In silico Phylogenetic Analysis of hAT Transposable Elements in Plants


1
Izmir International Biomedicine and Genome Institute, Dokuz Eylül University, Izmir 35340, Turkey
2
Izmir International Biomedicine and Genome Center (IBG), Izmir 35340, Turkey
*
Author to whom correspondence should be addressed.
Genes 2018, 9(6), 284; https://doi.org/10.3390/genes9060284
Submission received: 27 March 2018
/
Revised: 16 May 2018
/
Accepted: 4 June 2018
/
Published: 6 June 2018
(This article belongs to the Special Issue DNA Damage Responses in Plants)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Transposable elements of the hAT family exhibit a cross-kingdom distribution. The plant hAT transposons are proposed to play a critical role in plant adaptive evolution and DNA damage repair. The sequencing of an increasing number of plant genomes has facilitated the discovery of a plethora of hAT elements. This enabled us to perform an in-depth phylogenetic analysis of consensus hAT sequences in the fully-sequenced genomes of 11 plant species that represent diverse taxonomic divisions. Four putative nucleotide sequences were detected in cottonwood that were similar to the corresponding animal hAT elements, which are possibly sequence artifacts. Phylogenetic trees were constructed based both on the known and putative hAT sequences, by employing two different methods of phylogenetic inference. On the basis of the reconstructed phylogeny, plant hAT elements have rather evolved through kingdom-specific vertical gene transfer and gene amplifications within eudicotyledons, monocotyledons, and chlorophytes. Furthermore, the plant hAT sequences were searched for conserved DNA and amino acid sequence features. In this way, diagnostic sequence patterns were detected which allowed us to assign functional annotations to the plant hAT sequences.
1. Introduction
Transposable elements (TEs), or “jumping genes”, are mobile genetic elements capable of moving (transposing) from one location to another in the genome. The first elements, Activator (Ac) and Dissociation (Ds) were identified in maize (Zea mays) by McClintock [1]. Transposons are ubiquitous and fairly abundant in the genomes of eukaryotic and prokaryotic organisms, occupying over two-thirds of the human genome [2,3] and a significant fraction of some plant genomes [4,5,6].
Transposable elements can be broadly classified into two major classes [7] on the basis of their transposition mechanism, that is, retrotransposons, which transpose by means of a ‘copy-and-paste’ mechanism (Class I) and DNA transposons (Class II), which transpose directly, without involving the reverse transcription of an RNA intermediate.
The hAT family of Class II transposons, named after the three prototypic members, the maize transposon Activator (Ac), the Drosophila melanogaster element hobo and the Antirrhinum majus (napdragon) Tam3 transposon [8], transpose via a ‘cut-and-paste’ mechanism. The hAT transposons (i) require a TE-encoded transposase enzyme for insertion and excision; (ii) are flanked on both of their ends by terminal inverted repeats (TIRs), with a minimum length of 8 base pairs; and (iii) generate 4–8 bp target-site duplications upon insertion [9,10]. Members of the hAT family display a wide phylogenetic distribution across fungi, plants, and animals [11,12].
Transposable elements can be further divided into the autonomous elements, which encode the cellular machinery required to move independently, and the nonautonomous elements, which require the cellular apparatus of other autonomous TEs for their mobilization. For example, the nonautonomous element Ds depends on Ac to transpose (Ac/Ds TE system) [13].
In plants, transposon activity can dramatically affect the overall plant gene expression, structure, and function, often resulting in phenotypic changes, thereby contributing largely to adaptive plant evolution [14,15,16]. Of importance, transposon mobilization poses a major threat to the host genome by generating various types of DNA damage, including single-base mismatches and double-strand breaks (DBS). These transposition-inflicted DNA lesions lead to the activation of DNA damage repair pathways [17,18].
Despite the important role of the hAT family in the evolution of plants, to our knowledge, an in-depth study on the phylogeny of plant hAT elements is lacking. Previous phylogenetic studies of the plant hAT TEs, due to the scarcity of fully sequenced genomes, were restricted to few transposon sequences [11,12]. The current availability of completely sequenced and well-annotated plant genomes allowed us to perform a detailed phylogenetic analysis of the plant hAT transposons. To this end, the genomes of 11 plant species, representing diverse taxonomic divisions, including vascular plants, mosses, and green algae (Figure S1), were investigated for hAT homologs. Phylogenetic trees were reconstructed based on plant hAT DNA sequences, alone or together with fungi and plant hAT sequences, by employing two different methods of phylogenetic inference. Moreover, the plant hAT sequences were investigated for transposase amino acid sequences, as well as TIR sequence patterns, in order to define diagnostic sequence signatures that could be used to predict the functionality of these elements.
