Roles of Mitochondrial DNA Mutations in Stem Cell Ageing


1
Wellcome Centre for Mitochondrial Research, Institute of Neuroscience, Newcastle University, Newcastle upon Tyne NE2 4HH, UK
2
LLHW Centre for Aging and Vitality, Newcastle University Institute for Aging, The Medical School, Newcastle upon Tyne NE2 4HH, UK
*
Author to whom correspondence should be addressed.
Genes 2018, 9(4), 182; https://doi.org/10.3390/genes9040182
Submission received: 26 February 2018
/
Revised: 22 March 2018
/
Accepted: 26 March 2018
/
Published: 27 March 2018
(This article belongs to the Special Issue Mitochondria and Aging)
{kind=link}
{kind=link}
Abstract
:Mitochondrial DNA (mtDNA) mutations accumulate in somatic stem cells during ageing and cause mitochondrial dysfunction. In this review, we summarize the studies that link mtDNA mutations to stem cell ageing. We discuss the age-related behaviours of the somatic mtDNA mutations in stem cell populations and how they potentially contribute to stem cell ageing by altering mitochondrial properties in humans and in mtDNA-mutator mice. We also draw attention to the diverse fates of the mtDNA mutations with different origins during ageing, with potential selective pressures on the germline inherited but not the somatic mtDNA mutations.
1. Introduction
Ageing is a process where tissue gradually loses homeostasis and regeneration [1]. This process is systemic and closely associated to age-related changes in somatic stem cells [2]. These cells renew themselves and differentiate into tissue-specific daughter cells for tissue maintenance and regeneration. The age-related alterations in somatic stem cell properties include failure to generate functional progenies, depletion of the stem cell pool and cancerous transformation [3]. These changes largely affect mitotic tissue, such as blood, intestine and skin, where the stem cells actively produce progenies to maintain the high turnover of the tissue [4,5]. However, they also contribute to ageing post-mitotic tissue, such as brain and muscle, though stem cells in these tissues are considered quiescent under normal physiological conditions and activated in response to damage for repairing the tissue [6,7].
Mitochondria synthesize ATP via oxidative phosphorylation (OXPHOS) through five multi-subunit complexes. Mitochondria contain their own DNA (mtDNA), which encodes key subunits of these complexes. Replication of the mitochondrial genome is independent of the cell cycle [8]. In addition, mtDNA is susceptible to damage due to lack of histone protection and proximity to oxidative stress [9]. Due to these reasons, compared with the nuclear DNA, mtDNA is more prone to mutations. Multiple copies of mtDNA reside in a cell. Mutations of mtDNA usually occur as a proportion of the total copies and once they reach a threshold, mitochondria will display respiratory chain deficiency, a consequence of which is potentially excessive production of reactive oxygen species (ROS) [10]. mtDNA is maternally transmitted through germline with a bottleneck effect, where only a small portion of mtDNA molecules are distributed into each primordial germ cell, which are amplified in later oogenesis. As a result, mature oocytes may have very different levels of mtDNA mutations [11].
Ageing is accompanied by a reduction of mitochondrial function, resulting in respiratory chain defects which are thought to be associated with the accumulation of somatic mtDNA mutations [12]. The age-related change in mitochondria may in turn accelerate the ageing process [13]. Although the significance of mtDNA mutations in various parenchymal cells in normal ageing and age-related degenerative diseases has been broadly studied [14], the findings might not be able to be extrapolated to stem cells, as they are distinct from somatic cells in terms of biological and metabolic characteristics. With an increasing amount of research linking mtDNA mutations to stem cell ageing in the last decade, we discuss how the somatic mtDNA mutations behave during ageing in stem cell populations and how they potentially influence phenotypes of the stem cells with the evidence from the recent studies. Specifically, we want to draw attention to an intriguing age-related phenomenon of the germline inherited mtDNA mutations in patients with mitochondrial disorders, as opposed to the accumulated somatic mtDNA mutations in normal individuals, as mtDNA mutations from different origins seem to have diverse fates with age.
