Atmospheric Dry Deposition of Water-Soluble Nitrogen to the Subarctic Western North Pacific Ocean during Summer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experiments
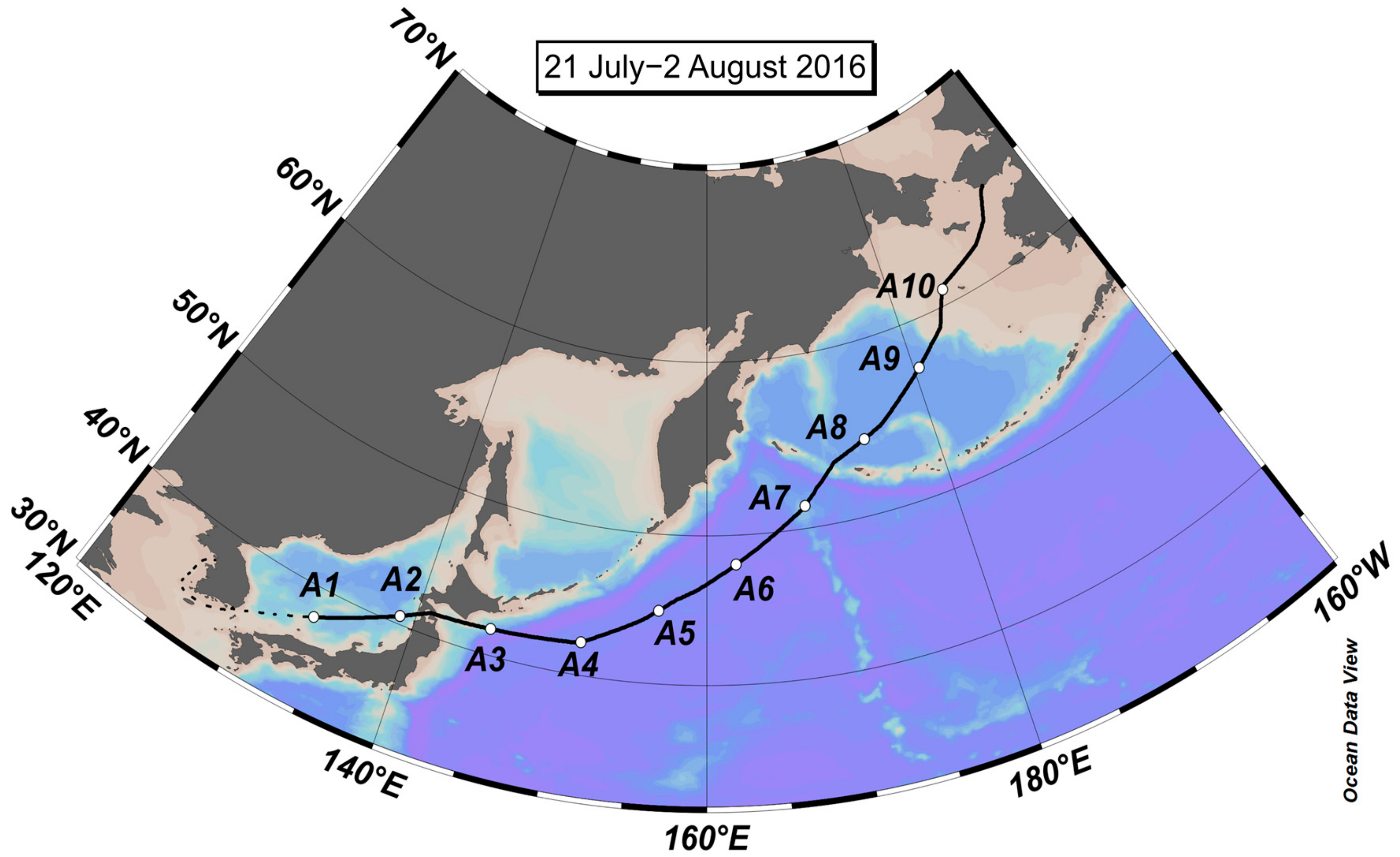
2.1. Aerosol Collection
2.2. Chemical Analysis
2.1.1. Ionic Species
2.1.2. Water-Soluble Total Nitrogen
2.3. Backward Trajectory Anzalysis
2.4. Dry Deposition Flux Estimate
3. Results and Discussion
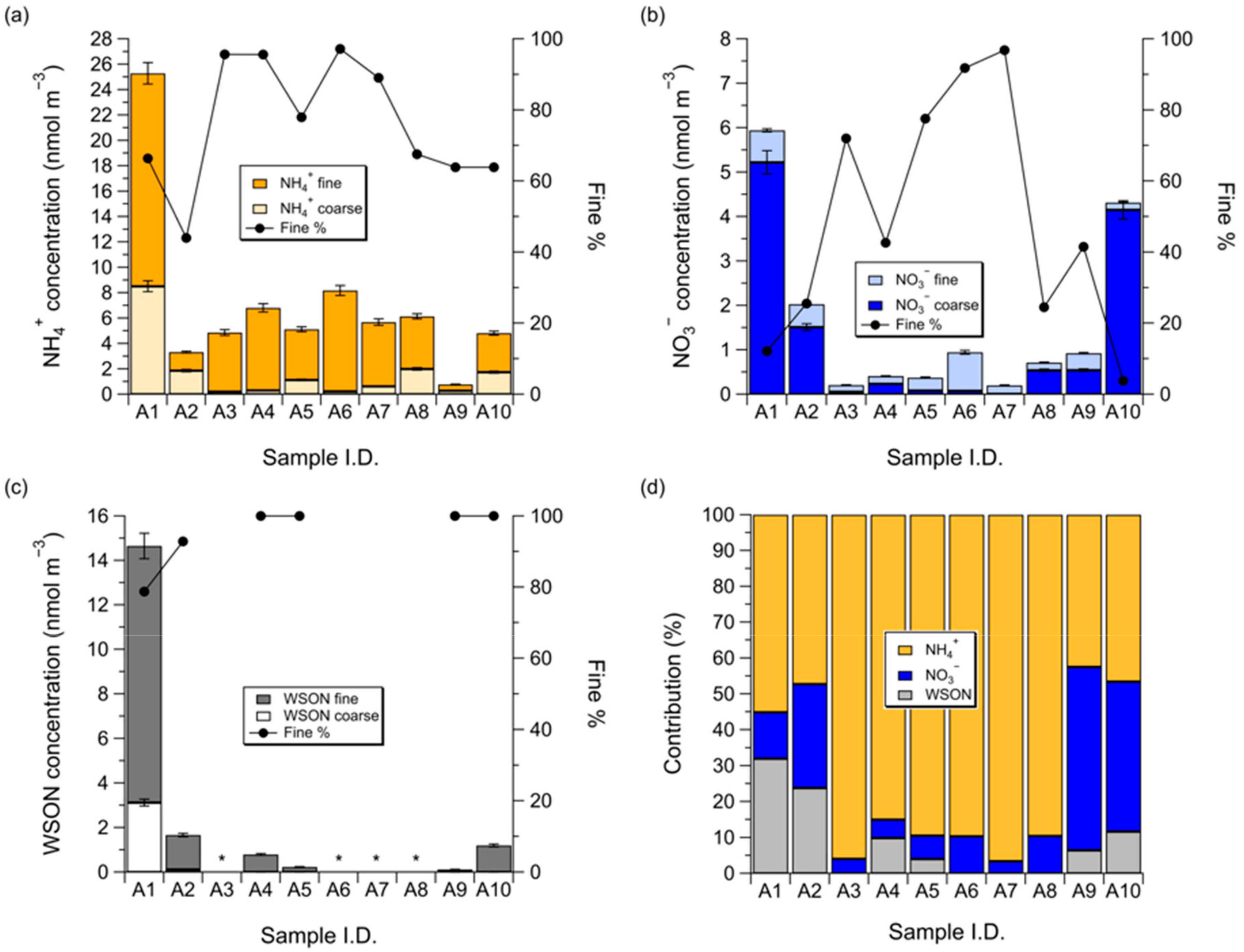
3.1. Concentrations of NH4+, NO3−, and WSON over the Subarctic Western North Pacific Ocean
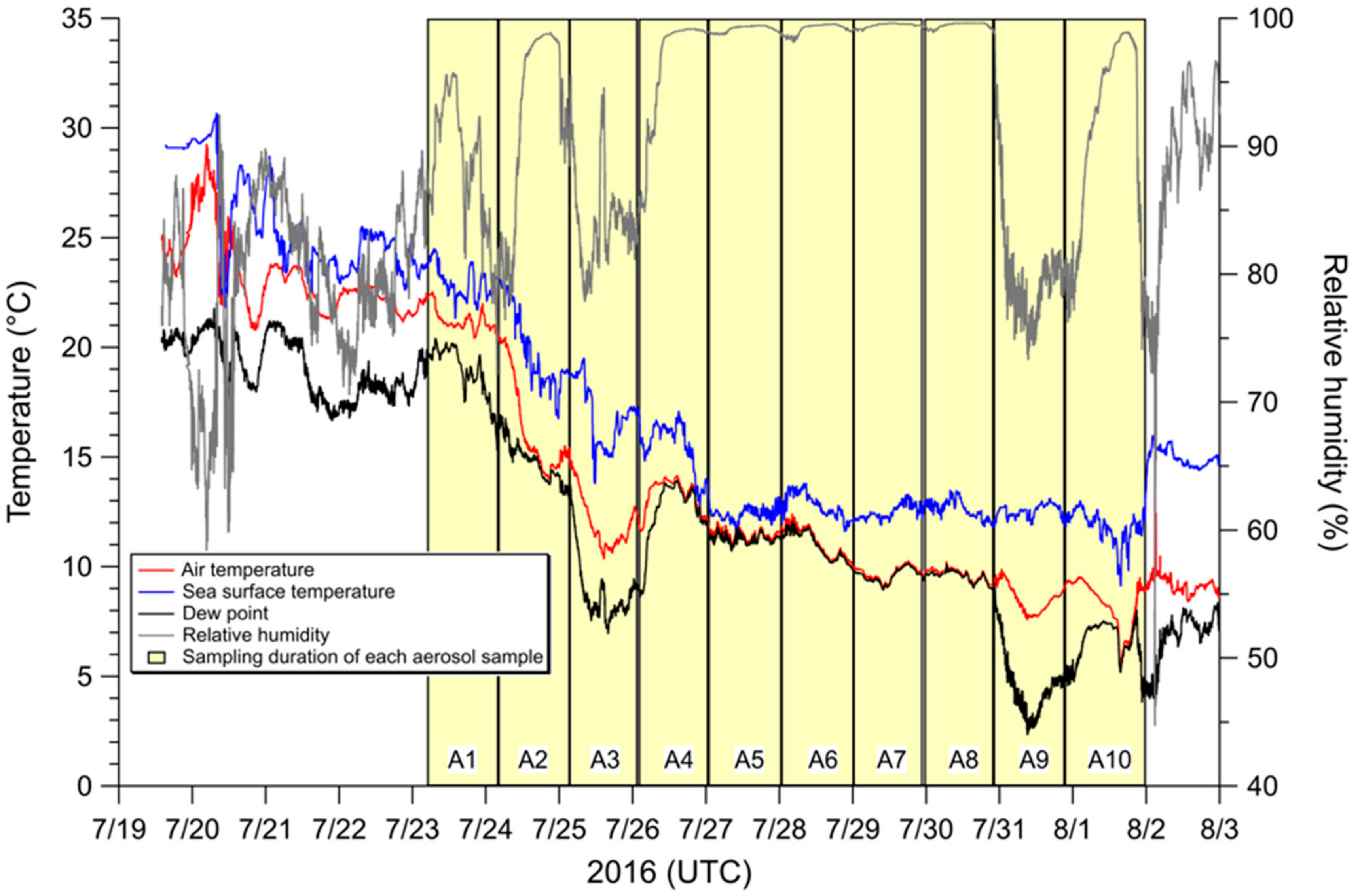
3.2. Influence of Sea Fog Events on Atmospheric Water-Soluble N Species
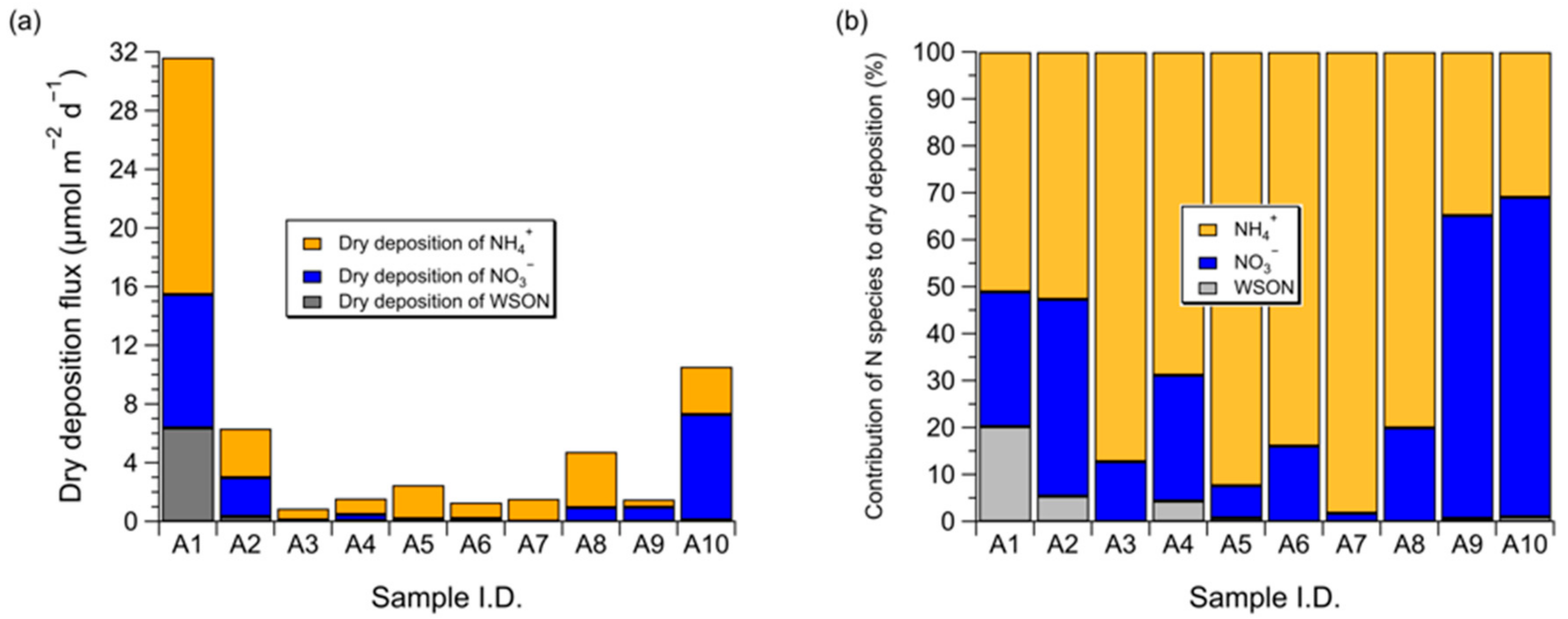
3.3. Dry Deposition Flux of Atmospheric Water-Soluble N to the Subarctic Western North Pacific Ocean
3.4. Potential Impact of Atmospheric Water-Soluble N on New Primary Production over the Subarctic Western North Pacific Ocean
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elser, J.J.; Bracken, M.E.S.; Cleland, E.E.; Gruner, D.S.; Harpole, W.S.; Hillebrand, H.; Ngai, J.T.; Seabloom, E.W.; Shurin, J.B.; Smith, J.E. Global analysis of nitrogen and phosphorus limitation of primary producers in freshwater, marine and terrestrial ecosystems. Ecol. Lett. 2007, 10, 1135–1142. [Google Scholar] [CrossRef]
- Uno, I.; Uematsu, M.; Hara, Y.; He, Y.J.; Ohara, T.; Mori, A.; Kamaya, T.; Murano, K.; Sadanaga, Y.; Bandow, H. Numerical study of the atmospheric input of anthropogenic total nitrate to the marginal seas in the western North Pacific region. Geophys. Res. Lett. 2007, 34, L17817–L17822. [Google Scholar] [CrossRef]
- Duce, R.A.; Liss, P.S.; Merrill, J.T.; Atlas, E.L.; Buat-Menard, P.; Hicks, B.B.; Miller, J.M.; Prospero, J.M.; Arimoto, R.; Church, T.M.; et al. The atmospheric input of trace species to the world ocean. Global Biogeochem. Cycles 1991, 5, 193–259. [Google Scholar] [CrossRef]
- Jickells, T. Atmospheric inputs of metals and nutrients to the oceans: Their magnitude and effects. Mar. Chem. 1995, 48, 199–214. [Google Scholar] [CrossRef]
- Duce, R.A.; LaRoche, J.; Altieri, K.; Arrigo, K.R.; Baker, A.R.; Capone, D.G.; Cornell, S.; Dentener, F.; Galloway, J.; Ganeshram, R.S.; et al. Impacts of atmospheric anthropogenic nitrogen on the open ocean. Science 2008, 320, 893–897. [Google Scholar] [CrossRef]
- Kim, I.N.; Lee, K.; Gruber, N.; Karl, D.M.; Bullister, J.L.; Yang, S.; Kim, T.W. Increasing anthropogenic nitrogen in the North Pacific Ocean. Science 2014, 346, 1102–1106. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, K.; Najjar, R.G.; Jeong, H.D.; Jeong, H.J. Increasing N abundance in the Northwestern Pacific Ocean due to atmospheric nitrogen deposition. Science 2011, 334, 505–509. [Google Scholar] [CrossRef]