2. Methods
2.1. Sequence Dataset
Collectively 272 consensus hAT DNA sequences of 10 Viridiplantae species were downloaded from the Repbase Update, a database of eukaryotic transposable elements [7,19], in October 2017 in FASTA format (Text S1). However, there are currently no hAT sequences available in Repbase for Physcomitrella patens. The scientific names of those species, along with their generic names and RefSeq Genome project accession codes (shown within parentheses, respectively), are as follows: Oryza sativa (rice; PRJNA122), Zea mays (maize; PRJNA249074), Triticum aestivum (wheat; PRJNA392179), Hordeum vulgare (barley; PRJEB13020), Arabidopsis thaliana (PRJNA10719), Nicotiana tabacum (tobacco; PRJNA208209), Medicago truncatula (barrel medic; PRJNA10791), Populus trichocarpa (black cottonwood; PRJNA17973), Chlamydomonas reinhardtii (PRJNA21061), and Volvox carteri (PRJNA50441) (Figure S1). Moreover, a total of 221 hAT sequences from diverse fungal and metazoan taxonomic groups (Table S1) were retrieved from Repbase (Text S1) [7,19]. For convenience, all hAT sequences under study are commenced by their genus and species initial letters.
The retrieved plant, animal, and fungi hAT DNA sequences were used as ‘probes’ to search the genomes of the 11 plant organisms available in NCBI GenBank [20], by applying reciprocal BLASTn [21] with default parameters, in order to identify novel hAT sequences in plants. Each novel sequence was manually examined and only the sequence hits with an E-value lower than e−10 were accepted and subsequently used for iterative database searching until new hits could not be found. In this way, four novel hAT homologous sequences were detected in P. trichocarpa. In a similar manner, the plant hAT sequences were used as ‘seeds’ to parse the corresponding fungal and animal genomes.
2.2. Phylogenetic Analyses
The full-length consensus hAT nucleotide sequences were aligned by employing MAFFT v.7 [22]. The resulting multiple sequence alignments were manually edited with JalView 2.10.3 [23]. The trimmed alignments were subsequently used for reconstructing phylogenetic trees by two different methods, a neighbor-joining method as implemented in the software package MEGA version 7.0.26 [24], and a maximum-likelihood method as implemented in PhyML 3.0 [25]. ModelTest [26] was used to estimate the best-fit model of nucleotide substitution, that is, GTR + G. Bootstrap analyses (100 pseudo-replicates) were performed in order to evaluate the robustness of the inferred trees. Phylogenetic tree data were rendered with Dendroscope version 3.5.9 [27].
2.3. Detection of Putative Terminal Inverted Repeats
Ιn order to identify putative TIRs 8 bp in the plant hAT sequences, the corresponding entire DNA sequences of those elements were provided as input to TIRfinder [28], a software tool for detecting inverted repeats in Class II elements. For this purpose, the consensus “(T/C)A(A/G)NG” proposed by Rubin et al. [12] was used as reference; the maximum number of mismatches was set to 4 so as to increase sensitivity and detect more novel putative TIRs. Consensus TIR sequences were generated with EBI’s Mview [29].
2.4. Analysis of Transposase Amino Acid Sequences
The translated transposase amino acid sequences of the plant hAT DNA sequences were obtained from Repbase in October 2017 in EMBL data format. The retrieved sequences were queried against the InterPro [30] v. 66.0 protein signature database using InterProScan [31], in order to identify their constituent domains. In the case of the transposase protein sequence of a plant hAT element, which was not available in Repbase, its corresponding DNA sequence was translated in all six open reading frames (ORFs) using EBI’s EMBOSS Sixpack [32]. The predicted sequences were then compared to the known transposase sequences by alignment with PROMALS3D [33], using structural information from the resolved tertiary structure of the house fly Hermes hAT transposase (PDB ID: 4D1Q, chain A) [34], so as to improve alignment accuracy; their protein domain arrangement was examined using InterProScan [31].
