Atmos. Environ. 2018, 173, 127–141. [Google Scholar] [CrossRef]
- Glasius, M.; La Cour, A.; Lohse, C. Fossil and nonfossil carbon in fine particulate matter: A study of five European cities. J. Geophys. Res. Atmos. 2011, 116, 1–11. [Google Scholar] [CrossRef]
- Sandrini, S.; Fuzzi, S.; Piazzalunga, A.; Prati, P.; Bonasoni, P.; Cavalli, F.; Bove, M.C.; Calvello, M.; Cappelletti, D.; Colombi, C.; et al. Spatial and seasonal variability of carbonaceous aerosol across Italy. Atmos. Environ. 2014, 99, 587–598. [Google Scholar] [CrossRef]
- Costabile, F.; Alas, H.; Aufderheide, M.; Avino, P.; Amato, F.; Argentini, S.; Barnaba, F.; Berico, M.; Bernardoni, V.; Biondi, R.; et al. First results of the “Carbonaceous Aerosol in Rome and Environs (CARE)” Experiment: Beyond current standards for PM10. Atmosphere 2017, 8, 249. [Google Scholar] [CrossRef]
- Pietrogrande, M.C.; Bacco, D.; Ferrari, S.; Ricciardelli, I.; Scotto, F.; Trentini, A.; Visentin, M. Characteristics and major sources of carbonaceous aerosols in PM2.5 in Emilia Romagna Region (Northern Italy) from four-year observations. Sci. Total Environ. 2016, 553, 172–183. [Google Scholar] [CrossRef]
- Khan, M.B.; Masiol, M.; Formenton, G.; Di Gilio, A.; de Gennaro, G.; Agostinelli, C.; Pavoni, B. Carbonaceous PM2.5 and secondary organic aerosol across the Veneto region (NE Italy). Sci. Total Environ. 2016, 542, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, I.; Bacco, D.; Rinaldi, M.; Bonafè, G.; Scotto, F.; Trentini, A.; Bertacci, G.; Ugolini, P.; Zigola, C.; Rovere, F.; et al. A three-year investigation of daily PM2.5 main chemical components in four sites: The routine measurement program of the Supersito Project (Po Valley, Italy). Atmos. Environ. 2017, 152, 418–430. [Google Scholar] [CrossRef]
- Costa, V.; Bacco, D.; Castellazzi, S.; Ricciardelli, I.; Vecchietti, R.; Zigola, C.; Pietrogrande, M.C. Characteristics of carbonaceous aerosols in Emilia-Romagna (Northern Italy) based on two fall/winter field campaigns. Atmos. Res. 2016, 167, 100–107. [Google Scholar] [CrossRef]
- Contini, D.; Vecchi, R.; Viana, M. Carbonaceous aerosols in the atmosphere. Atmosphere 2018, 9, 181. [Google Scholar] [CrossRef]
- Siciliano, T.; Siciliano, M.; Malitesta, C.; Proto, A.; Cucciniello, R.; Giove, A.; Iacobellis, S.; Genga, A. Carbonaceous PM10 and PM2.5 and secondary organic aerosol in a coastal rural site near Brindisi (Southern Italy). Environ. Sci. Pollut. Res. 2018, 25, 23929–23945. [Google Scholar] [CrossRef] [PubMed]
- Cesari, D.; Merico, E.; Dinoi, A.; Marinoni, A.; Bonasoni, P.; Contini, D. Seasonal variability of carbonaceous aerosols in an urban background area in Southern Italy. Atmos. Res. 2018, 200, 97–108. [Google Scholar] [CrossRef]
- Cesari, D.; De Benedetto, G.E.; Bonasoni, P.; Busetto, M.; Dinoi, A.; Merico, E.; Chirizzi, D.; Cristofanelli, P.; Donateo, A.; Grasso, F.M.; et al. Seasonal variability of PM2.5 and PM10 composition and sources in an urban background site in Southern Italy. Sci. Total Environ. 2018, 612, 202–213. [Google Scholar] [CrossRef]