- Doney, S.C.; Mahowald, N.; Lima, I.; Feely, R.A.; Mackenzie, F.T.; Lamarque, J.-F.; Rasch, P.J. Impact of anthropogenic atmospheric nitrogen and sulfur deposition on ocean acidification and the inorganic carbon system. Proc. Natl. Acad. Sci. USA 2007, 104, 14580–14585. [Google Scholar] [CrossRef]
- Baker, A.R.; Lesworth, T.; Adams, C.; Jickells, T.D.; Ganzeveld, L. Estimation of atmospheric nutrient inputs to the Atlantic Ocean from 50° N to 50° S based on large-scale field sampling: Fixed nitrogen and dry deposition of phosphorus. Glob. Biogeochem. Cycles 2010, 24, GB3006–GB3021. [Google Scholar] [CrossRef]
- Dentener, F.; Drevet, J.; Lamarque, J.F.; Bey, I.; Eickhout, B.; Fiore, A.M.; Hauglustaine, D.; Horowitz, L.W.; Krol, M.; Kulshrestha, U.C.; et al. Nitrogen and sulfur deposition on regional and global scales: A multimodel evaluation. Glob. Biogeochem. Cycles 2006, 20, GB4003–GB4023. [Google Scholar] [CrossRef]
- Okin, G.S.; Baker, A.R.; Tegen, I.; Mahowald, N.M.; Dentener, F.J.; Duce, R.A.; Galloway, J.N.; Hunter, K.; Kanakidou, M.; Kubilay, N.; et al. Impacts of atmospheric nutrient deposition on marine productivity: Roles of nitrogen, phosphorus, and iron. Glob. Biogeochem. Cycles 2011, 25, GB2022–GB2031. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, L.; Pan, Y.; Wang, Y.; Paulot, F.; Henze, D.K. Atmospheric nitrogen deposition to the northwestern Pacific: Seasonal variation and source attribution. Atmos. Chem. Phys. 2015, 15, 10905–10924. [Google Scholar] [CrossRef]
- Nakamura, T.; Matsumoto, K.; Uematsu, M. Chemical characteristics of aerosols transported from Asia to the East China Sea: An evaluation of anthropogenic combined nitrogen deposition in autumn. Atmos. Environ. 2005, 39, 1749–1758. [Google Scholar] [CrossRef]
- Matsumoto, K.; Minami, H.; Uyama, Y.; Uematsu, M. Size partitioning of particulate inorganic nitrogen species between the fine and coarse mode ranges and its implication to their deposition on the surface ocean. Atmos. Environ. 2009, 43, 4259–4265. [Google Scholar] [CrossRef]
- Jung, J.; Furutani, H.; Uematsu, M. Atmospheric inorganic nitrogen in marine aerosol and precipitation and its deposition to the North and South Pacific Oceans. J. Atmos. Chem. 2012, 68, 157–181. [Google Scholar] [CrossRef]
- Jung, J.; Furutani, H.; Uematsu, M.; Kim, S.; Yoon, S. Atmospheric inorganic nitrogen input via dry, wet, and sea fog deposition to the subarctic western North Pacific Ocean. Atmos. Chem. Phys. 2013, 13, 411–428. [Google Scholar] [CrossRef]