Selected amino acid sequences, corresponding to “active” transposases (see Results) were aligned with PROMALS3D [33], as before, and their three-dimensional structure and function were further examined by employing Phyre2 [35]. Moreover, ungapped sequence blocks, representing highly conserved regions of proteins, were extracted from the multiple alignment with the usage of Utopia suite’s CINEMA alignment editor [36] and submitted to WebLogo3 [37] to generate consensus amino acid sequences.
3. Results
3.1. Homologous hAT Sequences in Plants
A total of 276 plant hAT nucleotide sequences, including the four putative ones, were investigated in this study. Despite extensive homology-based searches, no moss (Physcomitrella) hAT homologous sequences were detected. Four putative Populus trichocarpa hAT homologous sequences were detected. These newly identified sequences were arbitrarily named Pt URR1L, Pt URR1aL, Pt Charlie3L, Pt Chap4L, (where “L” stands for “like”) by virtue of similarity to the Xenopus tropicalis sequences, Xt URR1, Xt URR1a, Xt Chap4a/4b, and Xt Charlie3, respectively (Table S2, Figure S2). Regarding these cottonwood sequences, they are vector-contaminated DNA sequences, based on the fact that they are in the same BAC clone ISB1-145J20 (Table S2). Of importance, the presence of sequence artifacts has an immense impact on the public databases, often resulting to misleading results and interpretation errors [38,39]. The number of hAT elements in the corresponding plant species is shown within parentheses: Oryza sativa (179), Zea mays (26), Arabidopsis thaliana (23), Chlamydomonas reinhardtii (13), Volvox carteri (11), Populus trichocarpa (9), Medicago truncatula (7), Hordeum vulgare (2), Triticum aestivum (1), and Nicotiana tabacum (1). Conversely, putative plant hAT sequences were not found in the fungal and animal genomes.
The members of the plant hAT family appear to vary greatly among individual organisms, both in abundance and length. In particular, the consensus rice hAT sequences appear to be the most prolific among plants, whereas a single consensus hAT element was identified in tobacco and wheat. Moreover, C. reinhardtii Gulliver is the longest consensus hAT element (7144 bp) and O. sativa DEBOAT (110 bp) the shortest one.
3.2. Phylogenetic Reconstruction
The entire length hAT nucleotide sequences were used in this study in order to extract as much information as possible from the individual sequences. To unravel the evolutionary relationships of hAT elements among kingdoms, fungal and metazoan hAT transposon sequences were also investigated along with their plant homologs. The neighbor-joining (NJ) method, which is based on a hierarchical clustering algorithm [40], was used to build an ‘interkingdom’ tree (Figure 1 and Figure S2). A maximum-likelihood (ML) method, a heuristic approach for finding the optimal tree that fits the observed data, was employed to assess the evolutionary relationships among plant hAT elements (Figure 2). The overall topology of the phylogenetic trees reconstructed with the NJ and ML methods is pretty congruent, especially in the major clades (Figure 2 and Figure S2). In both phylograms, three discrete, highly supported monophyletic clades are distinguished that correspond to the three kingdoms, suggesting that the emergence of hAT TEs followed the fungi–plant–animal divergence, according to the universal tree of life.
The hAT sequences of the cereal plants (rice, maize, barley, and wheat) appear to cluster together. These elements have probably emerged after the monocotyledons-eudicotyledons divergence through monocot lineage-specific or species-specific gene expansion (Figure 2 and Figure S2). The consensus O. sativa hAT sequences appear to be the most abundant among the plant species under investigation, leading to the suggestion that multiple transposition events, mutations, and insertions/deletions might have taken place during the course of evolution that gave rise to the contemporary rice hAT elements.
The well-supported Clade 1 in the trees, which was reconstructed with both methods, includes hAT sequences from Oryza sativa Clade 1a and Zea mays Clade 1b (Figure 2 and Figure S2). Clade 1 sequences might have been derived by an ancestral hAT sequence that existed before the rice-maize split (~70 million years ago [41]) through a series of species-specific gene duplications.