- Dinoi, A.; Cesari, D.; Marinoni, A.; Bonasoni, P.; Riccio, A.; Chianese, E.; Tirimberio, G.; Naccarato, A.; Sprovieri, F.; Andreoli, V.; et al. Inter-Comparison of Carbon Content in PM2.5 and PM10 Collected at Five Measurement Sites in Southern Italy. Atmosphere 2017, 8, 243. [Google Scholar] [CrossRef]
- Bernardoni, V.; Calzolai, G.; Chiari, M.; Fedi, M.; Lucarelli, F.; Nava, S.; Piazzalunga, A.; Riccobono, F.; Taccetti, F.; Valli, G.; et al. Radiocarbon analysis on organic and elemental carbon in aerosol samples and source apportionment at an urban site in Northern Italy. J. Aerosol Sci. 2013, 56, 88–99. [Google Scholar] [CrossRef]
- Gilardoni, S.; Vignati, E.; Cavalli, F.; Putaud, J.P.; Larsen, B.R.; Karl, M.; StenstrÃm, K.; Genberg, J.; Henne, S.; Dentener, F. Better constraints on sources of carbonaceous aerosols using a combined 14C-macro tracer analysis in a European rural background site. Atmos. Chem. Phys. 2011, 11, 5685–5700. [Google Scholar] [CrossRef]
- Eurostat Population Density by Metropolitan Regions. Available online: http://appsso.eurostat.ec.europa.eu/nui/submitViewTableAction.do (accessed on 9 May 2018).
- Cattani, G.; Di Menno di Bucchianico, A.; Gaeta, A.; Leone, G. QUALITÀ DELL’ ARIA. In XIII Rapporto Qualità Dell’ambiente Urbano; ISPRA Istituto Superiore per la Protezione e la Ricerca Ambientale: Rome, Italy, 2017; ISBN 9788844808587. [Google Scholar]
- European Parliament and Council. Directive (EU) 2016/2284 of the european parliament and of the council of 14 December 2016 on on the reduction of national emissions of certain atmospheric pollutants, amending Directive 2003/35/EC and repealing Directive 2001/81/EC. Off. J. Eur. Union 2016, 344, 1–31. [Google Scholar]
- Di Vaio, P.; Magli, E.; Barbato, F.; Caliendo, G.; Cocozziello, B.; Corvino, A.; De Marco, A.; Fiorino, F.; Frecentese, F.; Onorati, G.; et al. Chemical composition of PM10at urban sites in Naples (Italy). Atmosphere 2016, 7, 163. [Google Scholar] [CrossRef]
- Riccio, A.; Chianese, E.; Agrillo, G.; Esposito, C.; Ferrara, L.; Tirimberio, G. Source apportion of atmospheric particulate matter: A joint Eulerian/Lagrangian approach. Environ. Sci. Pollut. Res. 2014, 21, 13160–13168. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Chianese, E.; Monaco, D.; Costagliola, M.A.; Perretta, G.; Prati, M.V.; Agrillo, G.; Esposito, A.; Gasbarra, D.; Shindler, L.; et al. Real-world automotive particulate matter and PAH emission factors and profile concentrations: Results from an urban tunnel experiment in Naples, Italy. Atmos. Environ. 2016, 141, 379–387. [Google Scholar] [CrossRef]
- Riccio, A.; Chianese, E.; Tirimberio, G.; Prati, M.V. Emission factors of inorganic ions from road traffic: A case study from the city of Naples (Italy). Transp. Res. Part D Transp. Environ. 2017, 54, 239–249. [Google Scholar] [CrossRef]
- Draxler, R.R.; Rolph, G.D. HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory). Available online: http://www.arl.noaa.gov/HYSPLIT.php (accessed on 9 June 2019).
- Grange, S.K. Technical Note: Averaging Wind Speeds and Directions. Available online: http://rgdoi.net/10.13140/RG.2.1.3349.2006 (accessed on 9 June 2019).