- Luo, L.; Kao, S.-J.; Bao, H.; Xiao, H.; Xiao, H.; Yao, X.; Gao, H.; Li, J.; Lu, Y. Sources of reactive nitrogen in marine aerosol over the Northwest Pacific Ocean in spring. Atmos. Chem. Phys. 2018, 18, 6207–6222. [Google Scholar] [CrossRef]
- Prospero, J.M.; Barrett, K.; Church, T.; Dentener, F.; Duce, R.A.; Galloway, J.N.; Levy II, H.; Moody, J.; Quinn, P. Atmospheric deposition of nutrients to the North Atlantic Basin. Biogeochemistry 1996, 35, 27–73. [Google Scholar] [CrossRef]
- Spokes, L.J.; Yeatman, S.G.; Cornell, S.E.; Jickells, T.D. Nitrogen deposition to the eastern Atlantic Ocean. The importance of south-easterly flow. Tellus Ser. B 2000, 52, 37–49. [Google Scholar] [CrossRef]
- Baker, A.R.; Kelly, S.D.; Biswas, K.F.; Witt, M.; Jickells, T.D. Atmospheric deposition of nutrients to the Atlantic Ocean. Geophys. Res. Lett. 2003, 30, 2296–2299. [Google Scholar] [CrossRef]
- Baker, A.R.; Weston, K.; Kelly, S.D.; Voss, M.; Streu, P.; Cape, J.N. Dry and wet deposition of nutrients from the tropical Atlantic atmosphere: Links to primary productivity and nitrogen fixation. Deep Sea Res. Part I 2007, 54, 1704–1720. [Google Scholar] [CrossRef]
- Sandroni, V.; Raimbault, P.; Migon, C.; Garcia, N.; Gouze, E. Dry atmospheric deposition and diazotrophy as sources of new nitrogen to northwestern Mediterranean oligotrophic surface waters. Deep Sea Res. Part I 2007, 54, 1859–1870. [Google Scholar] [CrossRef]
- Loughner, C.P.; Tzortziou, M.; Shroder, S.; Pickering, K.E. Enhanced dry deposition of nitrogen pollution near coastlines: A case study covering the Chesapeake Bay estuary and Atlantic Ocean coastline. J. Geophys. Res. Atmos. 2016, 121, 14221–14238. [Google Scholar] [CrossRef]
- St-Laurent, P.; Friedrichs, M.A.M.; Najjar, R.G.; Martins, D.K.; Herrmann, M.; Miller, S.K.; Wilkin, J. Impacts of atmospheric nitrogen deposition on surface waters of the western North Atlantic mitigated by multiple feedbacks. J. Geophys. Res. Oceans 2017, 122, 8406–8426. [Google Scholar] [CrossRef]
- Gao, Y.; Arimoto, R.; Zhou, M.Y.; Merrill, J.T.; Duce, R.A. Relationships between the dust concentrations over eastern Asia and the remote North Pacific. J. Geophys. Res. 1992, 97, 9867–9872. [Google Scholar] [CrossRef]
- Uematsu, M.; Hattori, H.; Nakamura, T.; Narita, Y.; Jung, J.; Matsumoto, K.; Nakaguchi, Y.; Kumar, M.D. Atmospheric transport and deposition of anthropogenic substances from the Asia to the East China Sea. Mar. Chem. 2010, 120, 108–115. [Google Scholar] [CrossRef]
