Composition of Clean Marine Air and Biogenic Influences on VOCs during the MUMBA Campaign

 ,
, .jpg) , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Methods
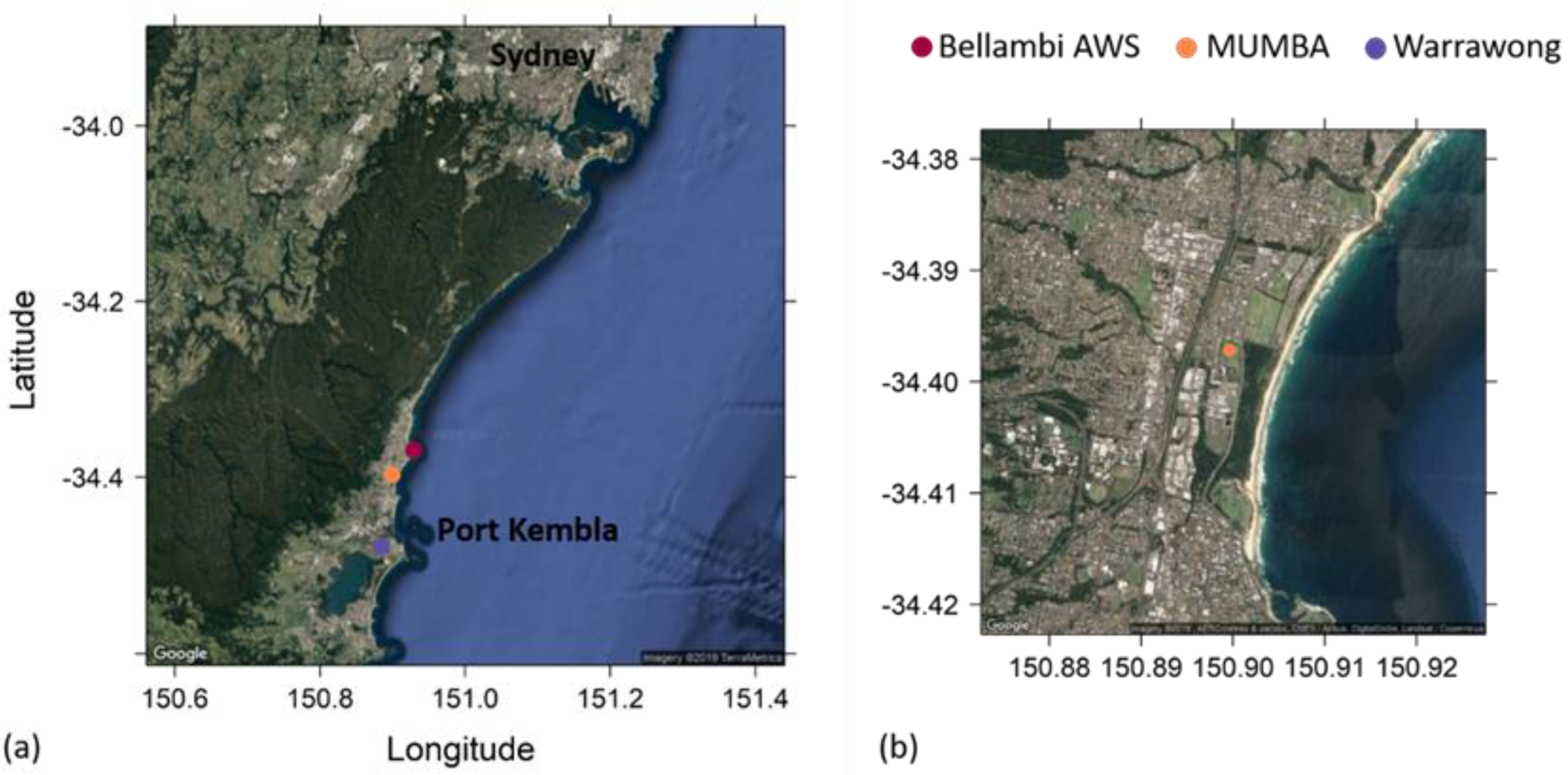
2.1. The MUMBA Campaign
2.2. The Proton Transfer Reaction-Mass Spectrometer (PTR-MS)
2.2.1. PTR-MS Operation
2.2.2. PTR-MS Data Processing and Detection Limits
2.3. Fourier Transform Infrared Spectrometer (FTIR)
- 2097–2242 cm−1, optimised for N2O and CO, also fitting CO2;
- 2150–2310 cm−1, fitting CO, N2O, H2O and CO2 isotopologues;
- 3001–3150 cm−1, fitting CH4 and H2O;
- 3520–3775 cm−1, fitting CO2 and H2O.
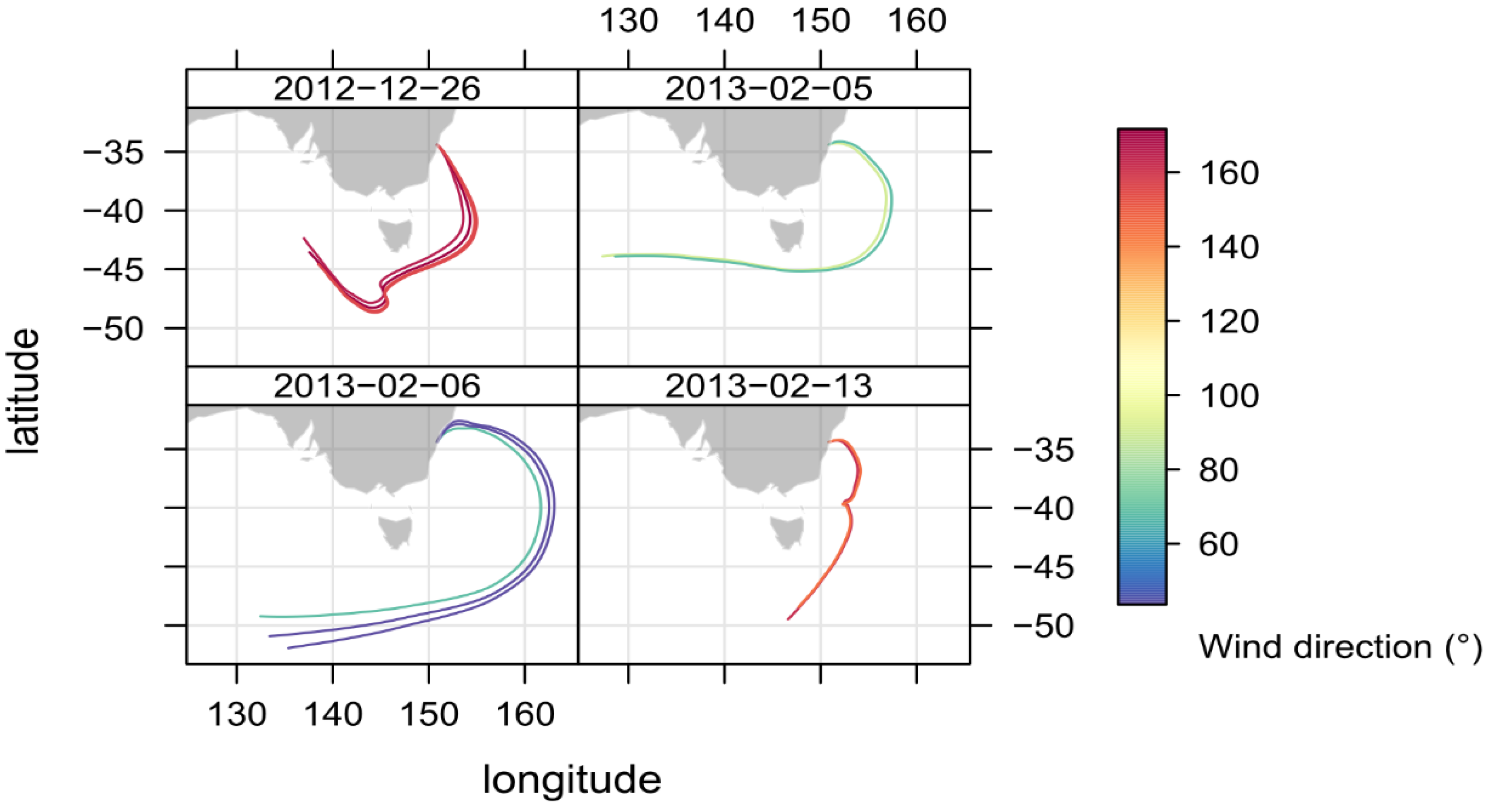
2.4. Identifying Episodes of Clean Marine Air at the MUMBA Site
3. Results
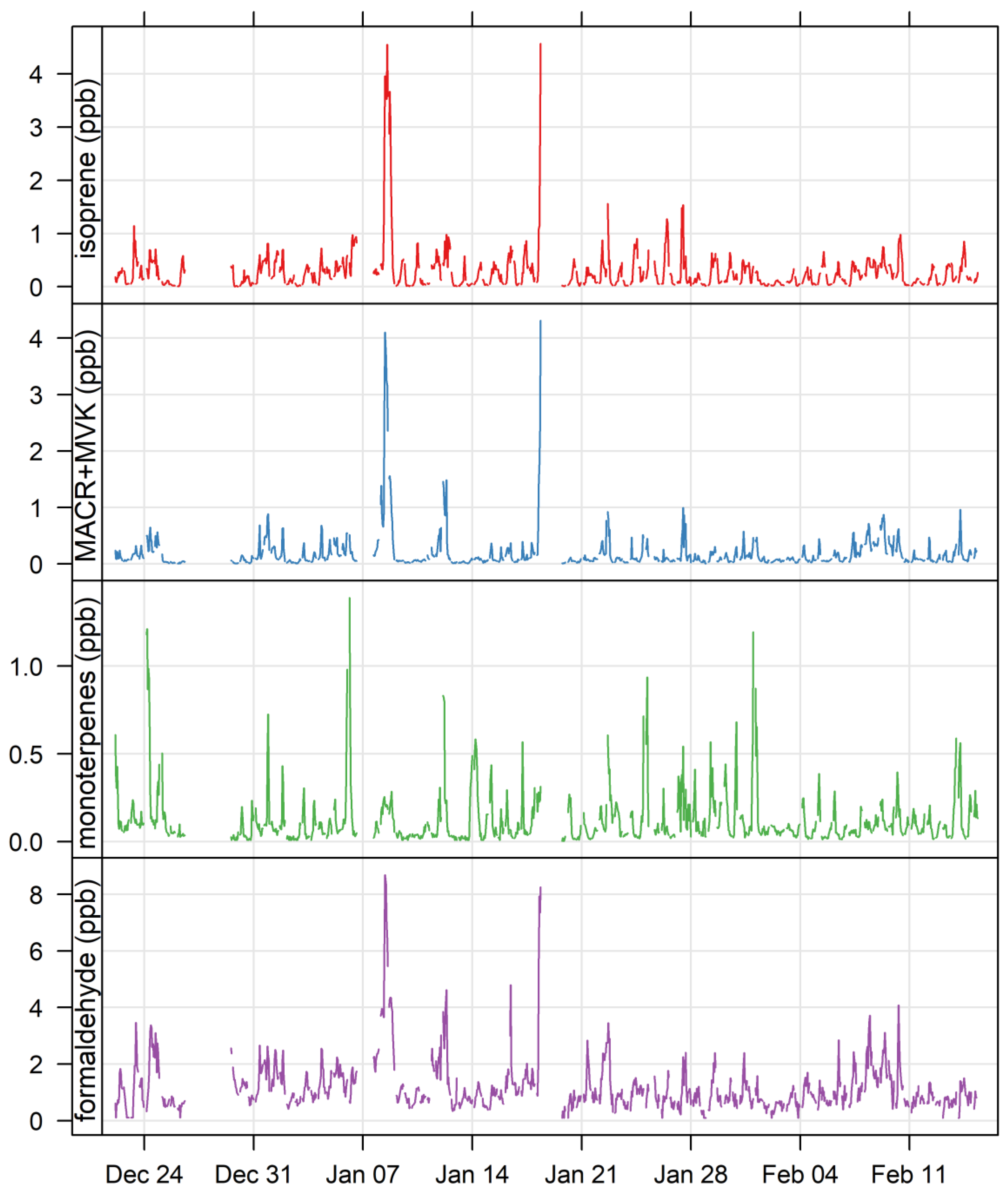
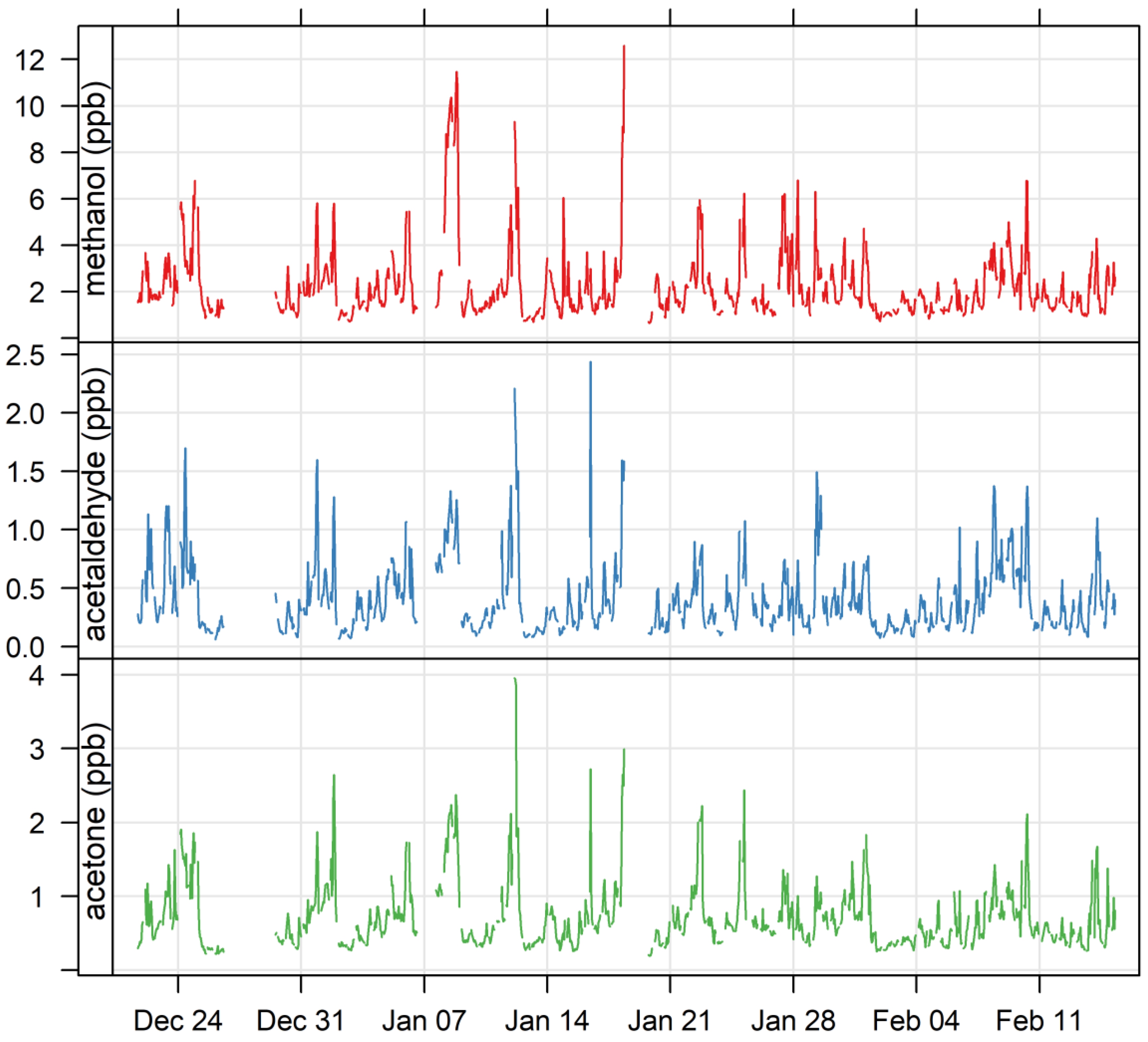
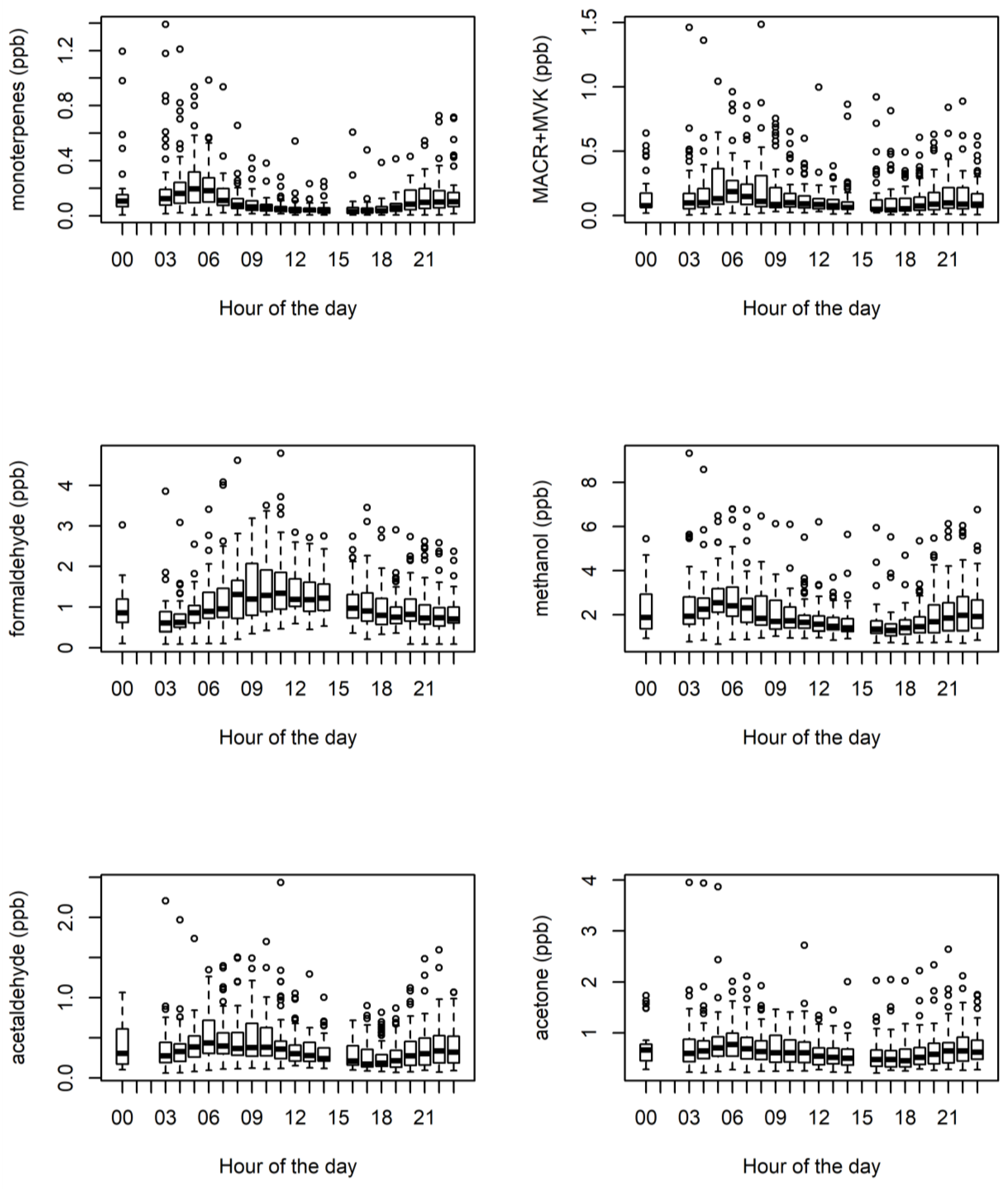
3.1. Ambient VOCs Measured during the MUMBA Campaign
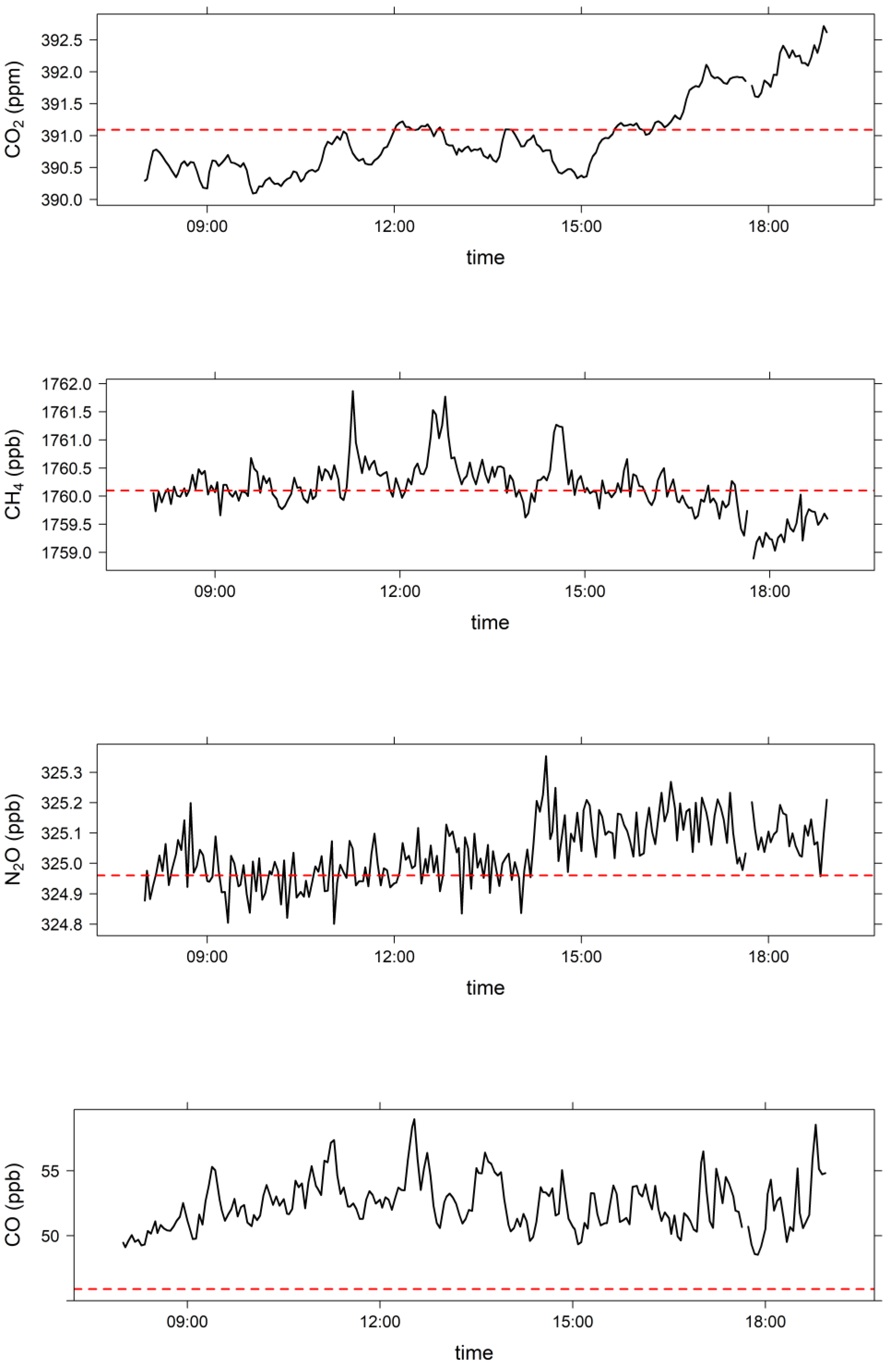
3.2. Comparison of Clean Marine Air at MUMBA with Measurements at Cape Grim, Tasmania
3.3. Comparison of VOCs in Clean Marine Air at MUMBA with other Measurements in the Literature
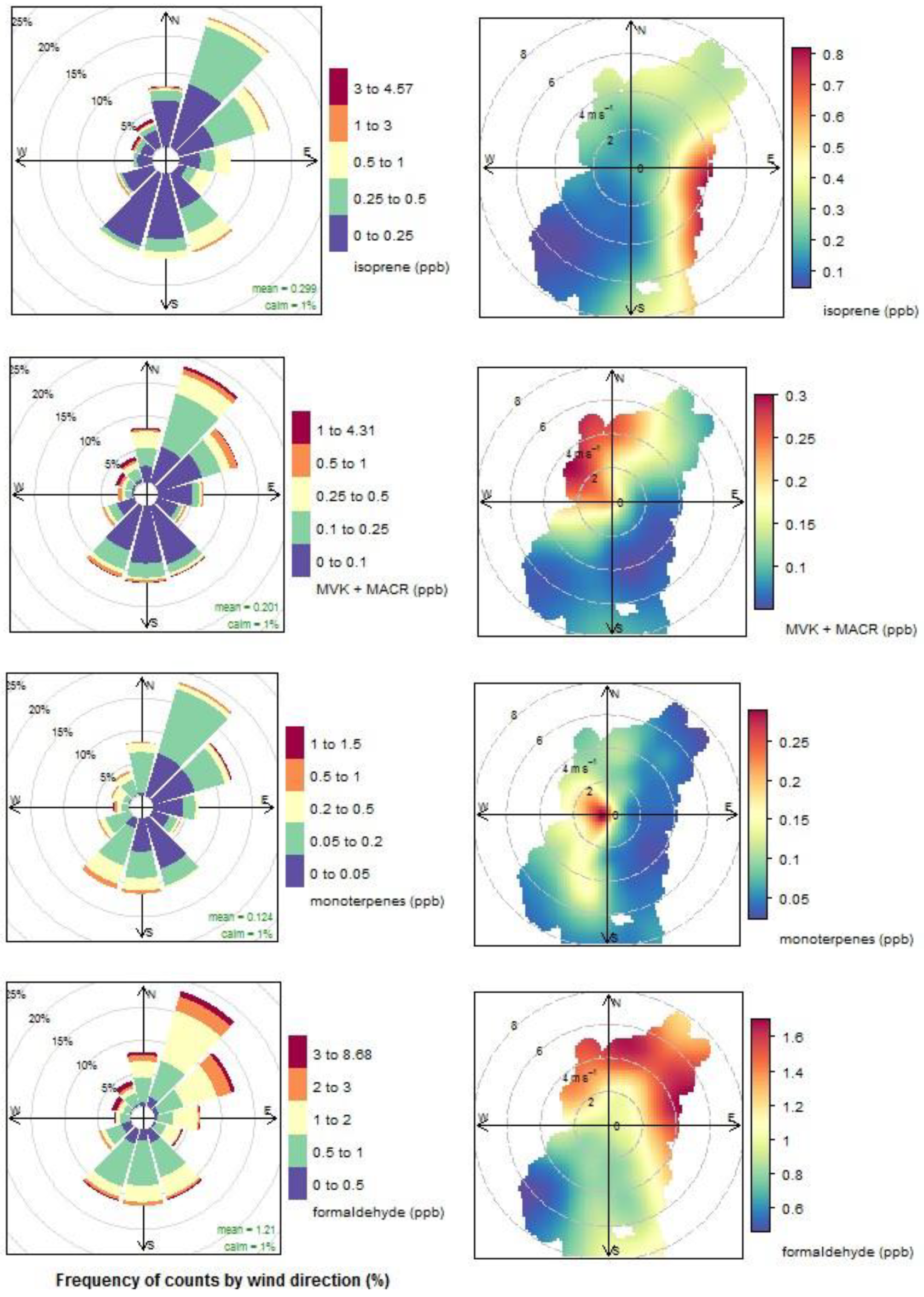
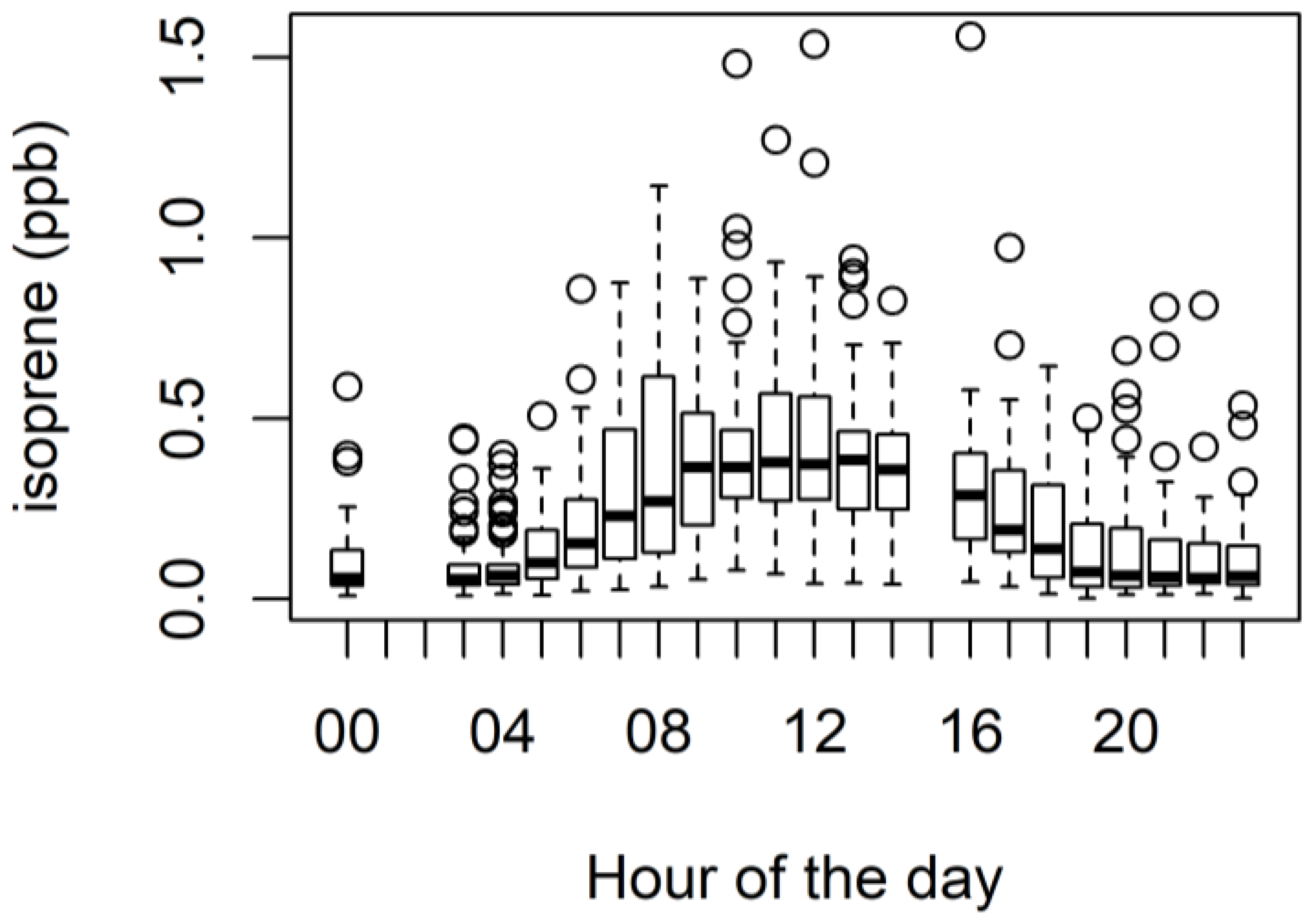
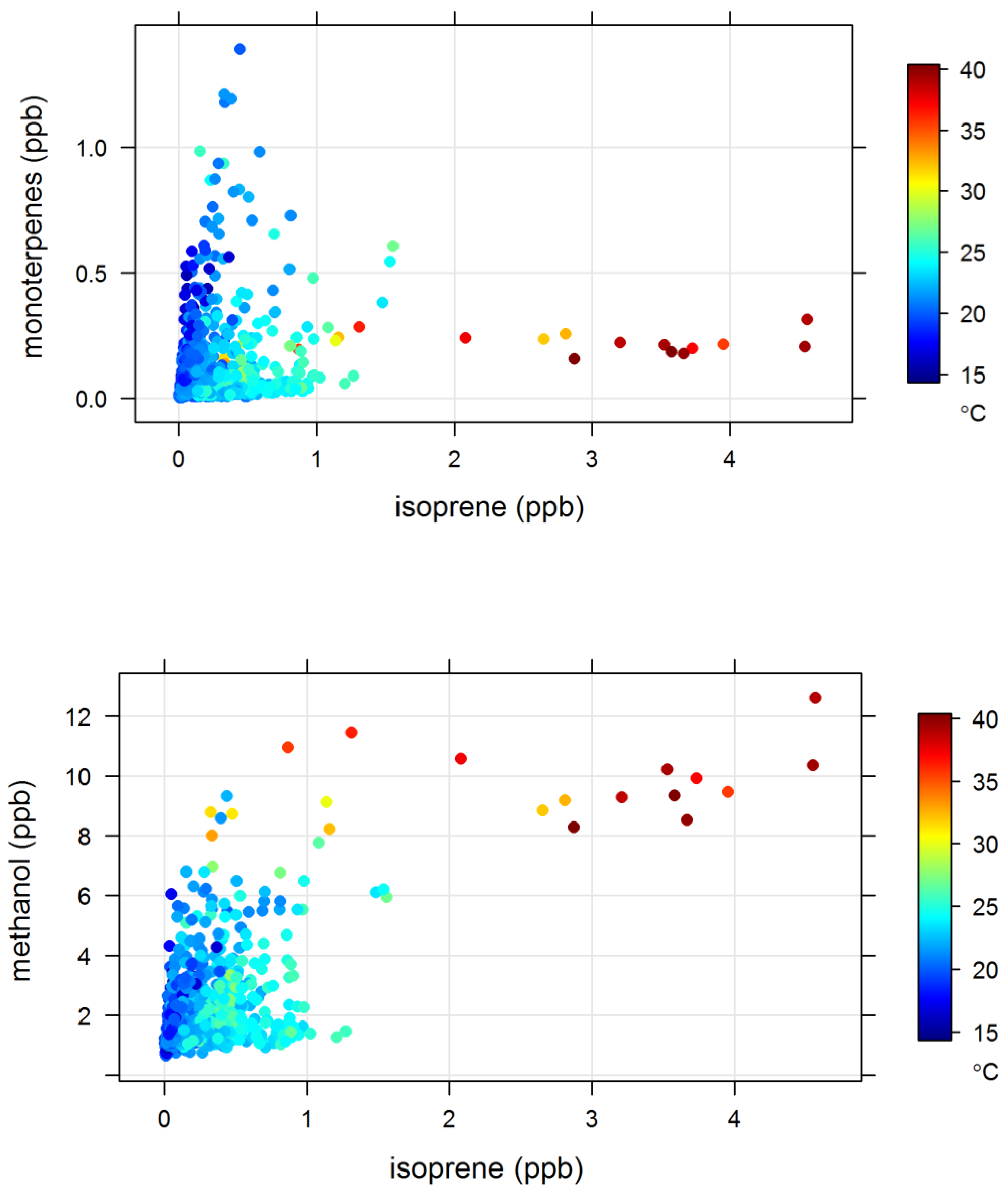
3.4. Biogenic VOCs of Terrestrial Origin Measured during MUMBA
4. Discussion
4.1. VOCs in Clean Marine Air
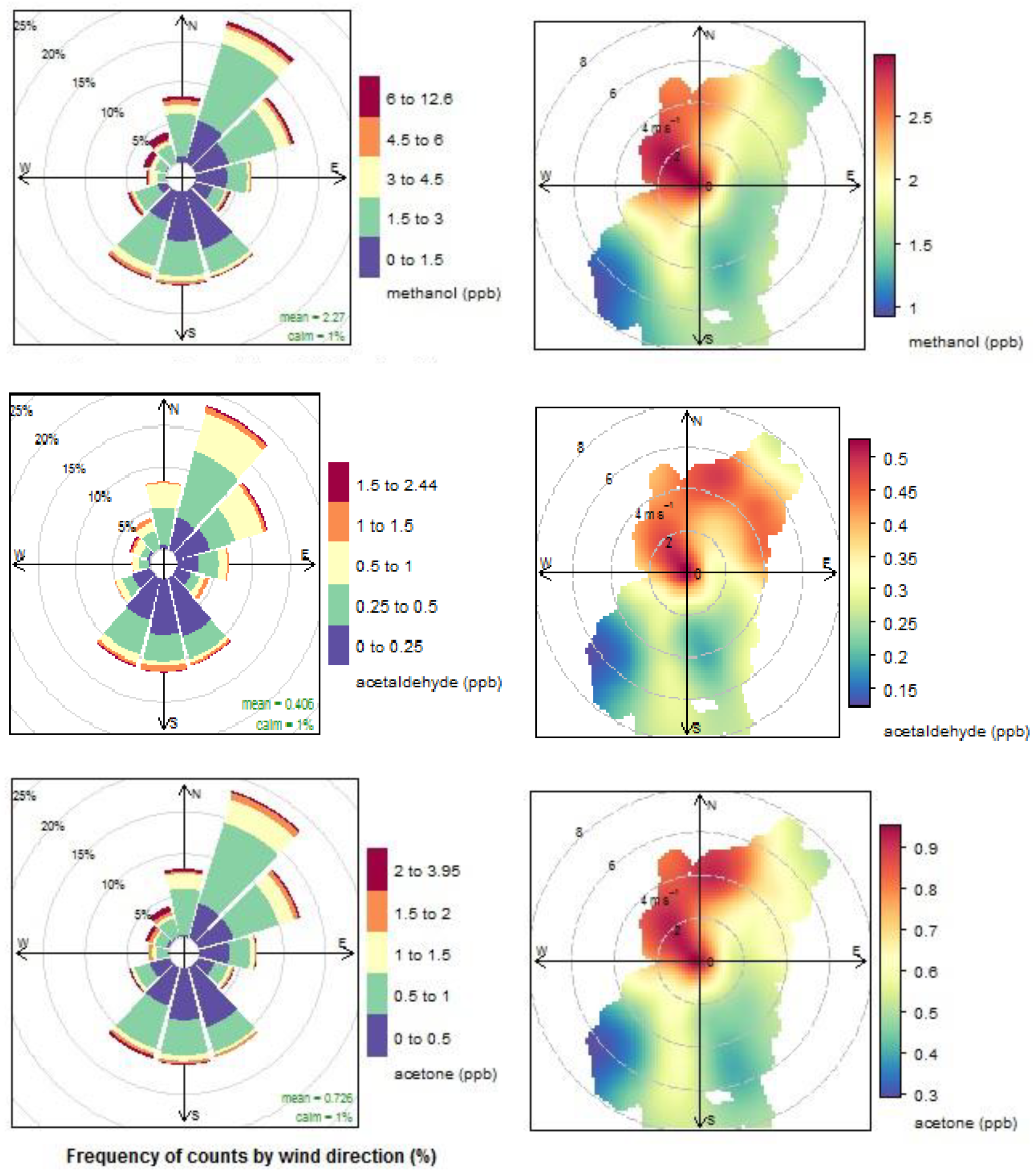
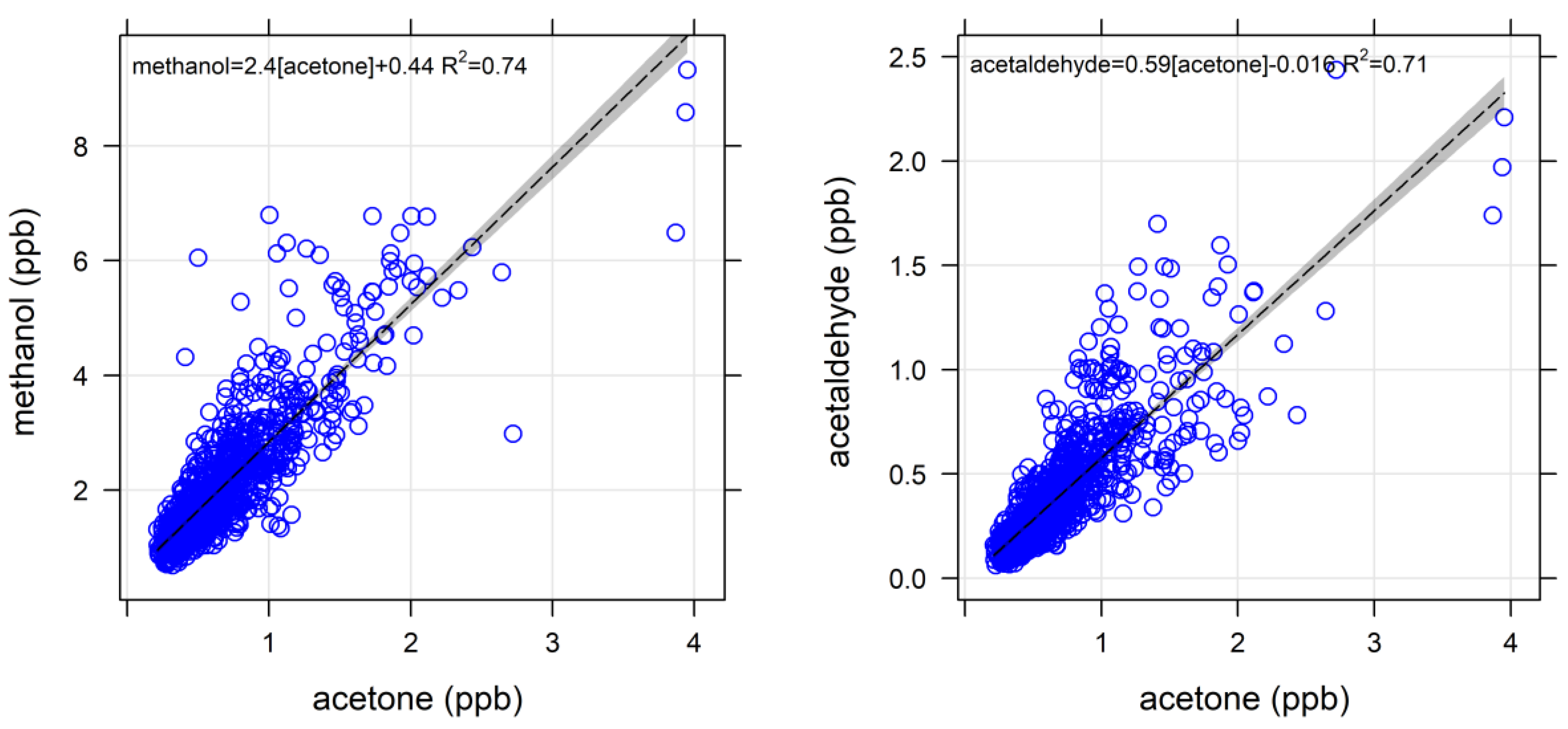
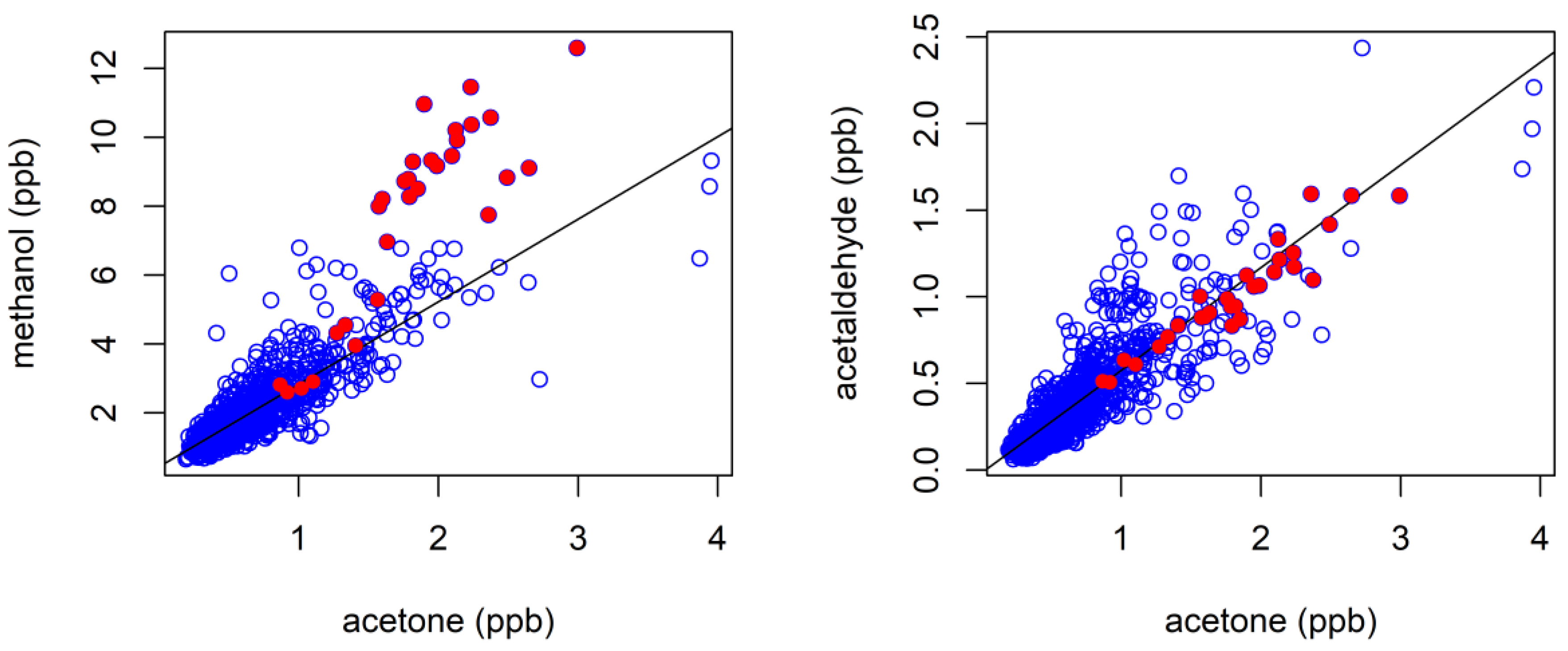
4.1.1. Oxygenated VOCs
4.1.2. Dimethyl Sulphide
4.1.3. Acetonitrile
4.1.4. Aromatic compounds
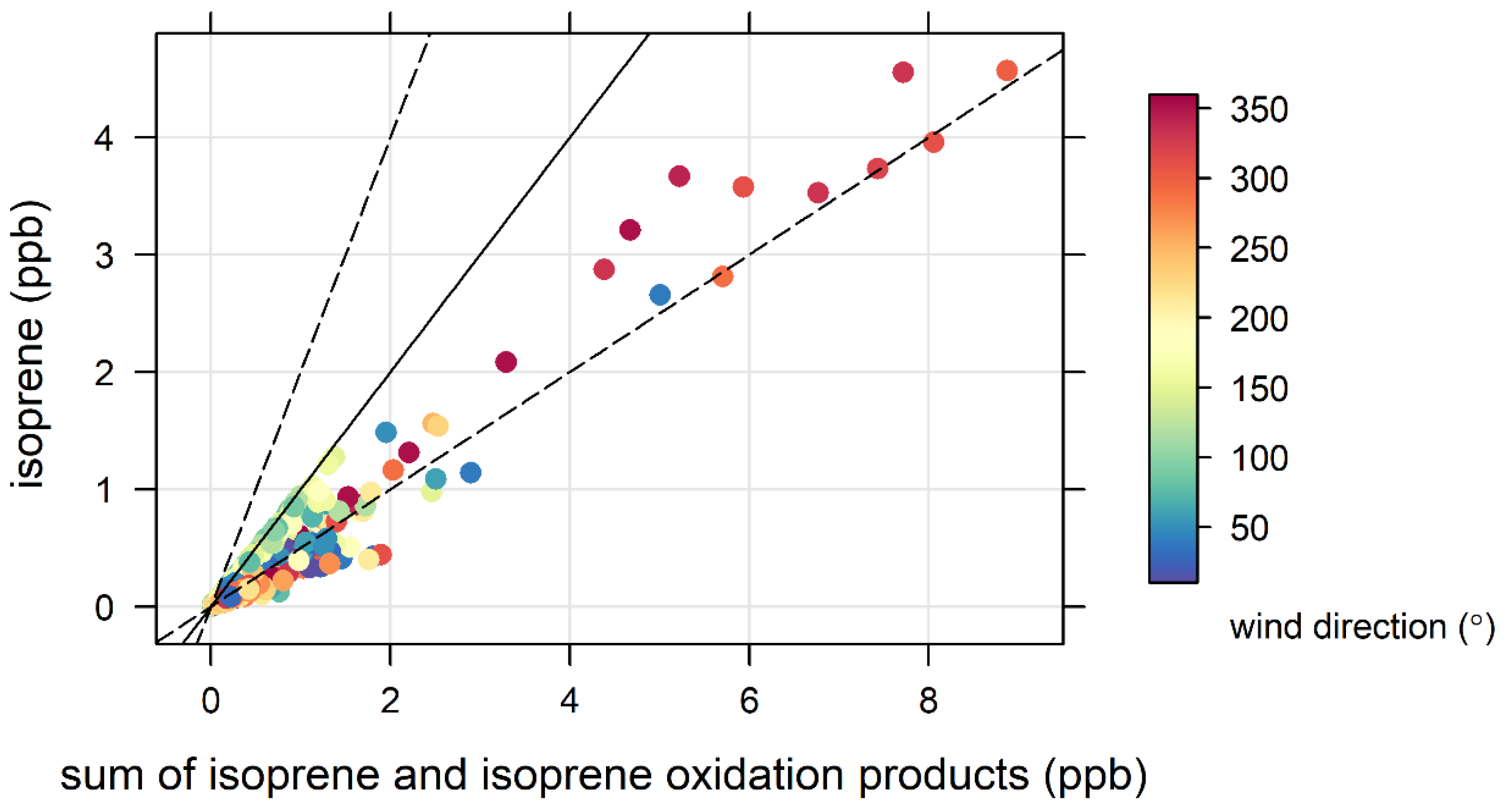
4.2. Terrestrial Biogenic VOCs during MUMBA
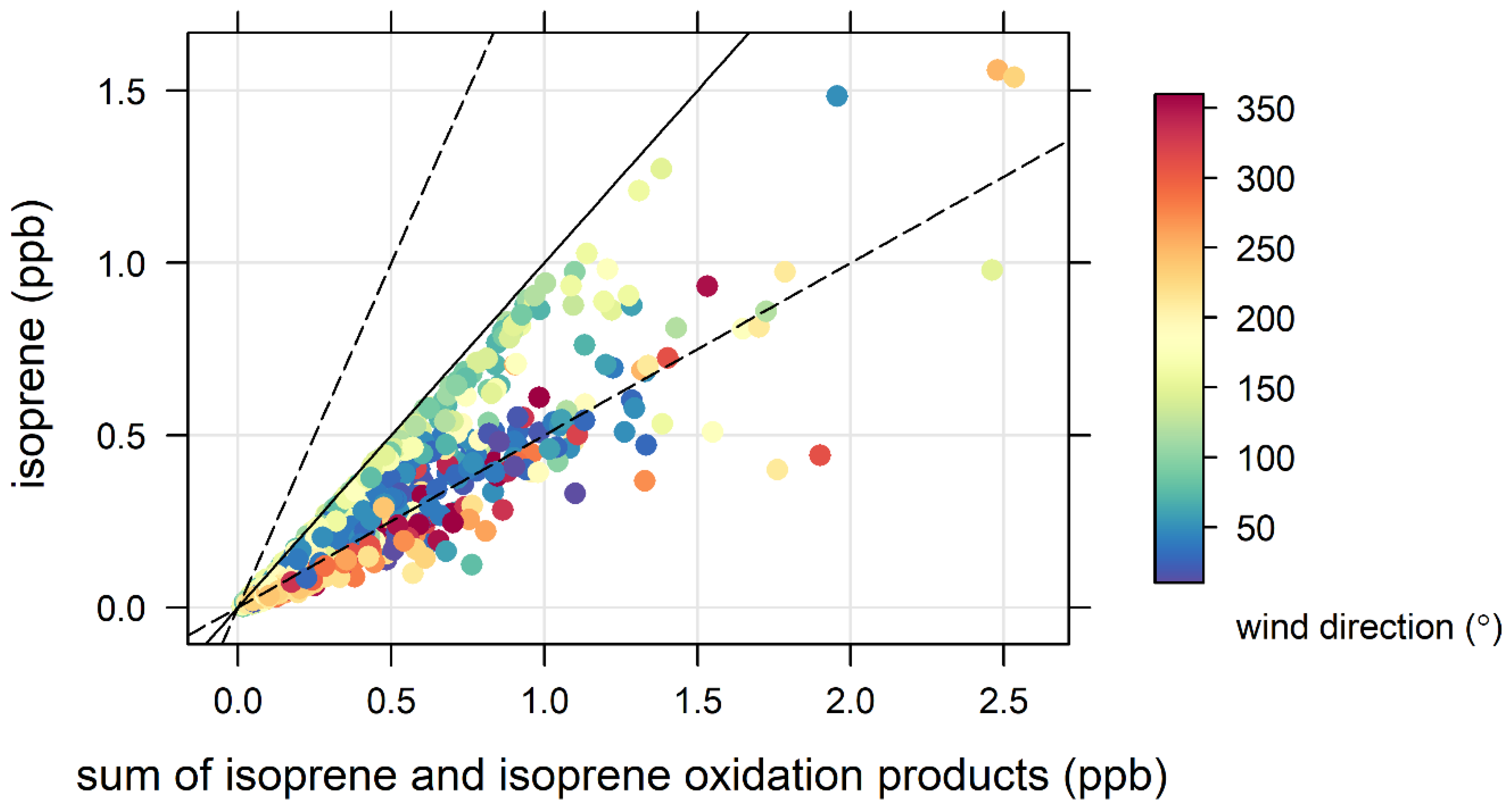
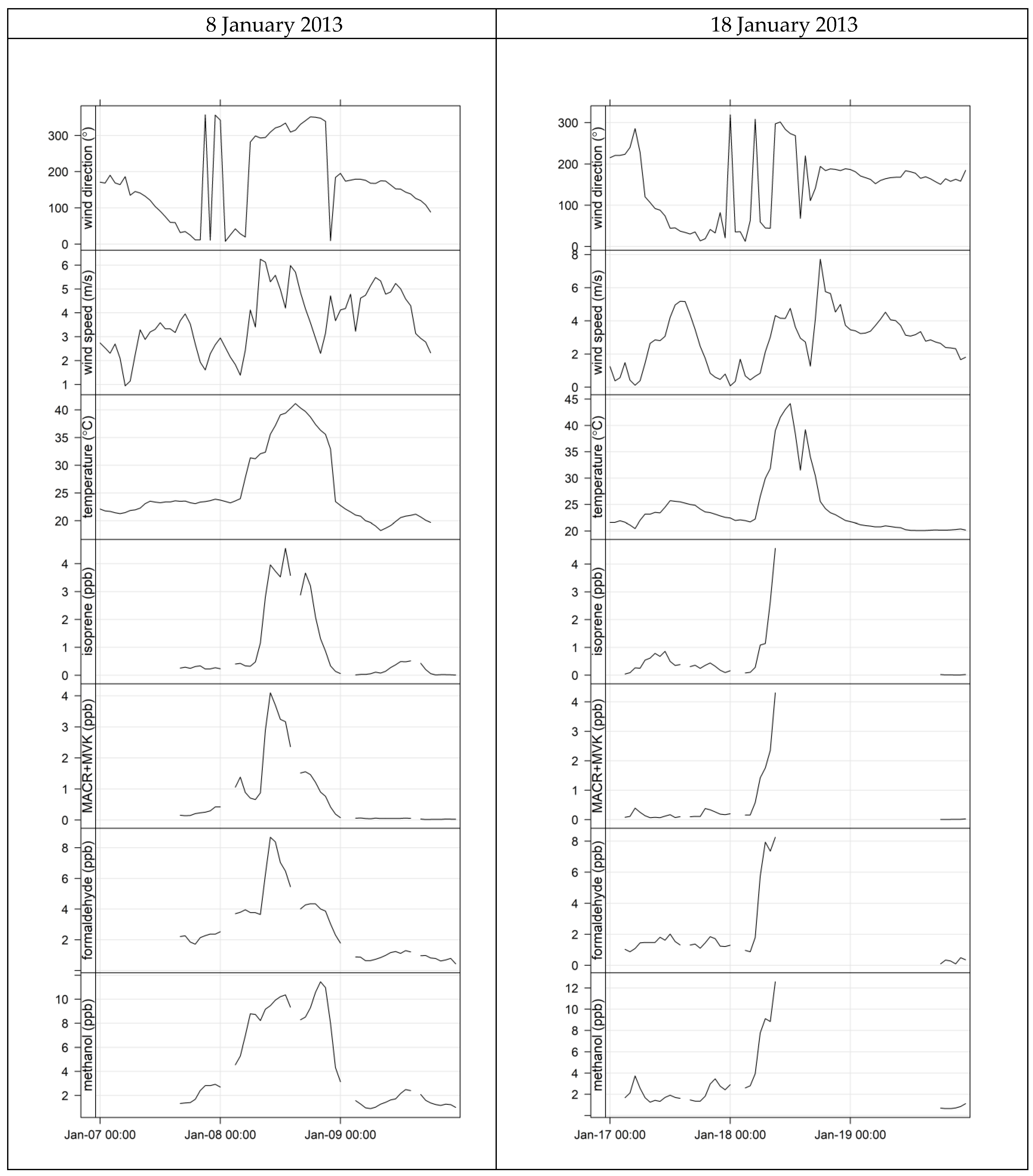
- the proximity of Puckey’s Estate to the measurement site leaves little time for oxidation to take place, and
- the escarpment is a source of isoprene; however, given its distance from the site (~10 km) and the average wind speed observed during the campaign (2.8 m s−1 or ~10 km/h), there is enough time (~1 hour) for oxidation of a substantial fraction of the isoprene to occur before the air reaches the site.
4.3. Characterisation of Terrestrial Biogenic VOCs on Atypically Hot Days
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | South-Easterly Winds | North-Easterly Winds | ||
|---|---|---|---|---|
| December 26th 08:00–18:59 | February 13th 14:00–17:59 | February 5th 12:00–17:59 | February 6th 13:00–18:59 | |
| Formaldehyde | 590 ± 80 | 600 ± 130 | 770 ± 170 | 910 ± 390 |
| Methanol | 1340 ± 170 | 1020 ± 70 | 1190 ± 110 | 1120 ± 170 |
| Acetonitrile | 56± 5 | 65 ± 4 | 66 ± 3 | 70 ± 4 |
| Acetaldehyde | 190 ± 40 | 120 ± 30 | 200 ± 40 | 170 ± 40 |
| Acetone | 260 ± 30 | 270 ± 10 | 320 ± 20 | 330 ± 30 |
| Dimethyl sulphide | 50 ± 10 | 35 ± 8 | 38 ± 8 | 40 ± 20 |
| Isoprene | 370 ± 120 | 410 ± 30 | 380 ± 200 | 160 ± 30 |
| MACR + MVK | 40 ± 10 | 39 ± 9 | 54 ± 8 | 46 ± 7 |
| benzene | 20 ± 30 | 28 ± 5 | 40 ± 20 | 50 ± 20 |
| toluene | 30 ± 20 | 45 ± 3 | 90 ± 70 | 180 ± 170 |
| C8H10 | 23 ± 6 | 70 ± 40 | 80 ± 40 | 90 ± 30 |
| C9H12 | 36 ± 7 | 50 ± 20 | 60 ± 20 | 90 ± 30 |
| monoterpenes | 33 ± 15 | 25 ± 9 | 23 ± 5 | 18 ± 6 |
| CO (ppb) | 52 ± 2 | 58 ± 6 | 60 ± 10 | 66 ± 4 |
| NOx | 990 ± 210 | 1850 ± 220 | 3300 ± 1500 | 3700 ± 690 |
| Ozone (ppb) | 20.5 ± 1.1 | 20.3 ± 0.8 | 18 ± 2 | 22 ± 3 |

Appendix B

Appendix C


References
- Chameides, W.L.; Lindsay, R.W.; Richardson, J.; Kiang, C.S. The Role of Biogenic Hydrocarbons in Urban Photochemical Smog—Atlanta as a Case-Study. Science 1988, 241, 1473–1475. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A.; Dockery, D.W.; Schwartz, J. Review of Epidemiological Evidence of Health-Effects of Particulate Air-Pollution. Inhal. Toxicol. 1995, 7, 1–18. [Google Scholar] [CrossRef]
- von Schneidemesser, E.; Monks, P.S.; Gros, V.; Gauduin, J.; Sanchez, O. How important is biogenic isoprene in an urban environment? A study in London and Paris. Geophys. Res. Lett. 2011, 38, L19804. [Google Scholar] [CrossRef]
- Chan, C.K.; Yao, X. Air pollution in mega cities in China. Atmos. Environ. 2008, 42, 1–42. [Google Scholar] [CrossRef]