- Zenker, K.; Vonwiller, M.; Szidat, S.; Calzolai, G.; Giannoni, M.; Bernardoni, V.; Jedynska, A.D.; Henzing, B.; Meijer, H.A.J.; Dusek, U. Evaluation and Inter-Comparison of Oxygen-Based OC-EC Separation Methods for Radiocarbon Analysis of Ambient Aerosol Particle Samples. Atmosphere 2017, 8, 226. [Google Scholar] [CrossRef]
- Szidat, S.; Bench, G.; Bernardoni, V.; Calzolai, G.; Czimczik, C.I.; Derendorp, L. Intercomparison of 14C Analysis of Carbonaceous Aerosols: Exercise 2009. Radiocarbon 2013, 55, 1496–1509. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.; Viana, M.; Yttri, K.E.; Genberg, J.; Putaud, J.P. Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: The EUSAAR protocol. Atmos. Meas. Tech. 2010, 3, 79–89. [Google Scholar] [CrossRef]
- Synal, H.A.; Stocker, M.; Suter, M. MICADAS: A new compact radiocarbon AMS system. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2007, 259, 7–13. [Google Scholar] [CrossRef]
- Reimer, P.J.; Brown, T.A.; Reimer, R.W. Discussion: Reporting and Calibration of Post-Bomb 14C Data. Radiocarbon 2004, 46, 1299–1304. [Google Scholar]
- Stuiver, M.; Polach, H.A. Discussion: Reporting of 14 C data. Radiocarbon 1977, 19, 355–363. [Google Scholar] [CrossRef]
- Mann, W.B.W.B. An international reference material for radiocarbon dating. Radiocarbon 1983, 25, 519–527. [Google Scholar] [CrossRef]
- Mook, W.G.; Plicht, J.V.D. Reporting 14C Activities and Concentrations. Radiocarbon 1999, 41, 227–239. [Google Scholar] [CrossRef]
- Hammer, S.; Levin, I. Monthly mean atmospheric D14CO2 at Jungfraujoch and Schauinsland from 1986 to 2016. heiDATA 2017. [Google Scholar] [CrossRef]
- NOAA MADIS (Metorological Assimilation Data Ingest System). Available online: https://madis.ncep.noaa.gov (accessed on 11 February 2018).
- Centrofunzionale Multirischi della Protezione Civile Regione Campania Archivio Pluviometrici. Available online: http://centrofunzionale.regione.campania.it/#/pages/sensori/archivio-pluviometrici (accessed on 9 June 2019).
- Petrarca, S.; Spinelli, F.; Cogliani, E.; Mancini, M. Profilo Climatico dell’Italia; ENEA: Rome, Italy, 1999; Volume 6, ISBN 88-8286-081-7. [Google Scholar]
- Ilmeteo Archivio-Meteo Napoli. Available online: https://www.ilmeteo.it/portale/archivio-meteo/Napoli (accessed on 1 June 2018).
- Giugliano, M.; Lonati, G.; Butelli, P.; Romele, L.; Tardivo, R.; Grosso, M. Fine particulate (PM2.5-PM1) at urban sites with different traffic exposure. Atmos. Environ. 2005, 39, 2421–2431. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Start Date | Origin | C_code | # days | T (°C) | Wind Dir | Wind Speed (m/s) | Precip (mm) | F14C (EC) | F14C (OC) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 24/11/16 | Saharan | Red | 2 ® | 13 | N | 1.7 | 30 | 0.229 ± 0.004 | 0.629 ± 0.006 |