- Akimoto, H. Global Air Quality and Pollution. Science 2003, 302, 1716–1719. [Google Scholar] [CrossRef]
- Galloway, J.N.; Townsend, A.R.; Erisman, J.W.; Bekunda, M.; Cai, Z.; Freney, J.R.; Martinelli, L.A.; Seitzinger, S.P.; Sutton, M.A. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 2008, 320, 889–892. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Huang, S.Z. Effects of atmospheric dry deposition on external nitrogen supply and new production in the northern South China Sea. Atmosphere 2018, 9, 386. [Google Scholar] [CrossRef]
- Cornell, S.E.; Jickells, T.D.; Cape, J.N.; Rowland, A.P.; Duce, R.A. Organic nitrogen deposition on land and coastal environments: A review of methods and data. Atmos. Environ. 2003, 37, 2173–2191. [Google Scholar] [CrossRef]
- Aneja, V.P.; Roelle, P.A.; Murray, G.C.; Southerland, J.; Erisman, J.W.; Fowler, D.; Asman, W.; Patni, N. Atmospheric nitrogen compounds II: Emissions, transport, transformation, deposition and assessment. Atmos. Environ. 2001, 35, 1903–1911. [Google Scholar] [CrossRef]
- Andreae, M.O.; Merlet, P. Emission of trace gases and aerosols from biomass burning. Glob. Biogeochem. Cycles 2001, 15, 955–966. [Google Scholar] [CrossRef]
- Jickells, T.D.; Kelly, S.D.; Baker, A.R.; Biswas, K.; Dennis, P.F.; Spokes, L.J.; Witt, M.; Yeatman, S.G. Isotopic evidence for a marine ammonia source. Geophys. Res. Lett. 2003, 30, 27-1–27-4. [Google Scholar] [CrossRef]
- Andreae, M.O.; Crutzen, P.J. Atmospheric aerosols: Biogeochemical sources and role in atmospheric chemistry. Science 1997, 276, 1052–1058. [Google Scholar] [CrossRef]
- Cape, J.N.; Cornell, S.E.; Jickells, T.D.; Nemitz, E. Organic nitrogen in the atmosphere—Where does it come from? A review of sources and methods. Atmos. Res. 2011, 102, 30–48. [Google Scholar] [CrossRef]
- Facchini, M.C.; Decesari, S.; Rinaldi, M.; Carbone, C.; Finessi, E.; Mircea, M.; Fuzzi, S.; Moretti, F.; Tagliavini, E.; Ceburnis, D.; et al. Important Source of Marine Secondary Organic Aerosol from Biogenic Amines. Environ. Sci. Technol. 2008, 42, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Kawamura, K.; Sawano, M. Size distributions of organic nitrogen and carbon in remote marine aerosols: Evidence of marine biological origin based on their isotopic ratios. Geophys. Res. Lett. 2010, 37, L06803–L06807. [Google Scholar] [CrossRef]
- Cornell, S.E. Atmospheric nitrogen deposition: Revisiting the question of the importance of the organic component. Environ. Pollut. 2011, 159, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ogawa, H.; Maripi, D.K.; Uematsu, M. Contribution of water soluble organic nitrogen to total nitrogen in marine aerosols over the East China Sea and western North Pacific. Atmos. Environ. 2006, 40, 7259–7264. [Google Scholar] [CrossRef]