The sequences Os hAT-N3/N3B/N3C/N3D/N3E, Os hAT-N10/N10B/N10C, Os hAT-N13/N13B/N13C, Os hAT-N17/N17B/N17C/N17D, Os hAT-N22/N22B, and Os hAT-N43/N43B, as well as Zm hAT-14/14N1/14N2 and Zm hAT-18/18N, form separate groups in both trees. Given that sequence duplications are very common in rice [42] and maize [43] genomes, the above sequences might reflect relatively recent duplication events that could have taken place in the corresponding species (Figure 2 and Figure S2).
The eudicotyledonous plant (Arabidopsis, tobacco, barrel medic, and cottonwood) hATs form a distinct group, albeit moderately supported with 52% bootstrap support (Figure 2), suggesting propagation of hAT genes after eudicots branched off from monocots, approximately 200 million years ago (MYA) [44].
Of note, Medicago truncatula SHATAG clusters consistently, with high confidence, with the P. trichocarpa hAT5/5B (Figure 2 and Figure S2). These three sequences might have originated from a common ancestor before the separation of the orders Fabales and Malpighiales in flowering plants; intraspecies diversification followed.
In the reconstructed phylogeny (Figure 2 and Figure S2), the green algae (Chlamydomonas and Volvox) hAT elements appear to form a well-supported monophyletic group, indicating that hAT transposons might have evolved within the chlorophyte lineage independently of those in vascular plants. The Gulliver element in C. reinhardtii appears to have a long branch; probably due to the multiple mutations accumulated at this sequence over time. In Volvox carteri, Vc hAT-2/2N1, Vc hAT-4/4N1, and Vc hAT-N1/N1A are likely duplicate sequences with significant similarity, given their very short branch lengths. This could be also indicative of the immense contribution of sequence duplications to the evolution of transposable elements in V. carteri.
The four novel Populus trichocarpa hAT sequences cluster with their orthologs in Xenopus tropicalis with 100% statistical confidence (Figure 1 and Figure S2). In the ML-based tree, the four ‘animal-like’ P. trichocarpa hAT sequences form their own well-supported group, very distantly related to the fellow plant hAT sequences (Figure 2). However, there is no evidence for horizontal gene transfer (HGT) events between P. trichocarpa and X. tropicalis.
3.3. Terminal Inverted Repeats Patterns
The output TIR sequences were manually inspected and a total of 207 putative TIRs of 8 bp minimum length were retrieved (Table S3). To minimize redundancy, a set of 153 nonidentical TIRs (Figure 3), was selected in order to generate a consensus. A sequence pattern of eight nucleotides “[T/C]AGNGNNG” was deduced, where the letters within brackets indicate alternative nucleotides at this position. This consensus is a modification and extension of the consensus “[T/C]A[A/G]NG” suggested in a previous study by Rubin et al., (2011) [12]. Moreover, in the hAT TIR patterns of plants, the nucleotide ‘G’ occurs more frequently in the third and fifth position (Figure 3).
3.4. Transposase Conserved Structural Features
Collectively, 69 transposase sequences were collected (Text S2). Of those, the sequences that contained the five amino acid key residues, D180, D248, R318, W319, and E572 (Hermes hAT transposase numbering; PDB ID: 4D1Q), essential for the function of transposases according to Hickman et al. [34] were subjected to Phyre2 [35] analysis (see Section 2.4). In this way, a total of 44 sequences, that harbored the five critical residues, were regarded as “active” transposases, whereas the sequences that lacked any of those residues were called “truncated” (Table S3). The “active” transposases consist of an N-terminal BED-type Zinc finger (138–199) [45], a site-specific DNA binding domain (227–271) [34], a catalytic domain with a long insertion (288–751) [34], and a highly conserved region (669–751) involved in dimerization in its C-terminus [34,46]; the numbers within the parentheses indicate the coordinates of each domain according to the maize Activator (AC) transposase sequence (Text S2). From the alignment of the “active” transposases, 17 conserved sequence blocks were identified (Figure 4). The five amino acids essential for the activity of hAT transposase enzymes [34] were also found unchanged in plant hAT transposases: D301, D367, R463, the aromatic residues [W/F] 464, and E719 (Figure 4). A series of highly conserved residues were also detected, apart from the known ones, including the conserved C401, H404, S723, and the invariant [F/W] 722 and R733 amino acids (Figure 4). These residues might also play an important role in maintaining the overall structure and function of plant hAT transposase enzymes.