- Parrish, D.D.; Singh, H.B.; Molina, L.; Madronich, S. Air quality progress in North American megacities: A review. Atmos. Environ. 2011, 45, 7015–7025. [Google Scholar] [CrossRef]
- Ma, J.Z.; Wang, W.; Chen, Y.; Liu, H.J.; Yan, P.; Ding, G.A.; Wang, M.L.; Sun, J.; Lelieveld, J. The IPAC-NC field campaign: A pollution and oxidization pool in the lower atmosphere over Huabei, China. Atmos. Chem. Phys. 2012, 12, 3883–3908. [Google Scholar] [CrossRef]
- Molina, L.T.; Kolb, C.E.; de Foy, B.; Lamb, B.K.; Brune, W.H.; Jimenez, J.L.; Ramos-Villegas, R.; Sarmiento, J.; Paramo-Figueroa, V.H.; Cardenas, B.; et al. Air quality in North America’s most populous city—Overview of the MCMA-2003 campaign. Atmos. Chem. Phys. 2007, 7, 2447–2473. [Google Scholar] [CrossRef]
- Molina, L.T.; Madronich, S.; Gaffney, J.S.; Apel, E.; de Foy, B.; Fast, J.; Ferrare, R.; Herndon, S.; Jimenez, J.L.; Lamb, B.; et al. An overview of the MILAGRO 2006 Campaign: Mexico City emissions and their transport and transformation. Atmos. Chem. Phys. 2010, 10, 8697–8760. [Google Scholar] [CrossRef] [Green Version]
- Carslaw, D.C.; Beevers, S.D.; Ropkins, K.; Bell, M.C. Detecting and quantifying aircraft and other on-airport contributions to ambient nitrogen oxides in the vicinity of a large international airport. Atmos. Environ. 2006, 40, 5424–5434. [Google Scholar] [CrossRef]
- Wilson, R.C.; Fleming, Z.L.; Monks, P.S.; Clain, G.; Henne, S.; Konovalov, I.B.; Szopa, S.; Menut, L. Have primary emission reduction measures reduced ozone across Europe? An analysis of European rural background ozone trends 1996-2005. Atmos. Chem. Phys. 2012, 12, 437–454. [Google Scholar] [CrossRef]
- Goldstein, A.H.; Galbally, I.E. Known and Unexplored Organic Constituents in the Earth’s Atmosphere. Environ. Sci. Technol. 2007, 41, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, J.; Berresheim, H.; Borrmann, S.; Crutzen, P.J.; Dentener, F.J.; Fischer, H.; Feichter, J.; Flatau, P.J.; Heland, J.; Holzinger, R.; et al. Global air pollution crossroads over the Mediterranean. Science 2002, 298, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, D.; Anderson, T.; Covert, D.; Kotchenruther, R.; Trost, B.; Danielson, J.; Simpson, W.; Berntsen, T.; Karlsdottir, S.; Blake, D.; et al. Transport of Asian air pollution to North America. Geophys. Res. Lett. 1999, 26, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Fehsenfeld, F.C.; Ancellet, G.; Bates, T.S.; Goldstein, A.H.; Hardesty, R.M.; Honrath, R.; Law, K.S.; Lewis, A.C.; Leaitch, R.; McKeen, S.; et al. International Consortium for Atmospheric Research on Transport and Transformation (ICARTT): North America to Europe—Overview of the 2004 summer field study. J. Geophys. Res. Atmos. 2006, 111. [Google Scholar] [CrossRef]
- Cope, M.E.; Hess, G.D.; Lee, S.; Tory, K.; Azzi, M.; Carras, J.; Lilley, W.; Manins, P.C.; Nelson, P.; Ng, L.; et al. The Australian Air Quality Forecasting System. Part I: Project description and early outcomes. J. Appl. Meteorol. 2004, 43, 649–662. [Google Scholar] [CrossRef]
- Hess, G.D.; Tory, K.J.; Cope, M.E.; Lee, S.; Puri, K.; Manins, P.C.; Young, M. The Australian Air Quality Forecasting System. Part II: Case study of a Sydney 7-day photochemical smog event. J. Appl. Meteorol. 2004, 43, 663–679. [Google Scholar] [CrossRef]
- Tory, K.J.; Cope, M.E.; Hess, G.D.; Lee, S.; Puri, K.; Manins, P.C.; Wong, N. The Australian Air Quality Forecasting System. Part III: Case study of a Melbourne 4-day photochemical smog eventt. J. Appl. Meteorol. 2004, 43, 680–695. [Google Scholar] [CrossRef]
- Cope, M.E.; Hess, G.D.; Lee, S.; Tory, K.J.; Burgers, M.; Dewundege, P.; Johnson, M. The Australian Air Quality Forecasting System: Exploring first steps towards determining the limits of predictability for short-term ozone forecasting. Bound.-Layer Meteor. 2005, 116, 363–384. [Google Scholar] [CrossRef]
- Cheung, H.C.; Morawska, L.; Ristovski, Z.D. Observation of new particle formation in subtropical urban environment. Atmos. Chem. Phys. 2011, 11, 3823–3833. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.C.; Morawska, L.; Ristovski, Z.D.; Wainwright, D. Influence of medium range transport of particles from nucleation burst on particle number concentration within the urban airshed. Atmos. Chem. Phys. 2012, 12, 4951–4962. [Google Scholar] [CrossRef] [Green Version]