2. Somatic mtDNA Mutations in Normal Ageing Humans
Respiratory chain defects have been observed in a variety of tissues in normal ageing humans, including the tissue traditionally considered post-mitotic, such as skeletal muscle [15], heart [16] and brain [17]; tissue with substantial mitotic potential after injury, for example, the liver [18]; as well as the typical mitotic tissue such as the epithelium of the stomach [19], small intestine [20] and colon (Figure 1) [21,22]. Various somatic mtDNA mutations were found to clonally expand and accumulate to high levels with age in the respiratory chain deficient areas of the tissue, regardless of their pathogenicity [21,23,24,25,26,27]. In addition, Shin et al. found increased mtDNA heterogeneity in CD34+ marked haematopoietic stem cell (HSC) and the progenitor cells both in the bone marrow and the peripheral blood from the adult donors compared to the homogenous umbilical cord blood [26,28]. In the intestinal crypt where all the cells derive from the stem cells located at the base of the crypt, the same somatic mtDNA mutation was determined at different regions along the OXPHOS defective crypt, indicating that they originated from a communal stem cell that harboured the mutation [20]. Monoclonal conversion where a single stem cell with the accumulated somatic mtDNA mutation passes the mutation to its progenies to occupy the entire crypt has also been observed in the stomach [19]. These studies provide robust evidence that in mitotic tissue, the accumulation of the somatic mtDNA mutation during ageing initiates in stem cells. However, whether the mutations aggregate in the stem cell of post-mitotic tissue, such as neural stem cells and satellite cells (muscle stem cell) and how they influence the fate of their progenies in aged humans remains unclear.
The accumulation of the somatic mtDNA mutation during ageing can be entirely achieved by random genetic drift, which takes place early in life in contrast to the damage to mtDNA by excessive ROS production from the respiratory chain defect [29]. The pathogenicity of the acquired mtDNA mutations is markedly higher than in germline variants, suggesting that the purifying selective mechanism against germline mtDNA mutations is absent from the somatic mtDNA mutations [30].
The occurrence and the spread of the somatic mtDNA mutations during ageing are notably tissue-specific, potentially influenced by the mitotic capability of the tissue. In contrast to miscellaneous mtDNA point mutations found in the colonic stem cell populations, no large-scale deletions have been detected in any OXPHOS deficient crypts [21]. Post-mitotic brain and muscle preferentially accumulate mtDNA deletions instead of point mutations with age [31,32]. It has been proposed that neurons, which are highly energy consuming and sensitive to ROS, tend to acquire mtDNA deletions during the process of repairing the mtDNA that are injured by ROS [33]. Whereas in mitotic tissue, mtDNA mutations are likely to arise from errors during replication [31]. Furthermore, the stem cell niche is involved in dispersing the somatic mtDNA mutations specifically in mitotic tissue during ageing. Gland and crypt fission from the stem cell niche in the stomach and colon, respectively, have been shown to spread the mutation from a single unit to form a patch [19,34].
However, limited numbers of human studies delve into how somatic mtDNA mutations affect the function of stem cells and their progenies to contribute to the ageing phenotype of the tissue, and the findings are controversial. Respiratory chain deficiency due to accumulated somatic mtDNA mutations in colonic stem cells diminished the cell population of the crypt by weakening the cell proliferation and enhancing the apoptosis of the stem cell progenies [35]. In contrast, the level of the lineage markers labelling all the differentiated daughter cells derived from the OXPHOS deficient stem cell were shown to be normal when compared to OXPHOS normal epithelium of the gastric unit and the small intestinal crypt, indicating that the functionality of the progenies were not affected [19,20]. Additionally, the patch of hepatic progenitor cells and their descendants with OXPHOS defects did not show any synthetic, metabolic or proliferative impairment [18]. Due to restricted sources of human tissue samples, difficulties in tracing the stem cell dynamic condition in humans, and shortage of the stem cell markers, the mechanism by which somatic mtDNA mutations influence stem cell ageing still remains unknown in humans, and scientists have pursued animal models for investigation.
3. Ageing of Somatic Stem Cell Populations in mtDNA-Mutator Mice
The development of an mtDNA-mutator mouse model highlights the potentially important role of somatic mtDNA mutations in ageing. These mice carry a knock-in mutation in the exonuclease domain of the mtDNA polymerase γ (Polg), compromising the proofreading ability of Polg, which results in an accumulation of mtDNA mutations with age [36,37]. The mutant mice have a shortened lifespan and display a series of progeroid phenotypes, such as weight loss, kyphosis, hair greying and loss, impaired hearing, thin subcutaneous fat, osteoporosis, sarcopenia and sterility, which mimics the signs of normal human ageing [36,37]. In addition, the development of mitotic tissue including intestine, thymus and testicle, is largely affected in the mutant mice [37]. Mutator mice suffer progressive anaemia due to defective haematopoiesis, which is the main cause of death [36,37,38]. These findings provide evidence that links the accumulation of somatic mtDNA mutations to stem cell ageing.