3.5. Autonomous Elements
Nonautonomous elements, which are typically derived from autonomous TEs by internal deletion, do not contain the minimal sequences required for transposition, that is, an active-like transposase and TIR sequences [10]. Nonautonomous TEs (e.g., Os_TEMPINDAS) are often denoted by the addition of the letter ‘N’ into the name of their corresponding autonomous element (e.g., Os_TEMPINDAS-N1). Of note, in the reconstructed ML-based phylogram (Figure 2), several autonomous hAT elements appear to cluster with their corresponding nonautonomous derivatives with high confidence; for example, At ATHAT7/At ATHATN7, Zm ZhAT14/Zm hAT-14N1, Os hAT-6/Os hAT-6N1, and Vc hAT-4/Vc hAT-4N1.
In our study, the hAT elements which were found to encode a candidate active transposase and also possess putative TIR sequences were considered as “autonomous.” In this way, a total of 35 potential autonomous plant hAT elements were identified (Table S3), representing 12.6% of the total elements.
The founding member of the hAT family, Zm AC, is an autonomous element, as expected. The species, Chlamydomonas and Volvox are predicted to carry a single autonomous element, Cr Gulliver and Vc hAT-1, respectively. The cereal plants Triticum and Hordeum, although they encode transposases that contain the key amino acid residues required for their function, are probably not active elements since they lack putative TIRs (Table S3).
It is also worth mentioning that the nonautonomous plant hAT elements, the number of which appears to markedly surpass the one of autonomous elements (Table S3), can potentially recruit the enzymatic machinery of the autonomous hAT elements for their transposition, such as the maize Ac/Ds TE system [10,13].
4. Conclusions
The members of the hAT family exhibit a wide distribution across all eukaryotic kingdoms. In the present study, the taxonomic distribution, evolution, and predicted functionality of the plant hAT family was investigated. Collectively, 276 nonredundant, concrete, plant hAT sequences were found (till October 2017). These sequences represent complete consensus hAT sequences [19] derived from each organism. However, there is a great discrepancy between the actual number of individual copies of hAT transposable elements in each species and the corresponding consensus sequences [47,48].
Phylogenetic analyses were performed with the full length of the hAT sequences so as to include all the available evolutionary information that is present in these sequences. Based on the reconstructed phylogeny, the hAT family of transposable elements has likely propagated in Viridiplantae through vertical gene transfer and subsequent gene proliferation within eudicotyledons, monocotyledons, and chlorophytes. The overall number of consensus hAT elements differs greatly among the individual plant genomes, ranging from a single consensus sequence in tobacco and wheat to 179 in rice, likely due to intraspecies gene duplications.
In the transposases encoded by the plat hAT sequences, conserved protein segments and a series of sequentially invariant/conserved amino acid residues were identified. Furthermore, a TIR sequence pattern “(T/C)AGNGNNG” was also defined. The transposase protein blocks and characteristic amino acid residues, as well as the TIR pattern, could be used as diagnostic signatures for the identification of other plant hAT sequences. In our study, the elements that were found to encode a putative, likely active, transposase flanked by TIRs were referred to as “autonomous”. A total of 35 candidate autonomous elements were detected which, apparently, represent a small fraction of plant hAT elements. These likely active hAT elements can be co-opted by their host plant organisms, through an evolutionary process called ‘molecular domestication’ or ‘exaptation’ to carry out functions beneficial to the host [49,50].
Notably, four putative plant sequences of enigmatic origin were detected in cottonwood that shared a high degree of nucleotide identity with the amphibian hAT elements. These four animal-like hAT sequences which are likely nonautonomous, as they do not possess either a transposase-like sequence or TIRs, are probably sequence artifacts.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/9/6/284/s1. Text S1. Sequences analyzed in the present study in FASTA format. Text S2. Transposase amino acid sequences analyzed in the present study in FASTA format. Figure S1. A cladogram derived from NCBI’s Taxonomy database [51] showing the evolutionary relationships among plant species. Figure S2. Full view of the tree shown in Figure 1. The branches, the length of which reflects evolutionary distance, are colored according to the plant taxonomic group. Bootstrap values greater than 50% are shown at the nodes. The scale bar at the upper right indicates the length of nucleotide substitutions per position. Table S1. Taxonomy of the animal and fungi species under investigation based on NCBI’s Taxonomy database [51]. The abbreviated names of the species investigated in this study are shown within parentheses. Table S2 Accession numbers of the putative Populus trichocarpa hAT sequences. Table S3. The plant hAT sequences along with their putative TIRs and encoded transposases. The sequences indicated by red are the ones with identical TIRs that were not included in Figure 3. The elements that contain both a putative TIR and an active-like transposase are referred to as autonomous.
