APC and MUTYH Analysis in FAP Patients: A Novel Mutation in APC Gene and Genotype-Phenotype Correlation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Mutation Analysis
2.3. In Silico Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Galiatsatos, P.; Foulkes, W.D. Familial adenomatous polyposis. Am. J. Gastroenterol. 2006, 101, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, M.H.; Vasen, H.F.A. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): A review of the literature. Crit. Rev. Oncol./Hematol. 2007, 61, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Half, E.; Bercovich, D.; Rozen, P. Familial adenomatous polyposis. Orphanet. J. Rare Dis. 2009, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Leoz, M.L.; Carballal, S.; Moreira, L.; Ocaña, T.; Balaguer, F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl. Clin. Genet. 2015, 8, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.; Findeis, S.; Lee, M.J. Familial adenomatous polyposis. J. Pediatr. Genet. 2016, 5, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, F.; Viel, A.; Bignamia, M. Role of MUTYH in human cancer. Mutat. Res. 2013, 743–744, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Lipton, L.; Tomlinson, I. The genetics of FAP and FAP-like syndromes. Fam. Cancer 2006, 5, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Talseth-Palmer, B.A.; Wijnen, J.T.; Andreassen, E.K.; Barker, D.; Jagmohan-Changur, S.; Tops, C.M.; Meldrum, C.; Dutch Cancer Genetics Group; Spigelman, A.; Hes, F.J.; et al. The importance of a large sample cohort for studies on modifier genes influencing disease severity in FAP patients. Hered. Cancer Clin. Pract. 2013, 11, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UMD- APC Mutations Database. Available online: http://www.umd.be/APC/ (accessed on 12 December 2017).
- The UMD- MUTYH Mutations Database. Available online: http://www.umd.be/MUTYH/ (accessed on 12 December 2017).
- Rouleau, E.; Zattara, H.; Lefol, C.; Noguchi, T.; Briaux, A.; Buecher, B.; Bourdon, V.; Sobol, H.; Lidereau, R.; Olschwang, S. First large rearrangement in the MUTYH gene and attenuated familial adenomatous polyposis syndrome. Clin. Genet. 2011, 80, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.E.; Thomas, C.B.; Thibodeau, S.N.; Ferber, M.J.; Halling, K.C. APC germline mutations in individuals being evaluated for familial adenomatous polyposis: A review of the Mayo Clinic experience with 1591 consecutive tests. J. Mol. Diagn. 2013, 15, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Heinen, C.D. Genotype to phenotype: Analyzing the effects of inherited mutations in colorectal cancer families. Mutat. Res. 2010, 693, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrezan, G.T.; da Silva, F.C.; Santos, E.M.; Krepischi, A.C.V.; Waddington Achatz, M.I.; Aguiar Junior, S.; Rossi, B.M.; Carraro, D.M. Mutational spectrum of the APC and MUTYH genes and genotype-phenotype correlations in Brazilian FAP, AFAP, and MAP patients. Orphanet. J. Rare Dis. 2013, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Dodaro, C.; Grifasi, C.; Florio, J.; Santangelo, M.L.; Duraturo, F.; De Rosa, M.; Izzo, P.; Renda, A. The role of mutation analysis of the APC gene in the management of FAP patients. A controversial issue. Ann. Ital. Chir. 2016, 87, 321–325. [Google Scholar] [PubMed]
- Nielsen, M.; Joerink-van de Beld, M.C.; Jones, N.; Vogt, S.; Tops, C.M.; Vasen, H.F.A.; Sampson, J.R.; Arets, S.; Hes, F.J. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH-associated polyposis. Gastroenterology 2009, 136, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Vogt, S.; Uhlhaas, S.; Stienen, D.; Kurth, I.; Hameister, H.; Mangold, E.; Kötting, J.; Kaminsky, E.; Propping, P.; et al. Analysis of rare APC variants at the mRNA level: Six pathogenic mutations and literature review. J. Mol. Diagn. 2009, 11, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Selvaggi, F.; De Paola, M.L.; Sciaudone, G.; Guadagni, I.; Parisi, M.; Pellino, G.; Molinari, A.M.; Cioffi, M. A novel frameshift mutation in exon 12 of the adenomatous polyposis coli gene in an Italian family with familial adenomatous polyposis and desmoid tumour. J. Mol. Genet. Med. 2010, 4, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Görgens, H.; Krüger, S.; Kuhlisch, E.; Pagenstecher, C.; Höhl, R.; Schackert, H.K.; Müller, A. Microsatellite stable colorectal cancers in clinically suspected hereditary nonpolyposis colorectal cancer patients without vertical transmission of disease are unlikely to be caused by biallelic germline mutations in MYH. J. Mol. Diagn. 2006, 8, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Human Genome Variation Society (HGVS). Available online: http://www.hgvs.org (accessed on 12 December 2017).