- Keywood, M.; Selleck, P.; Reisen, F.; Cohen, D.; Chambers, S.; Cheng, M.; Cope, M.; Crumeyrolle, S.; Dunne, E.; Emmerson, K.; et al. Comprehensive aerosol and gas data set from the Sydney Particle Study. Earth Syst. Sci. Data Discuss. 2019, 2019, 1–34. [Google Scholar] [CrossRef]
- Cainey, J.M.; Keywood, M.; Grose, M.R.; Krummel, P.; Galbally, I.E.; Johnston, P.; Gillett, R.W.; Meyer, M.; Fraser, P.; Steele, P.; et al. Precursors to Particles (P2P) at Cape Grim 2006: Campaign overview. Environ. Chem. 2007, 4, 143–150. [Google Scholar] [CrossRef]
- Fletcher, C.A.; Johnson, G.R.; Ristovski, Z.D.; Harvey, M. Hygroscopic and volatile properties of marine aerosol observed at Cape Grim during the P2P campaign. Environ. Chem. 2007, 4, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Modini, R.L.; Ristovski, Z.D.; Johnson, G.R.; He, C.; Surawski, N.; Morawska, L.; Suni, T.; Kulmala, M. New particle formation and growth at a remote, sub-tropical coastal location. Atmos. Chem. Phys. 2009, 9, 7607–7621. [Google Scholar] [CrossRef] [Green Version]
- Ristovski, Z.D.; Suni, T.; Kulmala, M.; Boy, M.; Meyer, N.K.; Duplissy, J.; Turnipseed, A.; Morawska, L.; Baltensperger, U. The role of sulphates and organic vapours in growth of newly formed particles in a eucalypt forest. Atmos. Chem. Phys. 2010, 10, 2919–2926. [Google Scholar] [CrossRef] [Green Version]
- Suni, T.; Kulmala, M.; Hirsikko, A.; Bergman, T.; Laakso, L.; Aalto, P.P.; Leuning, R.; Cleugh, H.; Zegelin, S.; Hughes, D.; et al. Formation and characteristics of ions and charged aerosol particles in a native Australian Eucalypt forest. Atmos. Chem. Phys. 2008, 8, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Nguyen Duc, H.; Chang, L.T.-C.; Trieu, T.; Salter, D.; Scorgie, Y. Source Contributions to Ozone Formation in the New South Wales Greater Metropolitan Region, Australia. Atmosphere 2018, 9, 443. [Google Scholar] [CrossRef]
- Utembe, S.R.; Rayner, P.J.; Silver, J.D.; Guerette, E.A.; Fisher, J.A.; Emmerson, K.M.; Cope, M.; Paton-Walsh, C.; Griffiths, A.D.; Duc, H.; et al. Hot Summers: Effect of Extreme Temperatures on Ozone in Sydney, Australia. Atmosphere 2018, 9, 25. [Google Scholar] [CrossRef]
- Chang, L.T.-C.; Scorgie, Y.; Duc, H.N.; Monk, K.; Fuchs, D.; Trieu, T. Major Source Contributions to Ambient PM2.5 and Exposures within the New South Wales Greater Metropolitan Region. Atmosphere 2019, 10, 138. [Google Scholar] [CrossRef]
- Cope, M.E.; Keywood, M.; Emmerson, K.; Galbally, I.; Boast, K.; Chambers, S.; Cheng, M.; Crumeyrolle, S.; Dunne, E.; Fedele, R.; et al. The Sydney Particle Study; The Centre for Australian Weather and Climate Research: Melbourne, Australia, 2014.
- Guenther, A.B.; Jiang, X.; Heald, C.L.; Sakulyanontvittaya, T.; Duhl, T.; Emmons, L.K.; Wang, X. The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): An extended and updated framework for modeling biogenic emissions. Geosci. Model Dev. 2012, 5, 1471–1492. [Google Scholar] [CrossRef]
- Emmerson, K.M.; Galbally, I.E.; Guenther, A.B.; Paton-Walsh, C.; Guerette, E.A.; Cope, M.E.; Keywood, M.D.; Lawson, S.J.; Molloy, S.B.; Dunne, E.; et al. Current estimates of biogenic emissions from eucalypts uncertain for southeast Australia. Atmos. Chem. Phys. 2016, 16, 6997–7011. [Google Scholar] [CrossRef] [Green Version]
- Emmerson, K.M.; Cope, M.E.; Galbally, I.E.; Lee, S.; Nelson, P.F. Isoprene and monoterpene emissions in south-east Australia: Comparison of a multi-layer canopy model with MEGAN and with atmospheric observations. Atmos. Chem. Phys. 2018, 18, 7539–7556. [Google Scholar] [CrossRef]
- Emmerson, K.M.; Palmer, P.I.; Thatcher, M.; Haverd, V.; Guenther, A.B. Sensitivity of isoprene emissions to drought over south-eastern Australia: Integrating models and satellite observations of soil moisture. Atmos. Environ. 2019, 209, 112–124. [Google Scholar] [CrossRef]
- Galbally, I.E.; Lawson, S.J.; Weeks, I.A.; Bentley, S.T.; Gillett, R.W.; Meyer, M.; Goldstein, A.H. Volatile organic compounds in marine air at Cape Grim, Australia. Environ. Chem. 2007, 4, 178–182. [Google Scholar] [CrossRef]