Author Contributions
G.K. and A.P. designed and supervised the study. G.K. and A.P. performed data analyses and wrote the manuscript. All authors reviewed and approved of the final manuscript.
Funding
Funding for article processing fee: Fully waived by Izmir Biomedicine and Genome Center (IBG).
Acknowledgments
The authors also would like to extend their thanks to Izmir Biomedicine and Genome Center (IBG), Izmir, Turkey for its support in implementing the project.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mc, C.B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [Green Version]
- De Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Karakulah, G. Rtfadb: A database of computationally predicted associations between retrotransposons and transcription factors in the human and mouse genomes. Genomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Pritham, E.J. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Karakulah, G.; Suner, A. Plantenrichment: A tool for enrichment analysis of transposable elements in plants. Genomics 2017, 109, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.D.; Atkinson, P.W.; O’Brochta, D.A. The hermes transposable element from the house fly, Musca domestica, is a short inverted repeat-type element of the hobo, Ac, and Tam3 (hAT) element family. Genet. Res. 1994, 64, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H., Jr. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Lopez, M.; Garcia-Perez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Kempken, F.; Windhofer, F. The hAT family: A versatile transposon group common to plants, fungi, animals, and man. Chromosoma 2001, 110, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.; Lithwick, G.; Levy, A.A. Structure and evolution of the hAT transposon superfamily. Genetics 2001, 158, 949–957. [Google Scholar] [PubMed]
- Doring, H.P.; Starlinger, P. Molecular genetics of transposable elements in plants. Annu. Rev. Genet. 1986, 20, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, N. Transposons and genome evolution in plants. Proc. Natl. Acad. Sci. USA 2000, 97, 7002–7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessler, S.R. Plant transposable elements. A hard act to follow. Plant Physiol. 2001, 125, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Hedges, D.J.; Deininger, P.L. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 2007, 616, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Schwacke, R.; Kunze, R. DNA damage-induced transcription of transposable elements and long non-coding rnas in arabidopsis is rare and atm-dependent. Mol. Plant 2016, 9, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Ostell, J.; Pruitt, K.D.; Sayers, E.W. Genbank. Nucleic Acids Res. 2018, 46, D41–D47. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [PubMed]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Gambin, T.; Startek, M.; Walczak, K.; Paszek, J.; Grzebelus, D.; Gambin, A. Tirfinder: A web tool for mining Class II transposons carrying terminal inverted repeats. Evol. Bioinform. Online 2013, 9, 17–27. [Google Scholar] [CrossRef]
- Brown, N.P.; Leroy, C.; Sander, C. Mview: A web-compatible database search or multiple alignment viewer. Bioinformatics 1998, 14, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.Y.; Dosztanyi, Z.; El-Gebali, S.; Fraser, M.; et al. Interpro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. Interproscan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Tang, M.; Grishin, N.V. PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, W30–W34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, A.B.; Perez, Z.N.; Zhou, L.; Musingarimi, P.; Ghirlando, R.; Hinshaw, J.E.; Craig, N.L.; Dyda, F. Molecular architecture of a eukaryotic DNA transposase. Nat. Struct. Mol. Biol. 2005, 12, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Bennett-Lovsey, R.M.; Herbert, A.D.; Sternberg, M.J.; Kelley, L.A. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program phyre. Proteins 2008, 70, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Pettifer, S.; Thorne, D.; McDermott, P.; Marsh, J.; Villeger, A.; Kell, D.B.; Attwood, T.K. Visualising biological data: A semantic approach to tool and database integration. BMC Bioinform. 2009, 10 (Suppl. 6), S19. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, T.L. Technical report. Vector DNA artifacts in the nucleotide sequence database. BioTechniques 1994, 16, 1124–1125. [Google Scholar] [PubMed]