- Den Dunnen, J.T. Sequence variant descriptions: HGVS nomenclature and Mutalyzer. Curr. Protoc. Hum. Genet. 2016, 90, 7.13.1–7.13.19. [Google Scholar] [CrossRef] [PubMed]
- Thery, J.C.; Krieger, S.; Gaildrat, P.; Revillion, F.; Buisine, M.P.; Killian, A.; Duponchel, C.; Rousselin, A.; Vaur, D.; Peyrat, J.P.; et al. Contribution of bioinformatics predictions and functional splicing assays to the interpretation of unclassified variants of the BRCA genes. Eur. J. Hum. Genet. 2011, 19, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Garre, P.; Briceño, V.; Xicola, R.M.; Doyle, B.J.; de la Hoya, M.; Sanz, J.; Llovet, P.; Pescador, P.; Puente, J.; Díaz-Rubio, E.; et al. Analysis of the oxidative damage repair genes NUDT1, OGG1, and MUTYH in patients from mismatch repair proficient HNPCC families (MSS-HNPCC). Clin. Cancer Res. 2011, 17, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Wachsmannova-Matelova, L.; Stevurkova, V.; Adamcikova, Z.; Holec, V.; Zajac, V. Different phenotype manifestation of familial adenomatous polyposis in families with APC mutation at codon 1309. Neoplasma 2009, 56, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.F.; Mallinson, E.K.L.; Bowen, J.; Lalloo, F.; Clancy, T.; Hill, J.; Evans, D.G.R. Genotype-phenotype correlation in colorectal polyposis. Clin. Genet. 2012, 81, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Konishi, M.; Iwama, T.; Utsunomiya, J.; Sugihara, K.I.; Miyaki, M. The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. Jpn. J. Clin. Oncol. 2000, 30, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.; Roos, A.; Muntinghe, F.L.; Enting, R.H.; de Vries, J.; Kleibeuker, J.H.; Witjes, M.J.H.; Links, T.P.; van Beek, A.P. Extra-intestinal manifestations of familial adenomatous polyposis. Ann. Surg. Oncol. 2008, 15, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Tekkis, P.P.; Neale, K.F.; Phillips, R.K.S.; Clark, S.K. Risk factors predicting intraabdominal desmoids in familial adenomatous polyposis: A single centre experience. Tech. Coloproctol. 2010, 14, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Sturt, N.J.H.; Gallagher, M.C.; Bassett, P.; Philp, C.R.; Neale, K.F.; Tomlinson, I.P.M.; Silver, A.R.J.; Phillips, R.K.S. Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut 2004, 53, 1832–1836. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, M.H.; De Vos Tot Nederveen Cappel, W.; Botma, A.; Nagengast, F.M.; Kleibeuker, J.H.; Mathus-Vliegen, E.M.; Dekker, E.; Dees, J.; Wijnen, J.; Vasen, H.F. Desmoid tumors in a Dutch cohort of patients with familial adenomatous polyposis. Clin. Gastroenterol. Hepatol. 2008, 6, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.T.; Miccoli, S.; Turchetti, D.; Bondavalli, D.; Viel, A.; Quaia, M.; Giacomini, E.; Gismondi, V.; Sanchez-Mete, L.; Stigliano, V.; et al. Type and frequency of MUTYH variants in Italian patients with suspected MAP: A retrospective multicenter study. J. Hum. Genet. 2017, 62, 309–315. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, V.G.; Minoprio, A.; Torreri, P.; Marinoni, I.; Bossa, C.; Petrucci, T.C.; Albertini, A.M.; Ranzani, G.N.; Bignami, M.; Mazzei, F. Functional analysis of MUTYH mutated proteins associated with familial adenomatous polyposis. DNA Repair 2010, 9, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.; Jones, N.; Christian, D.; Engel, C.; Nielsen, M.; Kaufmann, A.; Steinke, V.; Vasen, H.F.; Propping, P.; Sampson, J.R.; et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009, 137, 1976–1985. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Kim, H.; Cleary, S.; Cupples, C.; Gallinger, S.; Bristow, R. Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology 2008, 135, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Talseth-Palmer, B.A. The genetic basis of colonic adenomatous polyposis syndromes. Hered. Cancer Clin. Pract. 2017, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Lipsa, A.; Arunachal, G.; Ramadwar, M.; Sarin, R. Novel mutations and phenotypic associations identified through APC, MUTYH, NTHL1, POLD1, POLE gene analysis in Indian Familial Adenomatous Polyposis cohort. Sci. Rep. 2017, 7, 2214. [Google Scholar] [CrossRef] [PubMed]