- Lawson, S.J.; Galbally, I.; Dunne, E.; Gras, J. Measurement of VOCs in Marine Air at Cape Grim using Proton Transfer Reaction Mass Spectrometry; Bureau of Meteorology and CSIRO Marine and Atmospheric Research: Melbourne, Australia, 2011; pp. 23–32. [Google Scholar]
- Lewis, A.C.; Carpenter, L.J.; Pilling, M.J. Nonmethane hydrocarbons in Southern Ocean boundary layer air. J. Geophys. Res. Atmos. 2001, 106, 4987–4994. [Google Scholar] [CrossRef] [Green Version]
- Paton-Walsh, C.; Guérette, E.A.; Kubistin, D.; Humphries, R.; Stephen, R.W.; Dominick, D.; Galbally, I.; Buchholz, R.; Bhujel, M.; Chambers, S.; et al. The MUMBA campaign: Measurements of urban, marine and biogenic air. Earth Syst. Sci. Data 2017, 9, 349–362. [Google Scholar] [CrossRef]
- Paton-Walsh, C.; Guérette, É.A.; Emmerson, K.; Cope, M.; Kubistin, D.; Humphries, R.; Wilson, S.; Buchholz, R.; Jones, N.B.; Griffith, D.W.T.; et al. Urban air quality in a coastal city: Wollongong during the MUMBA campaign. Atmosphere 2018, 9, 500. [Google Scholar] [CrossRef]
- Dominick, D.; Wilson, S.R.; Paton-Walsh, C.; Humphries, R.; Guérette, E.A.; Keywood, M.; Kubistin, D.; Marwick, B. Characteristics of airborne particle number size distributions in a coastal-urban environment. Atmos. Environ. 2018, 186, 256–265. [Google Scholar] [CrossRef]
- Dominick, D.; Wilson, S.R.W.; Paton-Walsh, C.; Humphries, R.; Guérette, E.A.; Keywood, M.; Selleck, P.; Kubistin, D.; Marwick, B. Particle Formation Mechanisms at a complex environment. Atmosphere 2019, 10, 275. [Google Scholar] [CrossRef]
- Zhang, Y.; Jena, C.; Wang, K.; Paton-Walsh, C.; Guérette, É.-A.; Utembe, S.; Silver, J.D.; Keywood, M. Multiscale Applications of Two Online-Coupled Meteorology-Chemistry Models during Recent Field Campaigns in Australia, Part I: Model Description and WRF/Chem-ROMS Evaluation Using Surface and Satellite Data and Sensitivity to Spatial Grid Resolutions. Atmosphere 2019, 10, 189. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, K.; Jena, C.; Paton-Walsh, C.; Guérette, É.-A.; Utembe, S.; Silver, J.D.; Keywood, M. Multiscale Applications of Two Online-Coupled Meteorology-Chemistry Models During Recent Field Campaigns in Australia, Part II: Comparison of WRF/Chem and WRF/Chem-ROMS and Impacts of Air-Sea Interactions and Boundary Conditions. Atmosphere 2019, 10, 210. [Google Scholar] [CrossRef]
- Monk, K.; Guérette, E.-A.; Paton-Walsh, C.; Silver, J.D.; Emmerson, K.M.; Utembe, S.R.; Zhang, Y.; Griffiths, A.D.; Chang, L.T.-C.; Duc, H.N.; et al. Evaluation of regional air quality models over Sydney, Australia: Part 1 Meteorological model comparison. Atmosphere 2019, 10, 374. [Google Scholar] [CrossRef]
- Guérette, E.-A.; Monk, K.; Emmerson, K.M.; Utembe, S.R.; Zhang, Y.; Silver, J.D.; Duc, H.N.; Chang, L.T.-C.; Trieu, T.; Griffiths, A.D.; et al. Evaluation of regional air quality models over Sydney, Australia: Part 2 Model performance for surface ozone and PM2.5. Atmosphere 2019. in preparation for submission. [Google Scholar]
- Guérette, E.-A.; Paton-Walsh, C.; Kubistin, D.; Humphries, R.; Bhujel, M.; Buchholz, R.R.; Chambers, S.; Cheng, M.; Davy, P.; Dominick, D.; et al. Measurements of Urban, Marine and Biogenic Air (MUMBA): Characterisation of trace gases and aerosol at the urban, marine and biogenic interface in summer in Wollongong, Australia. In Supplement to: Paton-Walsh, Clare; Guérette, Elise-Andrée; Kubistin, Dagmar; Humphries, Ruhi; Wilson, Stephen R; Dominick, Doreena; Galbally, Ian; Buchholz, Rebecca R; Bhujel, Mahendra; Chambers, Scott; Cheng, Min; Cope, Martin; Davy, Perry; Emmerson, Kathryn; Griffith, David W T; Griffiths, Alan; Keywood, Melita; Lawson, Sarah; Molloy, Suzie; Rea, Geraldine; Selleck, Paul; Shi, Xue; Simmons, Jack; Velazco, Voltaire (2017): The MUMBA Campaign: Measurements of Urban, Marine and Biogenic Air. Earth System Science Data, 9(1), 349–362, https://doi.org/10.5194/essd-9-349-2017; PANGAEA, 2017. [Google Scholar] [CrossRef]
- Chambers, S.; Williams, A.G.; Zahorowski, W.; Griffiths, A.; Crawford, J. Separating remote fetch and local mixing influences on vertical radon measurements in the lower atmosphere. Tellus B 2011, 63, 843–859. [Google Scholar] [CrossRef]
- Chambers, S.D.; Williams, A.G.; Crawford, J.; Griffiths, A.D. On the use of radon for quantifying the effects of atmospheric stability on urban emissions. Atmos. Chem. Phys. 2015, 15, 1175–1190. [Google Scholar] [CrossRef] [Green Version]