- Zhang, Q.; Anastasio, C.; Jimenez-Cruz, M. Water-soluble organic nitrogen in atmospheric fine particles (PM2.5) from northern California. J. Geophys. Res. 2002, 107, AAC 3-1–AAC 3-9. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yamamoto, Y.; Nishizawa, K.; Kaneyasu, N.; Irino, T.; Yoshikawa-Inoue, H. Origin of the water-soluble organic nitrogen in the maritime aerosol. Atmos. Environ. 2017, 167, 97–103. [Google Scholar] [CrossRef]
- Jung, J.; Hong, S.-B.; Chen, M.; Hur, J.; Jiao, L.; Lee, Y.; Park, K.; Hahm, D.; Choi, J.-O.; Yang, E.J.; et al. Characteristics of biogenically-derived aerosols over the Amundsen Sea, Antarctica. Atmos. Chem. Phys. Discuss. 2019, 3. [Google Scholar] [CrossRef]
- Jung, J.; Furutani, H.; Uematsu, M.; Park, J. Distributions of atmospheric non-sea-salt sulfate and methanesulfonic acid over the Pacific Ocean between 48° N and 55° S during summer. Atmos. Environ. 2014, 99, 374–384. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Kawamura, K.; Jung, J.; Furutani, H.; Uematsu, M. Latitudinal distributions of organic nitrogen and organic carbon in marine aerosols over the western North Pacific. Atmos. Chem. Phys. 2011, 11, 3037–3049. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Fu, P.; Ono, K.; Tachibana, E.; Kawamura, K. Seasonal cycles of water-soluble organic nitrogen aerosols in a deciduous broadleaf forest in northern Japan. J. Geophys. Res. Atmos. 2014, 119, 1440–1454. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT atmospheric transport and dispersion modeling system. Bull. Amer. Meteor. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, J.; Liu, S. Characterization of nutrients in the atmospheric wet and dry deposition observed at the two monitoring sites over Yellow Sea and East China Sea. J. Atmos. Chem. 2007, 57, 41–57. [Google Scholar] [CrossRef]
- Altieri, K.E.; Hastings, M.G.; Gobel, A.R.; Peters, A.J.; Sigman, D.M. Isotopic composition of rainwater nitrate at Bermuda: The influence of air mass source and chemistry in the marine boundary layer. J. Geophys. Res. Atmos. 2013, 118, 11304–11316. [Google Scholar] [CrossRef]
- Lewis, J.M.; Koračin, D.; Redmond, K.T. Sea fog research in the United Kingdom and United States: A historical essay including outlook. Bull. Amer. Meteor. Soc. 2004, 85, 395–408. [Google Scholar] [CrossRef]
- Gultepe, I.; Tardif, R.; Michaelides, S.C.; Cermak, J.; Bott, A.; Bendix, J.; Müller, M.D.; Pagowski, M.; Hansen, B.; Ellrod, G.; et al. Fog Research: A review of past achievements and future perspectives. Pure Appl. Geophys. 2007, 164, 1121–1159. [Google Scholar] [CrossRef]