3.1. Age-Dependent Dysfunction of Stem Cells and Their Progenitors in Mitotic Tissue
The age-dependent accumulation of the somatic mtDNA mutations in the mtDNA-mutator mice either alters the properties of stem cells per se or the downstream progenitors. Studies initially focused on the haematopoietic system, which is a well-established model for stem cell research. Reports show that the mice suffer from abnormal haematopoiesis for both erythroid and lymphoid lineage [38,39,40] and that this consequence caused by the mtDNA mutation in the haematopoietic component is intrinsic, as transplanting the bone marrow from the homozygous mutants to the wildtype mice recapitulates the mutant phenotype [38]. Different stages of the progenitor cells through haematopoiesis have been shown to be more perturbed than the HSCs. However, HSCs themselves show notably compromised ability for repopulation and self-renewal after serial transplantation [39]. In addition, studies show that the development of the haematopoietic system is dysregulated during embryogenesis in the mtDNA mutator mice [40]. Intestinal epithelium is another beneficial model for somatic stem cell research as it allows us to visualise the gradual cradle-to-grave manifestations of the stem cell progenies in a straightforward fashion. Research on the small intestinal epithelium of the mutant mice reveals abnormal cell proliferation and increased apoptosis in the Lieberkühn crypt, where intestinal stem cells (ISC) and transit-amplifying cells are located [41]. Furthermore, the organoids consisting of ISCs and Paneth cells derived from the isolated crypts of the mutants did not grow efficiently in vitro [41]. These alterations resulted in prolonged cell migration and swollen morphology of the intestinal epithelium, leading to impaired fat absorption of the mtDNA mutator mice [41]. Studies have shown that, as with humans, the mtDNA mutations accumulating in the colonic crypts of the heterozygous mutator mice randomly distribute through the mitochondrial genome with no advantageous selection towards pathogenic mutations [42]. The clonal expansion of the mutations can be simulated by the model of random genetic drift [42]. In contrast, there is a strong rapid negative selection against nonsynonymous mtDNA mutations in the protein-coding regions through germline transmission [11,43], underlining the difference in the mechanisms of the selection during ageing and germline transmission.
3.2. Ageing of Progenitor Cells in Post-Mitotic Tissue
Compared to the severe symptoms due to mtDNA mutations in blood and other mitotic tissue, post-mitotic tissue seems less affected by mtDNA mutagenesis in the mtDNA mutator mice. Although respiratory chain function is preserved in the cerebrum, cerebellum and skeletal muscles, studies have shown direct alterations in the homeostasis of neural stem cells and muscle progenitors [40,44]. The number of the nestin-positive neuronal stem cells was decreased in the OXPHOS deficient subventricular zone of the adult mtDNA mutator mice, although the number of neurons was normal [40]. Neural stem cells (NSC) isolated from the embryos of the mutator mice showed strikingly reduced self-renewal ability, primarily because of the mtDNA mutagenesis, but not the secondary ROS, as these cells accumulated high levels of mtDNA point mutations, while barely showing respiratory chain deficiency [40]. Cardiac progenitor cells (CPC) isolated from the mtDNA mutator mice displayed attenuated proliferation and higher inclination to death [44]. Mutagenesis of mtDNA also blocked the metabolic transition of the mutant CPCs from glycolysis to OXPHOS as they differentiated, resulting in a massive cell death through differentiation [44]. Furthermore, myoblasts extracted from the mtDNA mutator mice were found to generate thinner myotubes in skeletal muscles [44]. These findings have underlined the functional change in somatic stem cells and precursors in the post-mitotic tissue due to age-related mtDNA mutagenesis.