- Savakis, C.; Doelz, R. Contamination of cDNA sequences in databases. Science 1993, 259, 1677–1678. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Wang, X.; Tang, H.; Paterson, A.H. Seventy million years of concerted evolution of a homoeologous chromosome pair, in parallel, in major poaceae lineages. Plant Cell 2011, 23, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, J.; Lin, W.; Li, S.; Li, H.; Zhou, J.; Ni, P.; Dong, W.; Hu, S.; Zeng, C.; et al. The genomes of Oryza sativa: A history of duplications. PLoS Biol. 2005, 3, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.E.; Langdale, J.A.; Kelly, S. The impact of widespread regulatory neofunctionalization on homeolog gene evolution following whole-genome duplication in maize. Genome Res. 2014, 24, 1348–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, K.H.; Gouy, M.; Yang, Y.W.; Sharp, P.M.; Li, W.H. Date of the monocot-dicot divergence estimated from chloroplast DNA sequence data. Proc. Natl. Acad. Sci. USA 1989, 86, 6201–6205. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L. The bed finger, a novel DNA-binding domain in chromatin-boundary-element-binding proteins and transposases. Trends Biochem. Sci. 2000, 25, 421–423. [Google Scholar] [CrossRef]
- Essers, L.; Adolphs, R.H.; Kunze, R. A highly conserved domain of the maize activator transposase is involved in dimerization. Plant Cell 2000, 12, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.I.; Kim, N.S. Transposable elements and genome size variations in plants. Genom. Inform. 2014, 12, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The b73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Sinzelle, L.; Izsvak, Z.; Ivics, Z. Molecular domestication of transposable elements: From detrimental parasites to useful host genes. Cell. Mol. Life Sci. 2009, 66, 1073–1093. [Google Scholar] [CrossRef] [PubMed]
- Volff, J.N. Turning junk into gold: Domestication of transposable elements and the creation of new genes in eukaryotes. BioEssays 2006, 28, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Federhen, S. The NCBI taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Unrooted neighbor-joining (NJ)-based tree of hAT transposon sequences. The major clades are collapsed for clarity. Taxa are represented by different coloring (inset). The four putative plant sequences are indicated by stars. The branch lengths depict evolutionary distance. Bootstrap values greater than 50% are shown at the nodes. The scale bar at the upper right indicates the length of nucleotide substitutions per site.
Figure 1.
Unrooted neighbor-joining (NJ)-based tree of hAT transposon sequences. The major clades are collapsed for clarity. Taxa are represented by different coloring (inset). The four putative plant sequences are indicated by stars. The branch lengths depict evolutionary distance. Bootstrap values greater than 50% are shown at the nodes. The scale bar at the upper right indicates the length of nucleotide substitutions per site.

Figure 2.
Unrooted maximum-likelihood (ML)-based tree of the plant hAT sequences. The tree is divided into two parts for clarity. Branch colors represent different taxonomic groups (insets). The conventions are the same as in Figure 1.
Figure 2.
Unrooted maximum-likelihood (ML)-based tree of the plant hAT sequences. The tree is divided into two parts for clarity. Branch colors represent different taxonomic groups (insets). The conventions are the same as in Figure 1.