- Wang, B.-H. Distributions and variations of sea fog in the world. In Sea Fog; China Ocean Press: Beijing, China, 1985; pp. 51–90. [Google Scholar]
- Sasakawa, M.; Ooki, A.; Uematsu, M. Aerosol size distribution during sea fog and its scavenge process of chemical substances over the northwestern North Pacific. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
- Jordan, C.E.; Talbot, R.W. Direct atmospheric deposition of water-soluble nitrogen to the Gulf of Maine. Glob. Biogeochem. Cycles 2000, 14, 1315–1329. [Google Scholar] [CrossRef]
- Matsumoto, K.; Uematsu, M. Free amino acids in marine aerosols over the western North Pacific Ocean. Atmos. Environ. 2005, 39, 2163–2170. [Google Scholar] [CrossRef]
- Neff, J.C.; Holland, E.A.; Dentener, F.J.; McDowell, W.H.; Russell, K.M. The origin, composition and rates of organic nitrogen deposition: A missing piece of the nitrogen cycle? Biogeochemistry 2002, 57, 99–136. [Google Scholar] [CrossRef]
- Cornell, S.; Mace, K.; Coeppicus, S.; Duce, R.; Huebert, B.; Jickells, T.; Zhuang, L.-Z. Organic nitrogen in Hawaiian rain and aerosol. J. Geophys. Res. 2001, 106, 7973–7983. [Google Scholar] [CrossRef]
- Zhang, Q.; Anastasio, C. Conversion of fogwater and aerosol organic nitrogen to ammonium, nitrate, and NOx during exposure to simulated sunlight and ozone. Environ. Sci. Technol. 2003, 37, 3522–3530. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Yao, X.H.; Gao, H.W.; Hsu, S.C.; Li, J.W.; Kao, S.J. Nitrogen speciation in various types of aerosols in spring over the northwestern Pacific Ocean. Atmos. Chem. Phys. 2016, 16, 325–341. [Google Scholar] [CrossRef]
- Adams, P.J.; Seinfeld, J.H.; Koch, D.M. Global concentrations of tropospheric sulfate, nitrate, and ammonium aerosol simulated in a general circulation model. J. Geophys. Res. D Atmos. 1999, 104, 13791–13823. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, G.S.; Bi, Y.F.; Liu, S.M. Nitrogen species in rainwater and aerosols of the Yellow and East China seas: Effects of the East Asian monsoon and anthropogenic emissions and relevance for the NW Pacific Ocean. Glob. Biogeochem. Cycles 2011, 25, GB3020–GB3033. [Google Scholar] [CrossRef]
- Kundu, S.; Kawamura, K.; Lee, M. Seasonal variation of the concentrations of nitrogenous species and their nitrogen isotopic ratios in aerosols at Gosan, Jeju Island: Implications for atmospheric processing and source changes of aerosols. J. Geophys. Res. 2010, 115, D20305–D20323. [Google Scholar] [CrossRef]