| 2 | 26/11/16 | West. Med./local | Green | 2 ® | 11 | NW | 1.4 | 0.283 ± 0.004 | 0.782 ± 0.007 | |
| 3 | 29/11/16 | Eu. cont. | LIGHT BLUE | 2 ® | 8 | NE | 6.5 | 0.354 ± 0.005 | 0.859 ± 0.008 | |
| 4 | 01/12/16 | Eu. cont. | LIGHT BLUE | 2 ® | 9 | NW | 0.9 | 0.382 ± 0.005 | 0.79 ± 0.006 | |
| 5 | 03/12/16 | West. Med./local | Green | 1 | 13 | NW | 0.6 | 0.453 ± 0.005 | 0.887 ± 0.007 | |
| 6 | 04/12/16 | West. Med./local | Green | 2 | 12 | N | 1.8 | 0.431 ± 0.005 | 0.841 ± 0.009 | |
| 7 | 10/12/16 | West. Med./local | Green | 1 | 8 | N | 0.9 | 0.363 ± 0.005 | 0.828 ± 0.007 | |
| 8 | 11/12/16 | West. Med./local | Green | 1 | 11 | NW | 0.7 | 0.4 | 0.328 ± 0.005 | 0.84 ± 0.008 |
| 9 | 12/12/16 | West. Med./local | Green | 1 | 12 | NE | 3.8 | 0.4 | 0.347 ± 0.005 | 0.838 ± 0.006 |
| 10 | 14/12/16 | Eu. cont. | LIGHT BLUE | 2 | 9 | N | 2.2 | 0.367 ± 0.005 | 0.828 ± 0.007 | |
| 11 | 17/12/16 | Eu. cont. | LIGHT BLUE | 2 ® | 7 | N | 2.1 | 0.501 ± 0.006 | 0.867 ± 0.011 | |
| 12 | 19/12/16 | Est. Med. | DARK BLUE | 1 | 8 | N | 2.3 | 2.6 | 0.512 ± 0.006 | 0.93 ± 0.007 |
| 13 | 20/12/16 | Est. Med. | DARK BLUE | 2 ® | 13 | NE | 3.1 | 3.2 | 0.48 ± 0.006 | 0.875 ± 0.007 |
| 14 | 22/12/16 | Est. Med. | DARK BLUE | 2 | 10 | NW | 2.1 | 0.414 ± 0.005 | 0.863 ± 0.007 | |
| 15 | 24/12/16 | Eu. cont. | LIGHT BLUE | 2 | 10 | NW | 1.2 | 0.317 ± 0.005 | 0.858 ± 0.008 | |
| 16 | 29/12/16 | Eu. cont. | LIGHT BLUE | 2 | 5 | NE | 5.2 | 0.485 ± 0.006 | 0.824 ± 0.009 | |
| 17 | 31/12/16 | Eu. cont. | LIGHT BLUE | 1 | 5 | NW | 2.5 | 0.491 ± 0.006 | 0.854 ± 0.007 | |
| 18 | 04/01/17 | Eu. cont. | LIGHT BLUE | 2 ® | 6 | N | 2.5 | 19.8 | 0.336 ± 0.004 | 0.787 ± 0.007 |
| Site | Year | Type | Thermal Protocol | PM2.5 (µg/m3) | OC (µg/m3) | EC (µg/m3) | TC (µg/m3) | n | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Napoli | 2016/2017 | Urban bkg. | EUSAAR2 | 29 ± 3 | 12 ± 1 | 2.2 ± 0.3 | 14 ± 2 | 18 | This study |
| South. | |||||||||
| Coastal | |||||||||
| Napoli | 2015/2016 | Urban bkg. | EUSAAR2 | 38 ± 3 | 12 ± 1 | 1.8 ± 0.1 | 14 ± 1 | 37 | [52] |
| South. | |||||||||
| Coastal | |||||||||
| Lecce | 2015/2016 | Urban bkg. | EUSAAR2 | 26 ± 2 | 9 ± 1 | 1.4 ± 0.1 | 10 ± 1 | 38 | [52] |
| South. | |||||||||
| Costal | |||||||||
| Lecce | 2013/2014 | Urban bkg. | NIOSH 5040 | 23 ± 2 | 7 ± 1 | 0.8 ± 0.1 | 8 ± 1 | 54 | [51] |
| South. | |||||||||
| Coastal | |||||||||
| Bologna Rimini Parma | 2012–2015 | Urban bkg. | EUSAAR2 | 33 ± 1 | 7.3 ± 0.3 | 1.9 ± 0.1 | 9.2 ± 0.4 | 207 (I) | [46] |
| North | |||||||||
| Inland | |||||||||
| Veneto | 2012/2013 (dec. feb) | Urban bkg. | NIOSH 5040 | 39(II) | 10 ± 1 | 1.8 ± 0.2 | 12 ± 1 | 120(III) | [45] |
| North | |||||||||
| Inland | |||||||||