4. Potential Mechanism Whereby Age-Related mtDNA Mutagenesis Affects Somatic Stem Cells
ROS is crucial for stem cell maintenance and is involved in manipulating stem cell differentiation, though too much ROS may be detrimental [45,46]. Mitochondrial DNA mutations are traditionally thought to affect the respiratory chain function of mitochondria, the main endogenous source of ROS, and interfere stem cell homeostasis [47]. The free radical theory of ageing based on mitochondria was initially proposed by Harman, where defective respiratory chain generates excessive ROS, damaging mtDNA and engendering somatic mtDNA mutagenesis with age, which further causes increased oxidative stress, forming a vicious cycle [48]. This theory was challenged when no obvious ROS elevation was found in different tissues of the mtDNA-mutator mice [37,49]. Analogous findings have been reported also in somatic stem cells [39]. However, treatment of antioxidant N-acetyl-l-cysteine (NAC) could rescue the impaired self-renewal ability of NSCs and abnormal haematopoiesis in the mutator mice [40]. The exact mechanism underlying these contradictory findings is as yet unknown. Despite an off-target possibility of NAC [50], these findings suggest that mtDNA mutagenesis may slightly alter the ROS/redox level (Figure 2), which is able to imbalance the quiescence and regeneration of the somatic stem cells [40]. This tiny change in the ROS/redox level might be insufficient to be detected, but it can be neutralized by NAC. However, the effect of the NAC on the stem cell population only seems to be effective if given during embryogenesis [40]. Of note, whether the effect of the antioxidant on the somatic stem cell benefits alleviating the ageing phenotype of either the stem cell or the tissue of the mutator mice remains controversial, as long-term treatment of NAC from embryogenesis did not rescue the aberrant erythropoiesis in the adult mutator mice [51]. In addition, calorie restriction, which reduces ROS and enhances antioxidative defence shows no effect on ameliorating the ageing phenotype of the mutator mice [52].
Recently, studies have reported another pre-ageing mouse model established by induced expression of mitochondrial-targeted endonuclease (Mito-Pstl) [53,54]. The main consequence of the double strand breaks of the mtDNA caused by the induction in these mice was mtDNA depletion [53,54]. In addition, mtDNA deletions accumulated in the post-mitotic brain and heart, but not in the lung and liver of these mice, though the expression of endonuclease was low in the brain and muscle but very high in the liver and lung [54]. Mitotic tissue was preferentially affected, with effects on the function of stem cells and progenitor cells. The differentiation of thymic progenitor cells was blocked at an early stage, leading to thymus shrinkage in these mice [54]. Satellite cells were lost in skeletal muscles of these mice, causing muscle wasting, but no change in the oxidative stress was detected in the muscle [53]. In addition, in the mitotic tissue that mainly displayed the ageing phenotype, no mtDNA depletion was found, implying that there might be another pathway that causes ageing rather than the secondary ROS generation caused by mtDNA depletion [54]. Together with the findings of the mtDNA-mutator mice, these observations suggest multi-factorial causes of ageing due to mtDNA mutagenesis or damage. In the cell model with the induction of the mtDNA endonuclease, elevated levels of ROS were detected, and this upregulated the ageing-related cell cycle arrest nuclear signalling pathway. This can be corrected by antioxidant NAC [54]. The absence of detectable changes in oxidative stress in either mtDNA-mutator mice or the Mito-Pstl mice is interesting, especially since many of the effects are reversed by NAC.
Accumulation of somatic mtDNA mutations can also cause dramatic alterations in mitochondrial dynamics, which is further able to perturb stem cell self-renewal and differentiation. During differentiation of the CPCs from the mtDNA mutator mouse, mitochondria displayed a series of abnormalities when compared to the WT mice, including imbalanced fusion and fission, reduced membrane potential, and poor development of the mitochondrial microstructure [44]. Mitochondrial dynamics can change mitochondrial morphology and regulate stem cell destiny by ROS signalling independent of generating ATP by OXPHOS [55]. Hence, though no change has been found in the ATP level in the stem cells from the mtDNA-mutator mice [39,41,44], the age-dependent accumulation of mtDNA mutations may still be able to alter the fate of stem cells secondarily through anomalous mitochondrial dynamics (Figure 2).
In addition, the age-related mutagenesis of somatic mtDNA may hamper stem cell metabolism as they differentiate (Figure 2). Stem cells are naturally glycolytic, and actively suppress OXPHOS to maintain quiescence. Differentiation of stem cells requires a metabolic transition from glycolysis to OXPHOS to increase ATP production [45]. Somatic mtDNA mutations prohibited this metabolic shift as CPCs differentiate, leading to substantial cell death [44]. This could also be the reason for the blockage of the differentiation at different stages through the early erythropoiesis [39].