- Spier, I.; Kerick, M.; Drichel, D.; Horpaopan, S.; Altmüller, J.; Laner, A.; Holzapfel, S.; Peters, S.; Adam, R.; Zhao, B.; et al. Exome sequencing identifies potential novel candidate genes in patients with unexplained colorectal adenomatous polyposis. Fam. Cancer 2016, 15, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Möslein, G.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Blanco, I.; Bülow, S.; Burn, J.; Capella, G.; et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008, 57, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanth, P.; Grimmett, J.; Champine, M.; Burt, R.; Samadder, N.J. Hereditary colorectal polyposis and cancer syndromes: A primer on diagnosis and management. Am. J. Gastroenterol. 2017, 112, 1509–1525. [Google Scholar] [CrossRef] [PubMed]



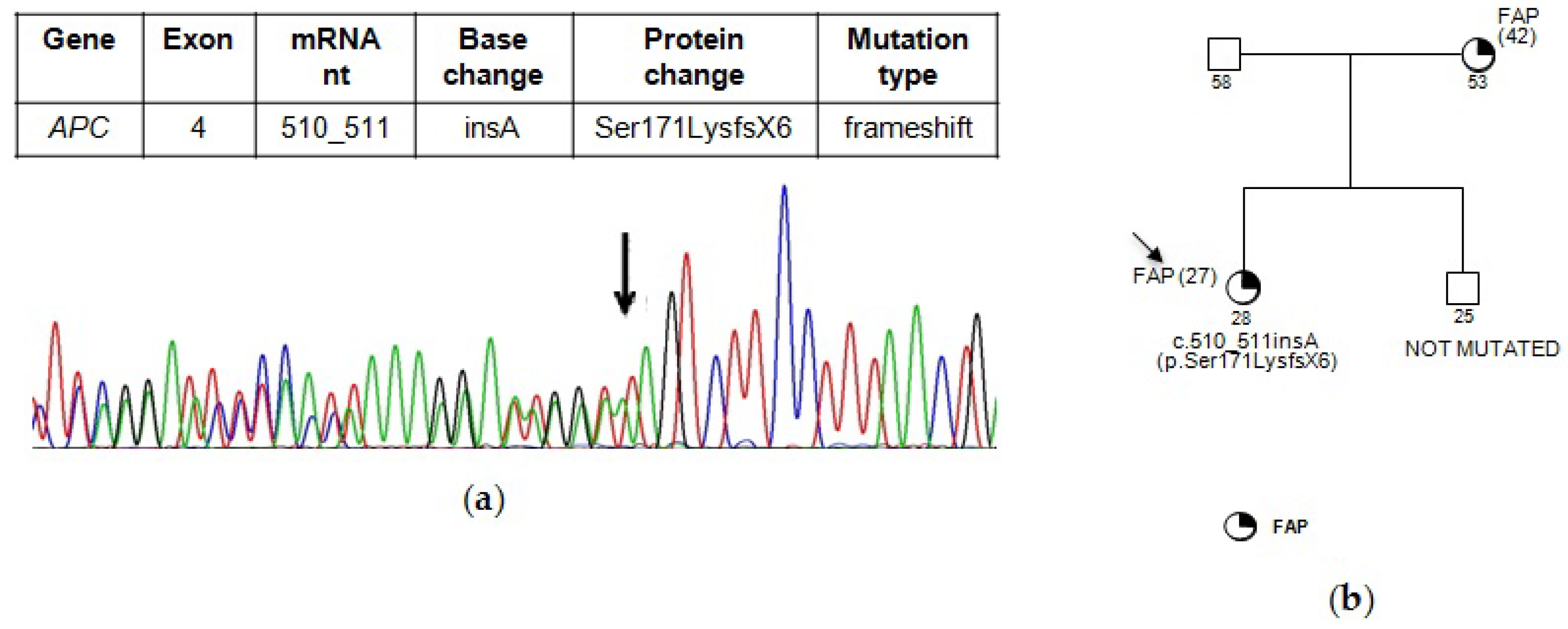{kind=link}
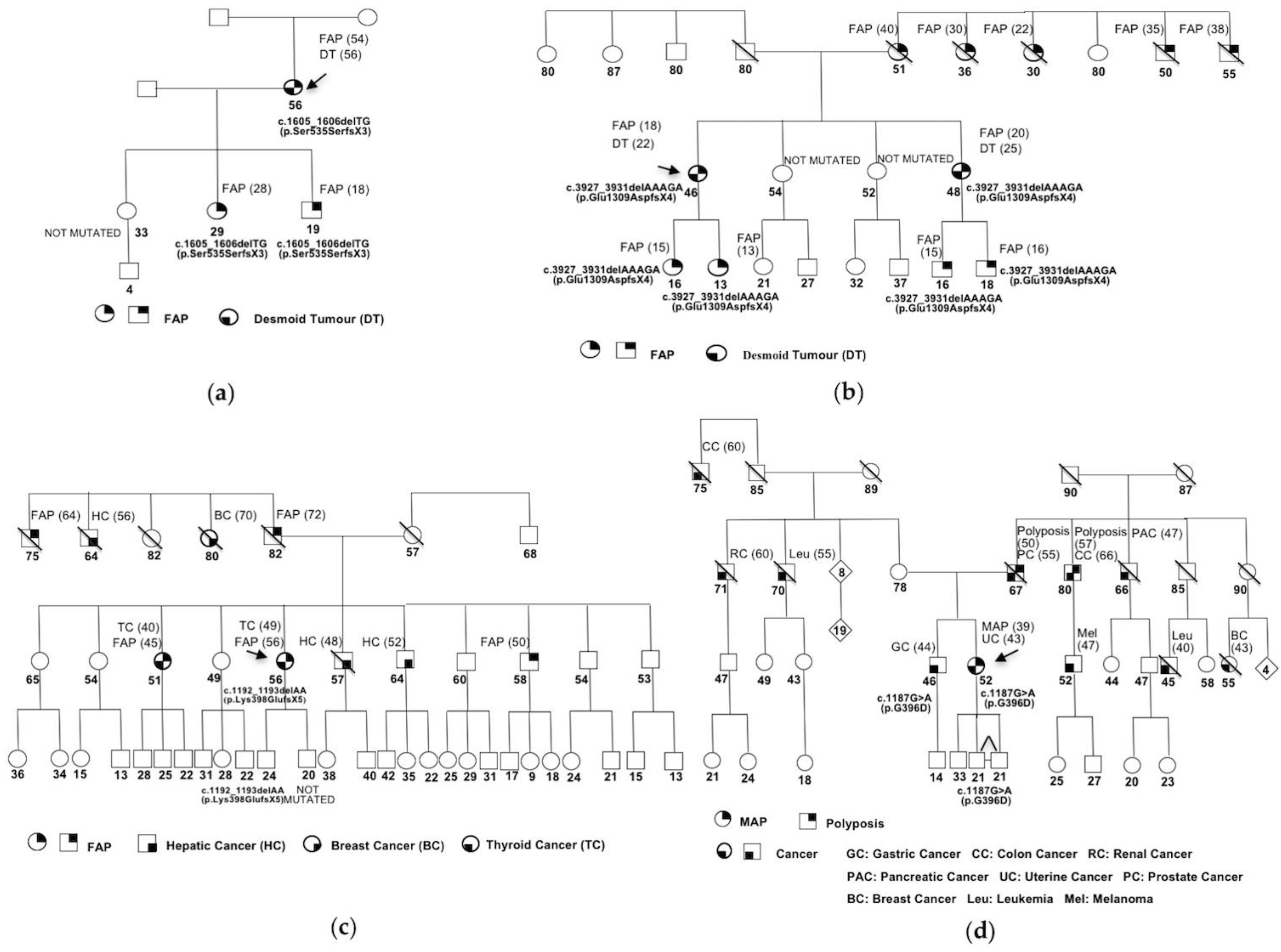{kind=link}
| Designation | Exon/Intron | PolyPhen | SIFT | Splice Site Finder (0–100) | MaxEntScan (0–12) | NNSPLICE (0–1) | GeneSplicer (0–24) | Ref. |
|---|---|---|---|---|---|---|---|---|
| IVS2+30 A>G (c.157+30 A>G) | 2 | - | - | SD:86,16/86,16 | SD:9,33/9,33 | SD:0,99/0,99 | SD:4,35/4,40 (+1.3%) | - |
| D271D (c.813 C>T) | 10 | - | - | SA:86,80/86,80 | SA:9,49/9,49 | SA:0,97/0,97 | SA:6,43/6,29 (−2.2%) | - |
| Q324H (c.1014G>C) | 12 | Benign (score 1.409) | Tolerated | - | - | - | - | [23] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Elia, G.; Caliendo, G.; Casamassimi, A.; Cioffi, M.; Molinari, A.M.; Vietri, M.T. APC and MUTYH Analysis in FAP Patients: A Novel Mutation in APC Gene and Genotype-Phenotype Correlation. Genes 2018, 9, 322. https://doi.org/10.3390/genes9070322
D’Elia G, Caliendo G, Casamassimi A, Cioffi M, Molinari AM, Vietri MT. APC and MUTYH Analysis in FAP Patients: A Novel Mutation in APC Gene and Genotype-Phenotype Correlation. Genes. 2018; 9(7):322. https://doi.org/10.3390/genes9070322
Chicago/Turabian StyleD’Elia, Giovanna, Gemma Caliendo, Amelia Casamassimi, Michele Cioffi, Anna Maria Molinari, and Maria Teresa Vietri. 2018. "APC and MUTYH Analysis in FAP Patients: A Novel Mutation in APC Gene and Genotype-Phenotype Correlation" Genes 9, no. 7: 322. https://doi.org/10.3390/genes9070322