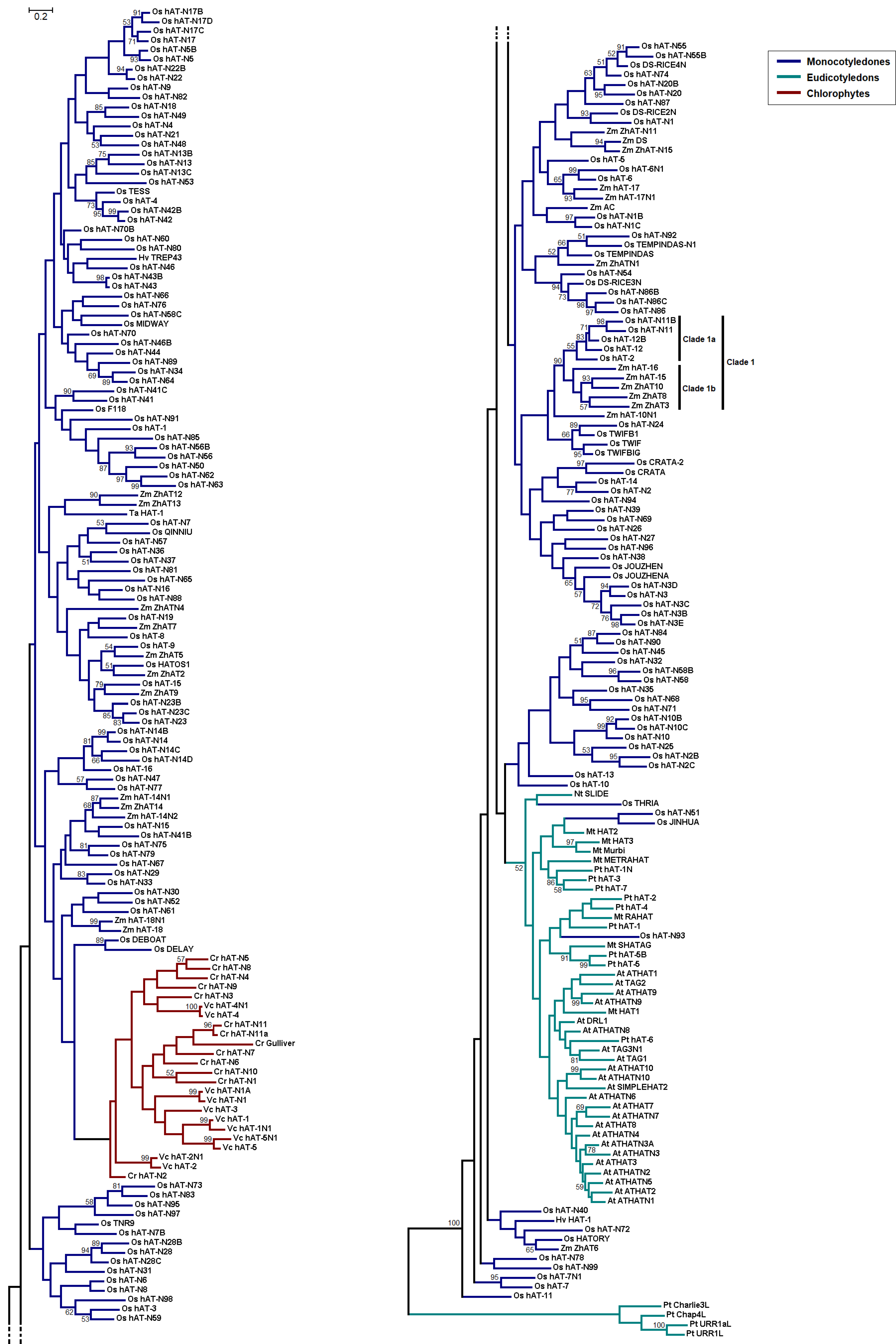
Figure 3.
Alignment of the putative terminal inverted repeats (TIR) sequences flanking the plant hAT sequences. The conserved nucleotides are highlighted in grey.
Figure 3.
Alignment of the putative terminal inverted repeats (TIR) sequences flanking the plant hAT sequences. The conserved nucleotides are highlighted in grey.

Figure 4.
Conserved sequence blocks derived from the plant hAT transposases. The amino acid residues are numbered according to maize Activator (AC). The height of each letter depicts the frequency of the corresponding residues at that position, with the most frequent being on the top. The invariant amino acids essential for the activity of hAT transposases are indicated by dots.
Figure 4.
Conserved sequence blocks derived from the plant hAT transposases. The amino acid residues are numbered according to maize Activator (AC). The height of each letter depicts the frequency of the corresponding residues at that position, with the most frequent being on the top. The invariant amino acids essential for the activity of hAT transposases are indicated by dots.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Karakülah, G.; Pavlopoulou, A. In silico Phylogenetic Analysis of hAT Transposable Elements in Plants. Genes 2018, 9, 284. https://doi.org/10.3390/genes9060284
AMA Style
Karakülah G, Pavlopoulou A. In silico Phylogenetic Analysis of hAT Transposable Elements in Plants. Genes. 2018; 9(6):284. https://doi.org/10.3390/genes9060284
Chicago/Turabian StyleKarakülah, Gökhan, and Athanasia Pavlopoulou. 2018. "In silico Phylogenetic Analysis of hAT Transposable Elements in Plants" Genes 9, no. 6: 284. https://doi.org/10.3390/genes9060284
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.