- Dunne, E.; Galbally, I.E.; Cheng, M.; Selleck, P.; Molloy, S.B.; Lawson, S.J. Comparison of VOC measurements made by PTR-MS, adsorbent tubes–GC-FID-MS and DNPH derivatization–HPLC during the Sydney Particle Study, 2012: A contribution to the assessment of uncertainty in routine atmospheric VOC measurements. Atmos. Meas. Tech. 2018, 11, 141–159. [Google Scholar] [CrossRef]
- Inomata, S.; Tanimoto, H.; Kameyama, S.; Tsunogai, U.; Irie, H.; Kanaya, Y.; Wang, Z. Technical Note: Determination of formaldehyde mixing ratios in air with PTR-MS: Laboratory experiments and field measurements. Atmos. Chem. Phys. 2008, 8, 273–284. [Google Scholar] [CrossRef]
- Baasandorj, M.; Millet, D.B.; Hu, L.; Mitroo, D.; Williams, B.J. Measuring acetic and formic acid by proton-transfer-reaction mass spectrometry: Sensitivity, humidity dependence, and quantifying interferences. Atmos. Meas. Tech. 2015, 8, 1303–1321. [Google Scholar] [CrossRef]
- Rothman, L.S.; Gordon, I.E.; Babikov, Y.; Barbe, A.; Chris Benner, D.; Bernath, P.F.; Birk, M.; Bizzocchi, L.; Boudon, V.; Brown, L.R.; et al. The HITRAN2012 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 2013, 130, 4–50. [Google Scholar] [CrossRef]
- Griffith, D.W.T. Synthetic Calibration and Quantitative Analysis of Gas-Phase FT-IR Spectra. Appl. Spectrosc. 1996, 50, 59–70. [Google Scholar] [CrossRef]
- Griffith, D.W.T.; Deutscher, N.M.; Caldow, C.; Kettlewell, G.; Riggenbach, M.; Hammer, S. A Fourier transform infrared trace gas and isotope analyser for atmospheric applications. Atmos. Meas. Tech. 2012, 5, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Hammer, S.; Griffith, D.W.T.; Konrad, G.; Vardag, S.; Caldow, C.; Levin, I. Assessment of a multi-species in situ FTIR for precise atmospheric greenhouse gas observations. Atmos. Meas. Tech. 2013, 6, 1153–1170. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, A.D.; Zahorowski, W.; Element, A.; Werczynski, S. A map of radon flux at the Australian land surface. Atmos. Chem. Phys. 2010, 10, 8969–8982. [Google Scholar] [CrossRef] [Green Version]
- Schery, S.D.; Huang, S. An estimate of the global distribution of radon emissions from the ocean. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.D.; Preunkert, S.; Weller, R.; Hong, S.-B.; Humphries, R.S.; Tositti, L.; Angot, H.; Legrand, M.; Williams, A.G.; Griffiths, A.D.; et al. Characterizing Atmospheric Transport Pathways to Antarctica and the Remote Southern Ocean Using Radon-222. Front. Earth Sci. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.D.; Williams, A.G.; Conen, F.; Griffiths, A.D.; Reimann, S.; Steinbacher, M.; Krummel, P.B.; Steele, L.P.; van der Schoot, M.V.; Galbally, I.E.; et al. Towards a Universal ‘Baseline’; Characterisation of Air Masses for High- and Low-Altitude Observing Stations Using Radon-222. Aerosol Air Qual. Res. 2016, 16, 885–899. [Google Scholar] [CrossRef]
- Crawford, J.; Chambers, S.D.; Cohen, D.D.; Williams, A.G.; Atanacio, A. Baseline characterisation of source contributions to daily-integrated PM2.5 observations at Cape Grim using Radon-222. Environ. Pollut. 2018, 243, 37–48. [Google Scholar] [CrossRef]
- Linfoot, S.; Johnson, M.; Young, M.; Angri, L.; Quigley, S.; Spencer, J.; Duc, H.; Trieu, T.; Xu, C. State of Knowledge: Ozone; State of NSW: Sydney, Austrilia, September 2010; ISBN 978-1-74232-835-5.
- Thoning, K.W.; Tans, P.P.; Komhyr, W.D. Atmospheric carbon dioxide at Mauna Loa Observatory: 2. Analysis of the NOAA GMCC data, 1974–1985. J. Geophys. Res. Atmos. 1989, 94, 8549–8565. [Google Scholar] [CrossRef]
- Hamilton, J.F.; Allen, G.; Watson, N.M.; Lee, J.D.; Saxton, J.E.; Lewis, A.C.; Vaughan, G.; Bower, K.N.; Flynn, M.J.; Crosier, J.; et al. Observations of an atmospheric chemical equator and its implications for the tropical warm pool region. J. Geophys. Res. Atmos. 2008, 113, 12. [Google Scholar] [CrossRef]
- Novelli, P.C.; Steele, L.P.; Tans, P.P. Mixing Ratios of Carbon-Monoxide in the Troposphere. J. Geophys. Res. Atmos. 1992, 97, 20731–20750. [Google Scholar] [CrossRef]