- Redfield, A.C. The biological control of chemical factors in the environment. Am. Sci. 1958, 46, 205–221. [Google Scholar]
- Baker, A.R.; Jickells, T.D.; Biswas, K.F.; Weston, K.; French, M. Nutrients in atmospheric aerosol particles along the Atlantic Meridional Transect. Deep Sea Res. Part II 2006, 53, 1706–1719. [Google Scholar] [CrossRef]
- Harrison, P.J.; Boyd, P.W.; Varela, D.E.; Takeda, S.; Shiomoto, A.; Odate, T. Comparison of factors controlling phytoplankton productivity in the NE and NW subarctic Pacific gyres. Prog. Oceanogr. 1999, 43, 205–234. [Google Scholar] [CrossRef]
- Nishioka, J.; Ono, T.; Saito, H.; Sakaoka, K.; Yoshimura, T. Oceanic iron supply mechanisms which support the spring diatom bloom in the Oyashio region, western subarctic Pacific. J. Geophys. Res. Oceans 2011, 116, C02021–C02037. [Google Scholar]
- Nishioka, J.; Ono, T.; Saito, H.; Nakatsuka, T.; Takeda, S.; Yoshimura, T.; Suzuki, K.; Kuma, K.; Nakabayashi, S.; Tsumune, D.; et al. Iron supply to the western subarctic Pacific: Importance of iron export from the Sea of Okhotsk. J. Geophys. Res. 2007, 112, C10012–C10026. [Google Scholar] [CrossRef]
- Lam, P.J.; Bishop, J.K.B. The continental margin is a key source of iron to the HNLC North Pacific Ocean. Geophys. Res. Lett. 2008, 35, L07608–L07612. [Google Scholar] [CrossRef]
- Duce, R.A.; Tindale, N.W. Atmospheric transport of iron and its deposition in the ocean. Limnol. Oceanogr. 1991, 36, 1715–1726. [Google Scholar] [CrossRef]
- Moore, J.K.; Doney, S.C.; Glover, D.M.; Fung, I.Y. Iron cycling and nutrient-limitation patterns in surface waters of the World Ocean. Deep Sea Res. Part II 2002, 49, 463–507. [Google Scholar] [CrossRef]
- Mahowald, N.M.; Baker, A.R.; Bergametti, G.; Brooks, N.; Duce, R.A.; Jickells, T.D.; Kubilay, N.; Prospero, J.M.; Tegen, I. Atmospheric global dust cycle and iron inputs to the ocean. Glob. Biogeochem. Cycles 2005, 19, GB4025–GB4039. [Google Scholar] [CrossRef]
- Yoon, J.-E.; Kim, K.; Macdonald, A.M.; Park, K.-T.; Kim, H.-C.; Yoo, K.-C.; Yoon, H.-I.; Yang, E.J.; Jung, J.; Lim, J.-H.; et al. Spatial and temporal variabilities of spring Asian dust events and their impacts on chlorophyll-a concentrations in the western North Pacific Ocean. Geophys. Res. Lett. 2017, 44, 1474–1482. [Google Scholar] [CrossRef]
- Matsumoto, K.; Honda, M.C.; Sasaoka, K.; Wakita, M.; Kawakami, H.; Watanabe, S. Seasonal variability of primary production and phytoplankton biomass in the western Pacific subarctic gyre: Control by light availability within the mixed layer. J. Geophys. Res. Oceans 2014, 119, 6523–6534. [Google Scholar] [CrossRef]