Furthermore, the quality control of mitochondria is perturbed in the stem cell population of the mtDNA-mutator mice, which may contribute to stem cell senescence. Autophagy is crucial for maintaining stem cell quiescence and stemness against ageing [56,57]. Studies have reported upregulated mitophagy rates in the CPCs [44] and inhibited autophagy in the early erythroid precursors of the mtDNA-mutator mice, the latter of which contributed to the malfunction of erythropoiesis [58]. Moreover, a lowered mitochondrial membrane potential (MMP) has been found in the HSCs, various haematopoietic progenitors, and the CPCs in the mtDNA-mutator mice [39,44], which is associated with mitophagy activation [59]. These observations indicate that mtDNA mutagenesis may affect stem cell function by interfering with the level of mitophagy/autophagy (Figure 2). However, how exactly mitophagy/autophagy responds to mtDNA mutagenesis in stem cells is uncertain, as the findings seem to be contradictory in different stem cell/progenitor populations. Of note, the loss of the MMP also occurs during apoptosis [60]. Consistently, strikingly increased apoptosis levels were found in the erythroid precursors and the ISC population (Figure 2) [39,41], which may subsequently lead to the depletion of the stem cell/progenitors. Surprisingly, no change in the apoptosis was observed in the HSC [39].
The effect of age-related mtDNA mutagenesis on somatic stem cell ageing seems highly tissue-specific. For haematopoiesis, it primarily affects the differentiation of downstream progenitor cells rather than HSCs themselves [39]. In contrast, it markedly affects NSCs but its impact fades during differentiation [40].
5. Behaviour of mtDNA Mutations Inherited through Germline with Age
mtDNA mutations from different origins seem to act differently with age, despite causing the same biochemical defect [14]. Mitochondrial disorders are caused by mtDNA mutations inherited through the germline or occurring during embryogenesis. As opposed to the acquired somatic mtDNA mutations that accumulate in stem cell populations with age, some of the inherited mtDNA mutations are lost in mitotic tissue over time. Pearson syndrome is a severe medical condition caused by a single, large-scale mtDNA deletion affecting the bone marrow haematopoietic compartment and causing pancytopenia in patients [61]. However, the anaemia and the vacuolated haematopoietic precursors in the bone marrow can be rescued by an age-related loss of the deletion and patients may subsequently develop Kearns-Sayer syndrome, which primarily affects post-mitotic tissue [62,63,64]. It has also been reported that the mutation level of the inherited m.3243A>G, the most common mtDNA point mutation that causes mitochondrial disease, decreased in blood and various epithelial tissues with age in patients and asymptomatic carriers [65,66,67,68]. The in silico simulation suggests that the loss of the mutation occurs in HSCs [69]. In addition, in a recently-developed mouse model of mitochondrial disease, the inherited m.5024C>T was lost in the blood of the mice with high mutation levels with age [70]. These studies imply that these inherited mtDNA mutations seem to be selected against in the stem cell population in contrast to the somatic mtDNA mutations, which are under no selective pressures [30]. However, no observations of the negative selection have been reported directly in stem cells in vivo, and the mechanism is as yet unexplored. Investigating the underlying mechanism of the mtDNA mutation with different origins reacting differentially to age is crucial for understanding both the process of normal ageing and development of the mitochondrial disease.
6. Conclusions
We have provided evidence of the somatic mtDNA mutations accumulating in stem cell populations in normal humans and discussed their tissue-specific ability to expand clonally during ageing. The premature ageing mtDNA-mutator mouse model gives insight into how acquired mtDNA mutations affect the function of the stem cells and progenitors in both the mitotic and post-mitotic tissue, as well as the potential mechanisms by which age-related mtDNA mutagenesis affects stem cell homeostasis. We also highlight the intriguing contrast in the selective pressures against the mtDNA mutation with different origins in the fast-renewing mitotic tissue. Stem cells, which are responsible for tissue maintenance and regeneration, might be involved in the selective process against the inherited mtDNA mutation. Recently, studies have reported that stem cells might actively regulate their identity by manipulating the quality control of the mitochondria, for example, by removing the dysfunctional mitochondria [56] or by unevenly segregating young and aged mitochondria [71]. The quality control system might lose the function during ageing, leading to the absence of selective pressures on the somatic mtDNA mutations, which in turn accelerates ageing. We believe that investigating the mechanism of the difference in the selective pressure on the somatic and the inherited mtDNA mutations has a profound influence on understanding the progress of normal ageing, ageing-related degenerative diseases and cancer formation [72,73]. Studies have now provided several methods for preventing the germline transmission of the inherited mtDNA mutations to enable producing healthy children from affected mothers [74,75,76]. However, no efficacious interventions have been found for diseases caused by mutations that occur during embryogenesis. Understanding the mechanism of the selective pressure on the inborn mtDNA mutation will also propel the development of the treatment for mitochondrial disorders, especially for those occurring during embryogenesis and those that are recessive with late-onset.