- Buchholz, R.R.; Paton-Walsh, C.; Griffith, D.W.T.; Kubistin, D.; Caldow, C.; Fisher, J.A.; Deutscher, N.M.; Kettlewell, G.; Riggenbach, M.; Macatangay, R.; et al. Source and meteorological influences on air quality (CO, CH4 & CO2) at a Southern Hemisphere urban site. Atmos. Environ. 2016, 126, 274–289. [Google Scholar] [CrossRef]
- Singh, H.; Chen, Y.; Staudt, A.; Jacob, D.; Blake, D.; Heikes, B.; Snow, J. Evidence from the Pacific troposphere for large global sources of oxygenated organic compounds. Nature 2001, 410, 1078–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.B.; Salas, L.J.; Chatfield, R.B.; Czech, E.; Fried, A.; Walega, J.; Evans, M.J.; Field, B.D.; Jacob, D.J.; Blake, D.; et al. Analysis of the atmospheric distribution, sources, and sinks of oxygenated volatile organic chemicals based on measurements over the Pacific during TRACE-P. J. Geophys. Res. Atmos. 2004, 109, D15S07. [Google Scholar] [CrossRef]
- Colomb, A.; Gros, V.; Alvain, S.; Sarda-Esteve, R.; Bonsang, B.; Moulin, C.; Klüpfel, T.; Williams, J. Variation of atmospheric volatile organic compounds over the Southern Indian Ocean (30–49°S). Environ. Chem. 2009, 6, 70–82. [Google Scholar] [CrossRef]
- Gilman, J.B.; Kuster, W.C.; Goldan, P.D.; Herndon, S.C.; Zahniser, M.S.; Tucker, S.C.; Brewer, W.A.; Lerner, B.M.; Williams, E.J.; Harley, R.A.; et al. Measurements of volatile organic compounds during the 2006 TexAQS/GoMACCS campaign: Industrial influences, regional characteristics, and diurnal dependencies of the OH reactivity. J. Geophys. Res. Atmos. 2009, 114, D00F06. [Google Scholar] [CrossRef]
- Lawson, S.J.; Selleck, P.W.; Galbally, I.E.; Keywood, M.D.; Harvey, M.J.; Lerot, C.; Helmig, D.; Ristovski, Z. Seasonal in situ observations of glyoxal and methylglyoxal over the temperate oceans of the Southern Hemisphere. Atmos. Chem. Phys. 2015, 15, 223–240. [Google Scholar] [CrossRef] [Green Version]
- Marandino, C.A.; De Bruyn, W.J.; Miller, S.D.; Prather, M.J.; Saltzman, E.S. Oceanic uptake and the global atmospheric acetone budget. Geophys. Res. Lett. 2005, 32, L15806. [Google Scholar] [CrossRef]
- Peters, E.; Wittrock, F.; Großmann, K.; Frieß, U.; Richter, A.; Burrows, J.P. Formaldehyde and nitrogen dioxide over the remote western Pacific Ocean: SCIAMACHY and GOME-2 validation using ship-based MAX-DOAS observations. Atmos. Chem. Phys. 2012, 12, 11179–11197. [Google Scholar] [CrossRef] [Green Version]
- Warneke, C.; de Gouw, J.A. Organic trace gas composition of the marine boundary layer over the northwest Indian Ocean in April 2000. Atmos. Environ. 2001, 35, 5923–5933. [Google Scholar] [CrossRef]
- Williams, J.; Custer, T.; Riede, H.; Sander, R.; Jöckel, P.; Hoor, P.; Pozzer, A.; Wong-Zehnpfennig, S.; Hosaynali Beygi, Z.; Fischer, H.; et al. Assessing the effect of marine isoprene and ship emissions on ozone, using modelling and measurements from the South Atlantic Ocean. Environ. Chem. 2010, 7, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.; Holzinger, R.; Gros, V.; Xu, X.; Atlas, E.; Wallace, D.W.R. Measurements of organic species in air and seawater from the tropical Atlantic. Geophys. Res. Lett. 2004, 31, L23S06. [Google Scholar] [CrossRef]
- Wisthaler, A.; Hansel, A.; Dickerson, R.R.; Crutzen, P.J. Organic trace gas measurements by PTR-MS during INDOEX 1999. J. Geophys. Res. Atmos. 2002, 107, 8024. [Google Scholar] [CrossRef]
- White, C.J.; Fox-Hughes, P. Seasonal climate summary southern hemisphere (summer 2012–13): Australia’s hottest summer on record and extreme east coast rainfall. Aust. Meteorol. Oceanogr. J. 2013, 63, 443–456. [Google Scholar] [CrossRef]
- Galbally, I.E.; Kirstine, W. The Production of Methanol by Flowering Plants and the Global Cycle of Methanol. J. Atmos. Chem. 2002, 43, 195–229. [Google Scholar] [CrossRef]
- Millet, D.B.; Jacob, D.J.; Custer, T.G.; de Gouw, J.A.; Goldstein, A.H.; Karl, T.; Singh, H.B.; Sive, B.C.; Talbot, R.W.; Warneke, C.; et al. New constraints on terrestrial and oceanic sources of atmospheric methanol. Atmos. Chem. Phys. 2008, 8, 6887–6905. [Google Scholar] [CrossRef] [Green Version]
- Millet, D.B.; Guenther, A.; Siegel, D.A.; Nelson, N.B.; Singh, H.B.; de Gouw, J.A.; Warneke, C.; Williams, J.; Eerdekens, G.; Sinha, V.; et al. Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations. Atmos. Chem. Phys. 2010, 10, 3405–3425. [Google Scholar] [CrossRef] [Green Version]