- Glibert, P.M.; Garside, C.; Fuhrman, J.A.; Roman, M.R. Time-dependent coupling of inorganic and organic nitrogen uptake and regeneration in the plume of the Chesapeake Bay estuary and its regulation by large heterotrophs. Limnol. Oceanogr. 1991, 36, 895–909. [Google Scholar] [CrossRef]
- Bronk, D.A.; See, J.H.; Bradley, P.; Killberg, L. DON as a source of bioavailable nitrogen for phytoplankton. Biogeosciences 2007, 4, 283–296. [Google Scholar] [CrossRef]
- Wedyan, M.A.; Fandi, K.G.; Al-Rousan, S. Bioavailability of atmospheric dissolved organic nitrogen in the marine aerosol over the Gulf of Aqaba. Aust. J. Basic Appl. Sci. 2007, 1, 208–212. [Google Scholar]
- Wong, C.S.; Waser, N.; Nojiri, Y.; Whitney, F.A.; Page, J.S.; Zeng, J. Seasonal cycles of nutrients and dissolved inorganic carbon at high and mid latitudes in the North Pacific Ocean during the Skaugran cruises: Determination of new production and nutrient uptake ratios. Deep Sea Res. Part II 2002, 49, 5317–5338. [Google Scholar] [CrossRef]
- Han, Y.; Huiwang, G.; Xiaohong, Y.; Zifa, W. Simulating dry deposition fluxes of PM10 and particulate inorganic nitrogen over the eastern China seas during a severe Asian dust event using WRF-Chem model. J. Ocean Univ. China 2012, 11, 301–314. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, J.; Han, B.; Rodriguez, B.; Miyazaki, Y.; Chung, H.Y.; Kim, K.; Choi, J.-O.; Park, K.; Kim, I.-N.; Kim, S.; et al. Atmospheric Dry Deposition of Water-Soluble Nitrogen to the Subarctic Western North Pacific Ocean during Summer. Atmosphere 2019, 10, 351. https://doi.org/10.3390/atmos10070351
Jung J, Han B, Rodriguez B, Miyazaki Y, Chung HY, Kim K, Choi J-O, Park K, Kim I-N, Kim S, et al. Atmospheric Dry Deposition of Water-Soluble Nitrogen to the Subarctic Western North Pacific Ocean during Summer. Atmosphere. 2019; 10(7):351. https://doi.org/10.3390/atmos10070351
Chicago/Turabian StyleJung, Jinyoung, Byeol Han, Blanca Rodriguez, Yuzo Miyazaki, Hyun Young Chung, Kitae Kim, Jung-Ok Choi, Keyhong Park, II-Nam Kim, Saewung Kim, and et al. 2019. "Atmospheric Dry Deposition of Water-Soluble Nitrogen to the Subarctic Western North Pacific Ocean during Summer" Atmosphere 10, no. 7: 351. https://doi.org/10.3390/atmos10070351
APA StyleJung, J., Han, B., Rodriguez, B., Miyazaki, Y., Chung, H. Y., Kim, K., Choi, J.-O., Park, K., Kim, I.-N., Kim, S., Yang, E. J., & Kang, S.-H. (2019). Atmospheric Dry Deposition of Water-Soluble Nitrogen to the Subarctic Western North Pacific Ocean during Summer. Atmosphere, 10(7), 351. https://doi.org/10.3390/atmos10070351