Acknowledgments
This work was supported by the Newcastle University Centre for Aging and Vitality supported by the Biotechnology and Biological Sciences Research Council (BBSRC), Engineering and Physical Sciences Research Council (EPSRC), Economic and Social Research Council (ESRC), and Medical Research Council (MRC) as part of the cross-council Lifelong Health and Wellbeing Initiative; the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z); UK National Institute for Health Research (NIHR) Biomedical Research Centre in Age and Age Related Diseases award to the Newcastle upon Tyne Hospitals National Health Service (NHS) Foundation; the UK NHS Highly Specialized Service for Rare Mitochondrial Disorders of Adults and Children.
Author Contributions
T.S., L.C.G. and D.M.T. wrote the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rando, T.A. Stem cells, ageing and the quest for immortality. Nature 2006, 441, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Rando, T.A. Emerging models and paradigms for stem cell ageing. Nat. Cell Biol. 2011, 13, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Rando, T.A. Manifestations and mechanisms of stem cell aging. J. Cell Biol. 2011, 193, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Rudnicki, M.A. A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev. Biol. 2000, 218, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Ming, G.-L.; Song, H. Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell 2015, 17, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.; Clayton, D.A. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell 1977, 11, 719–727. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.-G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 2008, 9, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Linnane, A.; Ozawa, T.; Marzuki, S.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 333, 642–645. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The mitochondrial basis of aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Müller-Höcker, J. Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: An age-related alteration. J. Neurol. Sci. 1990, 100, 14–21. [Google Scholar] [CrossRef]
- Müller-Höcker, J. Cytochrome c oxidase deficient cardiomyocytes in the human heart—an age-related phenomenon. A histochemical ultracytochemical study. Am. J. Pathol. 1989, 134, 1167–1173. [Google Scholar] [PubMed]
- Cottrell, D.A.; Blakely, E.L.; Johnson, M.A.; Ince, P.G.; Borthwick, G.M.; Turnbull, D.M. Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age. Neurobiol. Aging 2001, 22, 265–272. [Google Scholar] [CrossRef]
- Fellous, T.G.; Islam, S.; Tadrous, P.J.; Elia, G.; Kocher, H.M.; Bhattacharya, S.; Mears, L.; Turnbull, D.M.; Taylor, R.W.; Greaves, L.C.; et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology 2009, 49, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.A.C.; Greaves, L.C.; Gutierrez-Gonzalez, L.; Rodriguez-Justo, M.; Deheragoda, M.; Leedham, S.J.; Taylor, R.W.; Lee, C.Y.; Preston, S.L.; Lovell, M.; et al. Mechanisms of field cancerization in the human stomach: The expansion and spread of mutated gastric stem cells. Gastroenterology 2008, 134, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Gonzalez, L.; Deheragoda, M.; Elia, G.; Leedham, S.J.; Shankar, A.; Imber, C.; Jankowski, J.A.; Turnbull, D.M.; Novelli, M.; Wright, N.A.; et al. Analysis of the clonal architecture of the human small intestinal epithelium establishes a common stem cell for all lineages and reveals a mechanism for the fixation and spread of mutations. J. Pathol. 2009, 217, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C.; et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Investig. 2003, 112, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Barron, M.J.; Plusa, S.; Kirkwood, T.B.; Mathers, J.C.; Taylor, R.W.; Turnbull, D.M. Defects in multiple complexes of the respiratory chain are present in ageing human colonic crypts. Exp. Gerontol. 2010, 45, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA–deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: A useful biomarker or more? Exp. Gerontol. 