- Wittrock, F.; Richter, A.; Oetjen, H.; Burrows, J.P.; Kanakidou, M.; Myriokefalitakis, S.; Volkamer, R.; Beirle, S.; Platt, U.; Wagner, T. Simultaneous global observations of glyoxal and formaldehyde from space. Geophys. Res. Lett. 2006, 33, L16804. [Google Scholar] [CrossRef]
- Ayers, G.P.; Gillett, R.W. DMS and its oxidation products in the remote marine atmosphere: Implications for climate and atmospheric chemistry. J. Sea Res. 2000, 43, 275–286. [Google Scholar] [CrossRef]
- Dunne, E.; Galbally, I.E.; Lawson, S.; Patti, A. Interference in the PTR-MS measurement of acetonitrile at m/z 42 in polluted urban air—A study using switchable reagent ion PTR-MS. Int. J. Mass Spectrom. 2012, 319–320, 40–47. [Google Scholar] [CrossRef]
- Hamm, S.; Warneck, P. The interhemispheric distribution and the budget of acetonitrile in the troposphere. J. Geophys. Res. Atmos. 1990, 95, 20593–20606. [Google Scholar] [CrossRef]
- Guenther, A.; Hewitt, C.N.; Erickson, D.; Fall, R.; Geron, C.; Graedel, T.; Harley, P.; Klinger, L.; Lerdau, M.; McKay, W.A.; et al. A global model of natural volatile organic compound emissions. J. Geophys. Res. Atmos. 1995, 100, 8873–8892. [Google Scholar] [CrossRef]
- Jacob, D.J.; Field, B.D.; Jin, E.M.; Bey, I.; Li, Q.; Logan, J.A.; Yantosca, R.M.; Singh, H.B. Atmospheric budget of acetone. J. Geophys. Res. Atmos. 2002, 107, 4100. [Google Scholar] [CrossRef]
- Jacob, D.J.; Field, B.D.; Li, Q.; Blake, D.R.; de Gouw, J.; Warneke, C.; Hansel, A.; Wisthaler, A.; Singh, H.B.; Guenther, A. Global budget of methanol: Constraints from atmospheric observations. J. Geophys. Res. 2005, 110, D08303. [Google Scholar] [CrossRef]
- Fischer, E.V.; Jacob, D.J.; Millet, D.B.; Yantosca, R.M.; Mao, J. The role of the ocean in the global atmospheric budget of acetone. Geophys. Res. Lett. 2012, 39, L01807. [Google Scholar] [CrossRef]
- Khan, M.A.H.; Cooke, M.C.; Utembe, S.R.; Archibald, A.T.; Maxwell, P.; Morris, W.C.; Xiao, P.; Derwent, R.G.; Jenkin, M.E.; Percival, C.J.; et al. A study of global atmospheric budget and distribution of acetone using global atmospheric model STOCHEM-CRI. Atmos. Environ. 2015, 112, 269–277. [Google Scholar] [CrossRef]
- Winters, A.J.; Adams, M.A.; Bleby, T.M.; Rennenberg, H.; Steigner, D.; Steinbrecher, R.; Kreuzwieser, J. Emissions of isoprene, monoterpene and short-chained carbonyl compounds from Eucalyptus spp. in southern Australia. Atmos. Environ. 2009, 43, 3035–3043. [Google Scholar] [CrossRef]
- Kanawade, V.P.; Jobson, B.T.; Guenther, A.B.; Erupe, M.E.; Pressley, S.N.; Tripathi, S.N.; Lee, S.H. Isoprene suppression of new particle formation in a mixed deciduous forest. Atmos. Chem. Phys. 2011, 11, 6013–6027. [Google Scholar] [CrossRef] [Green Version]
- Kiendler-Scharr, A.; Wildt, J.; Maso, M.D.; Hohaus, T.; Kleist, E.; Mentel, T.F.; Tillmann, R.; Uerlings, R.; Schurr, U.; Wahner, A. New particle formation in forests inhibited by isoprene emissions. Nature 2009, 461, 381–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Carslaw, D.C.; Ropkins, K. Openair—An R package for air quality data analysis. Environ. Model. Softw. 2012, 27, 52–61. [Google Scholar] [CrossRef]
- Draxler, R.R.; Hess, G.D. An overview of the HYSPLIT_4 modelling system for trajectories, dispersion and deposition. Aust. Meteorol. Mag. 1998, 47, 295–308. [Google Scholar]










| Protonated Mass (m/z) | Species | Sensitivity (ncps/ppb) | Detection Limit for a 1-h Measurement (ppb) | |
|---|---|---|---|---|
| Water Dependency | Average | |||
| 31 | Formaldehyde | 4.6 − 0.11*[H2O] | 2.9 | 0.205 |
| 33 | Methanol | 5.9 + 0.7*[H2O] | 16.7 | 0.062 |
| 42 | Acetonitrile | 21.5 + 1.4*[H2O] | 43.6 | 0.002 |
| 45 | Acetaldehyde | 21.4 + 1.3*[H2O] | 41.7 | 0.018 |
| 59 | Acetone | 25.1 + 1.9*[H2O] | 54.5 | 0.013 |
| 63 | Dimethyl sulphide | Not determined | 36 | 0.010 |
| 69 | Isoprene | Not determined | 31.4 | 0.005 |
| 71 | Methacrolein | 25.9 + 1.9*[H2O] | 55.2 | 0.005 |
| 79 | Benzene | NA | 23.9 | 0.012 |
| 93 | Toluene | NA | 31.1 | 0.008 |
| 107 | m-xylene | NA | 34.9 | 0.016 |
| 121 | 1,3,5-trimethyl benzene | NA | 32.8 | 0.013 |
| 137 | α-pinene | NA | 15.7 | 0.016 |
| Parameters | Period | |||
|---|---|---|---|---|