| Italy (different sites) | 2005–2012 | Urban bkg. | NIOSH 5040/NIOSH-like | 33 ± 3 | 8 ± 1 | 2 ± 0.2 | 10 ± 1 | 16 sites | [42] |
| Italy (different sites) | 2005–2012 | Urban Kerbside | NIOSH 5040/NIOSH-like | 34 ± 12 | 9 ± 3 | 3.7 ± 0.9 | 13 ± 4 | 3 sites | [42] |
| Capo Granitola | 2015/2016 | Remote | EUSAAR2 | 10 ± 1 | 1.7 ± 0.3 | 1.7 ± 0.1 | 2 ± 0.3 | 14 | [52] |
| South | |||||||||
| Coastal | |||||||||
| Lamezia terme | 2015/2016 | Remote | EUSAAR2 | 7 ± 1 | 4 ± 0.3 | 0.6 ± 0.05 | 4.7 ± 0.3 | 38 | [52] |
| South | |||||||||
| Coastal | |||||||||
| Monte Curcio | 2015/2016 | Remote | EUSAAR2 | 3 ± 0.2 | 0.9 ± 0.1 | 0.05 ± 0.01 | 0.9 ± 0.3 | 31 | [52] |
| South | |||||||||
| Inland | |||||||||
| San Pietro Capofiume | 2012–2015 | Remote | EUSAAR2 | 26 ± 1 | 8 ± 1 | 1.3 ± 0.3 | 10 ± 2 | 47 | [46] |
| North | |||||||||
| Inland | |||||||||
| Italy (different sites) | 2005–2012 | Remote | NIOSH 5040/NIOSH-like | 22 ± 4 | 6 ± 2 | 0.9 ± 0.3 | 7 ± 1.6 | 6 sites | [42] |
| Inland |
| Site | Year | Season | PM | Fossil Fraction of EC (%) | Fossil Fraction of OC (%) | Ref. |
|---|---|---|---|---|---|---|
| Naples (Italy) | 2016/2017 | Late fall-winter | PM2.5 | 66 | 23 | This study |
| Budapest (Hungary) | 2014 | Winter | PM2.5 * | 63 | 30 | [37] |
| Ispra (Italy) remote | 2007 | Winter | PM2.5 | 51 | 18 | [54] |
| Barcelona (Spain) | 2009 | Winter | PM1 | 87 | 40 | [33] |
| Birmingham (UK) | 2007–2008 | Jun–sept and Jan–May | PM2.5 | 90 | 41 | [32] |
| Aveiro (Portugal) | 2002–2004 | Winter | PM2.5 | 17 | 20 | [29] |
| Zurich (Switzerland) | 2003 | Winter | PM10 | 75 | 32 | [30] |
| Goteborg (Sweden) | 2005 | Winter | PM10 | 87–91 | 35–45 | [31] |
| Milan (Italy) | 2009–2012 | Winter | PM10 | 84 | 37 | [53] |
| Oslo (Norway) | 2007 | Winter | PM10 * | 65 | 38 | [34] |
| Oslo (Norway) | 2007 | Winter | PM1 * | 48 | 39 | [34] |
| Zurich (Swtzerland) | 2008 | Winter | PM1–PM10 | 64 | 34 | [36] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirignano, C.; Riccio, A.; Chianese, E.; Ni, H.; Zenker, K.; D’Onofrio, A.; Meijer, H.A.J.; Dusek, U. High Contribution of Biomass Combustion to PM2.5 in the City Centre of Naples (Italy). Atmosphere 2019, 10, 451. https://doi.org/10.3390/atmos10080451
Sirignano C, Riccio A, Chianese E, Ni H, Zenker K, D’Onofrio A, Meijer HAJ, Dusek U. High Contribution of Biomass Combustion to PM2.5 in the City Centre of Naples (Italy). Atmosphere. 2019; 10(8):451. https://doi.org/10.3390/atmos10080451
Chicago/Turabian StyleSirignano, Carmina, Angelo Riccio, Elena Chianese, Haiyan Ni, Katrin Zenker, Antonio D’Onofrio, Harro A.J. Meijer, and Ulrike Dusek. 2019. "High Contribution of Biomass Combustion to PM2.5 in the City Centre of Naples (Italy)" Atmosphere 10, no. 8: 451. https://doi.org/10.3390/atmos10080451