2008, 43, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.G.; Kajigaya, S.; Tarnowka, M.; McCoy, J.P.; Levin, B.C.; Young, N.S. Mitochondrial DNA sequence heterogeneity in circulating normal human CD34 cells and granulocytes. Blood 2004, 103, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Luo, X.; Zhu, J.; Zhang, X.; Zhu, Y.; Cheng, H.; Xia, Z.; Su, N.; Zhang, N.; Zhou, J. Mitochondrial DNA 4977 bp deletion is a common phenomenon in hair and increases with age. Bosn. J. Basic Med. Sci. 2012, 12, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.G.; Kajigaya, S.; McCoy, J.P.; Levin, B.C.; Young, N.S. Marked mitochondrial DNA sequence heterogeneity in single CD34+ cell clones from normal adult bone marrow. Blood 2004, 103, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Elson, J.L.; Samuels, D.C.; Turnbull, D.M.; Chinnery, P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 2001, 68, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Elson, J.L.; Nooteboom, M.; Grady, J.P.; Taylor, G.A.; Taylor, R.W.; Mathers, J.C.; Kirkwood, T.B.L.; Turnbull, D.M. Comparison of mitochondrial mutation spectra in ageing human colonic epithelium and disease: Absence of evidence for purifying selection in somatic mitochondrial DNA point mutations. PLoS Genet. 2012, 8, e1003082. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Krishnan, K.J.; Taylor, G.; Elson, J.L.; Bender, A.; Taylor, R.W.; Morris, C.M.; Turnbull, D.M. The low abundance of clonally expanded mitochondrial DNA point mutations in aged substantia nigra neurons. Aging Cell 2009, 8, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Pallotti, F.; Chen, X.; Bonilla, E.; Schon, E.A. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. Am. J. Hum. Genet. 1996, 59, 591–602. [Google Scholar] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Lightowlers, R.N.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Preston, S.L.; Tadrous, P.J.; Taylor, R.W.; Barron, M.J.; Oukrif, D.; Leedham, S.J.; Deheragoda, M.; Sasieni, P.; Novelli, M.R.; et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc. Natl. Acad. Sci. USA 2006, 103, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Nooteboom, M.; Johnson, R.; Taylor, R.W.; Wright, N.A.; Lightowlers, R.N.; Kirkwood, T.B.L.; Mathers, J.C.; Turnbull, D.M.; Greaves, L.C. Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts. Aging Cell 2010, 9, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Logan, T.D.; Hochberg, M.L.; Shelat, S.G.; Yu, X.; Wilding, G.E.; Tan, W.; Kujoth, G.C.; Prolla, T.A.; Selak, M.A.; et al. Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction. Blood 2009, 114, 4045–4053. [Google Scholar] [CrossRef] [PubMed]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Hämäläinen, R.H.; Yatsuga, S.; Uutela, M.; Terzioglu, M.; Götz, A.; Forsström, S.; Salven, P.; Angers-Loustau, A.; Kopra, O.H.; et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in POLG mutator mice. Cell Metab. 2012, 15, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.G.; Magness, S.; Kujoth, G.C.; Prolla, T.A.; Maeda, N. Mitochondrial DNA polymerase editing mutation, POLGD257A, disturbs stem-progenitor cell cycling in the small intestine and restricts excess fat absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G914–G924. [Google Scholar] [CrossRef] [PubMed]
- Baines, H.L.; Stewart, J.B.; Stamp, C.; Zupanic, A.; Kirkwood, T.B.L.; Larsson, N.-G.; Turnbull, D.M.; Greaves, L.C. Similar patterns of clonally expanded somatic mtDNA mutations in the colon of heterozygous mtDNA mutator mice and ageing humans. Mech. Ageing Dev. 2014, 139, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Wredenberg, A.; Cansu, Z.; Trifunovic, A.; Larsson, N.-G. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008, 6, e10. [Google Scholar] [CrossRef] [PubMed]
- Orogo, A.M.; Gonzalez, E.R.; Kubli, D.A.; Baptista, I.L.; Ong, S.-B.; Prolla, T.A.; Sussman, M.A.; Murphy, A.N.; Gustafsson, Å.B. Accumulation of mitochondrial DNA mutations disrupts cardiac progenitor cell function and reduces survival. J. Biol. Chem. 2015, 290, 22061–22075. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.Q.; Suda, T. Reactive oxygen species and mitochondrial homeostasis as regulators of stem cell fate and function. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, R.H. Mitochondria and mtDNA integrity in stem cell function and differentiation. Curr. Opin. Gene. Dev. 2016, 38, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [PubMed]