| December 26th 08:00–18:59 | February 5th 12:00–17:59 | February 6th 13:00–18:59 | February 13th 14:00–17:59 | |
| Wind direction (°) | 159 ± 4 | 70 ± 20 | 49 ± 9 | 160 ± 10 |
| Wind speed (m s−1) | 5.0 ± 0.7 | 3.0 ± 0.6 | 5.6 ± 0.5 | 3.3 ± 0.6 |
| Temperature (°C) | 20.0 ± 0.5 | 23.4 ± 0.3 | 24.2 ± 0.6 | 23.1 ± 0.3 |
| Relative humidity (%) | 65 ± 3 | 76 ± 2 | 70 ± 3 | 68 ± 1 |
| Radon (mBq/m3) | 150 ± 20 | 120 ± 20 | 150 ± 30 | 150 ± 30 |
| Mass | Species | Number of Hours | Mean (ppb) | SD (ppb) | 1st Quartile (ppb) | Median (ppb) | 3rd Quartile (ppb) | Range Min–Max (ppb) |
|---|---|---|---|---|---|---|---|---|
| 31 | Formaldehyde | 1025 | 1.19 | 0.95 | 0.65 | 0.92 | 1.44 | 0.09–8.69 |
| 33 | Methanol | 1025 | 2.2 | 1.6 | 1.3 | 1.8 | 2.6 | 0.7–12.6 |
| 42 | Acetonitrile | 1018 | 0.076 | 0.023 | 0.061 | 0.071 | 0.085 | 0.037–0.217 |
| 45 | Acetaldehyde | 1025 | 0.41 | 0.31 | 0.19 | 0.32 | 0.54 | 0.06–2.44 |
| 59 | Acetone | 1025 | 0.71 | 0.46 | 0.41 | 0.59 | 0.84 | 0.19–3.95 |
| 69 | Isoprene | 1027 | 0.29 | 0.42 | 0.07 | 0.18 | 0.37 | 0.003–4.57 |
| 71 | MACR + MVK | 1025 | 0.20 | 0.35 | 0.05 | 0.09 | 0.20 | 0.006–4.31 |
| 79 | Benzene | 1027 | 0.113 | 0.094 | 0.049 | 0.087 | 0.149 | 0.004–0.816 |
| 93 | Toluene | 1027 | 0.31 | 0.34 | 0.09 | 0.20 | 0.37 | 0.004–2.67 |
| 107 | C8H10 | 1015 | 0.24 | 0.27 | 0.07 | 0.14 | 0.30 | 0.005–2.11 |
| 121 | C9H12 | 1027 | 0.15 | 0.15 | 0.06 | 0.10 | 0.17 | 0.007–1.71 |
| 137 | Monoterpenes | 1027 | 0.12 | 0.16 | 0.04 | 0.07 | 0.14 | 0.004–1.39 |
| n/a | Carbon monoxide | 1139 | 113 | 86 | 65 | 85 | 122 | 46–860 |
| n/a | Ozone | 1284 | 18.3 | 8.7 | 12.9 | 18.2 | 23.2 | 1.0–53.9 |
| n/a | NO | 1237 | 2.4 | 6.8 | 0.3 | 0.9 | 2.1 | 0–136.4 |
| n/a | NO2 | 1237 | 5.0 | 4.0 | 1.8 | 3.8 | 7.2 | 0.2–23.1 |
| n/a | NOx | 1237 | 7.5 | 9.3 | 2.6 | 4.9 | 9.1 | 0.2–156.2 |
| MUMBA 26 December 2012 (08:00 to 18:59) | Cape Grim Baseline 26 December 2012 | Cape Grim Baseline December 2012 | |
|---|---|---|---|
| CO2 (ppm) | 391.0 ± 0.6 | 391.09 | 391.16 ± 0.07 |
| CH4 (ppb) | 1760.1 ± 0.4 | 1760.1 | 1763.7 ± 3.4 |
| N2O (ppb) | 325.04 ± 0.08 | 324.96 | 325.1 ± 0.2 |
| CO (ppb) | 52.4 ± 1.7 | 45.9 | 48.6 ± 2.5 |
| O3 (ppb) | 20.5 ± 1.1 | 20 ± 4 * |
| Protonated Mass | Species | MUMBA 26 Dec 2012 | MUMBA 13 Feb 2013 | Cape Grim [35,36] (Southern Ocean) | Southern Hemisphere Mid-Latitude Oceans [68,70,74] | Tropical Oceans [67,71,73,75,76] |
|---|---|---|---|---|---|---|
| 31 | Formaldehyde | 590(80) | 600(130) | -- | -- | 211(144) |
| 33 | Methanol | 1340(170) | 1020(70) | 476(26)–633 | 546(139)–680(260) | 575(211)–890(400) |
| 42 | Acetonitrile | 56(5) | 65(4) | 25(1)–32 | 23(7)–50(10) | 110(20)–142(20) |
| 45 | Acetaldehyde | 190(40) | 120(30) | <4–53 | 290(100) | 178(30)–204(40) |
| 59 | Acetone | 260(30) | 270(10) | 61–118(5) | 127(38)–453(114) | 361(51)–530(200) |
| 63 | Dimethyl sulphide | 50(10) | 35(8) | ~80–95 | 77(34) | 50(50)–89(58) |
| 79 | Benzene | 20(10) | 28(5) | 4(1) | 10–80(40) | -- |
| 93 | Toluene | 30(20) | 45(3) | -- | 9 | 9(7) |
| 107 | C8H10 | 23(6) | 70(40) | -- | 10 | -- |
| 121 | C9H12 | 36(7) | 50(20) | -- | -- | -- |
| CO (ppb) | 52(2) | 58(6) | ~50 | 45(4)–54(4) | 64(6)–111(16) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guérette, É.-A.; Paton-Walsh, C.; Galbally, I.; Molloy, S.; Lawson, S.; Kubistin, D.; Buchholz, R.; Griffith, D.W.T.; Langenfelds, R.L.; Krummel, P.B.; et al. Composition of Clean Marine Air and Biogenic Influences on VOCs during the MUMBA Campaign. Atmosphere 2019, 10, 383. https://doi.org/10.3390/atmos10070383
Guérette É-A, Paton-Walsh C, Galbally I, Molloy S, Lawson S, Kubistin D, Buchholz R, Griffith DWT, Langenfelds RL, Krummel PB, et al. Composition of Clean Marine Air and Biogenic Influences on VOCs during the MUMBA Campaign. Atmosphere. 2019; 10(7):383. https://doi.org/10.3390/atmos10070383
Chicago/Turabian StyleGuérette, Élise-Andrée, Clare Paton-Walsh, Ian Galbally, Suzie Molloy, Sarah Lawson, Dagmar Kubistin, Rebecca Buchholz, David W.T. Griffith, Ray L. Langenfelds, Paul B. Krummel, and et al. 2019. "Composition of Clean Marine Air and Biogenic Influences on VOCs during the MUMBA Campaign" Atmosphere 10, no. 7: 383. https://doi.org/10.3390/atmos10070383