- Baines, H.L.; Turnbull, D.M.; Greaves, L.C. Human stem cell aging: Do mitochondrial DNA mutations have a causal role? Aging Cell 2014, 13, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.J.; Leoncini, S.; Pecorelli, A.; Wortmann, S.B.; Ahola, S.; Forsström, S.; Guerranti, R.; De Felice, C.; Smeitink, J.; Ciccoli, L.; et al. mtDNA mutagenesis impairs elimination of mitochondria during erythroid maturation leading to enhanced erythrocyte destruction. Nat. Commun. 2015, 6, 6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Someya, S.; Kujoth, G.C.; Kim, M.J.; Hacker, T.A.; Vermulst, M.; Weindruch, R.; Prolla, T.A. Effects of calorie restriction on the lifespan and healthspan of POLG mitochondrial mutator mice. PLoS ONE 2017, 12, e0171159. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pickrell, A.M.; Rossi, S.G.; Pinto, M.; Dillon, L.M.; Hida, A.; Rotundo, R.L.; Moraes, C.T. Transient systemic mtDNA damage leads to muscle wasting by reducing the satellite cell pool. Hum. Mol. Genet. 2013, 22, 3976–3986. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Pickrell, A.M.; Wang, X.; Bacman, S.R.; Yu, A.; Hida, A.; Dillon, L.M.; Morton, P.D.; Malek, T.R.; Williams, S.L.; et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ. 2016, 24, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Li-Harms, X.; Milasta, S.; Lynch, J.; Wright, C.; Joshi, A.; Iyengar, R.; Neale, G.; Wang, X.; Wang, Y.-D.; Prolla, T.A.; et al. Mito-protective autophagy is impaired in erythroid cells of aged mtDNA-mutator mice. Blood 2015, 125, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Larsson, N.-G.; Holme, E.; Kristiansson, B.; Oldfors, A.; Tulinius, M. Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr. Res. 1990, 28, 131–136. [Google Scholar] [CrossRef] [PubMed]
- McShane, M.A.; Hammans, S.R.; Sweeney, M.; Holt, I.J.; Beattie, T.J.; Brett, E.M.; Harding, A.E. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am. J. Hum. Genet. 1991, 48, 39–42. [Google Scholar] [PubMed]
- Nørby, S.; Lestienne, P.; Nelson, I.; Nielsen, I.M.; Schmalbruch, H.; Sjö, O.; Warburg, M. Juvenile Kearns-Sayre syndrome initially misdiagnosed as a psychosomatic disorder. J. Med. Genet. 1994, 31, 45–50. [Google Scholar] [CrossRef] [PubMed]
- ’t Hart, L.M.; Jansen, J.J.; Lemkes, H.H.; de Knijff, P.; Maassen, J.A. Heteroplasmy levels of a mitochondrial gene mutation associated with diabetes mellitus decrease in leucocyte DNA upon aging. Hum. Mutat. 1996, 7, 193–197. [Google Scholar] [CrossRef]
- Olsson, C.; Johnsen, E.; Nilsson, M.; Wilander, E.; Syvanen, A.C.; Lagerstrom-Fermer, M. The level of the mitochondrial mutation A3243G decreases upon ageing in epithelial cells from individuals with diabetes and deafness. Eur. J. Hum. Genet. 2001, 9, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Poulton, J.; Marchington, D.; Suomalainen, A. Decrease of 3243 A→G mtDNA mutation from blood in MELAS syndrome: A longitudinal study. Am. J. Hum. Genet. 2001, 68, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, A.L.; Andersen, P.H.; Kyvik, K.O.; Jeppesen, T.D.; Vissing, J.; Schwartz, M. Tissue specific distribution of the 3243A→G mtDNA mutation. J. Med. Genet. 2006, 43, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Rajasimha, H.K.; Chinnery, P.F.; Samuels, D.C. Selection against pathogenic mtDNA mutations in a stem cell population leads to the loss of the 3243A→G mutation in blood. Am. J. Hum. Genet. 2008, 82, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, J.H.; Baines, H.L.; Bratic, A.; Simard, M.-L.; Freyer, C.; Mourier, A.; Stamp, C.; Filograna, R.; Larsson, N.-G.; Greaves, L.C.; et al. A phenotype-driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 2016, 16, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Khrapko, K.; Vijg, J. Mitochondrial DNA mutations and aging: Devils in the details? Trends Genet. 2009, 25, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Popadin, K.; Gunbin, K.V.; Khrapko, K. Mitochondrial DNA mutations and cancer: Lessons from the parathyroid. Am. J. Pathol. 2014, 184, 2852–2854. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010, 465, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.-S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2012, 493, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sha, H.; Ji, D.; Zhang, H.L.; Chen, D.; Cao, Y.; Zhu, J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell 2014, 157, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
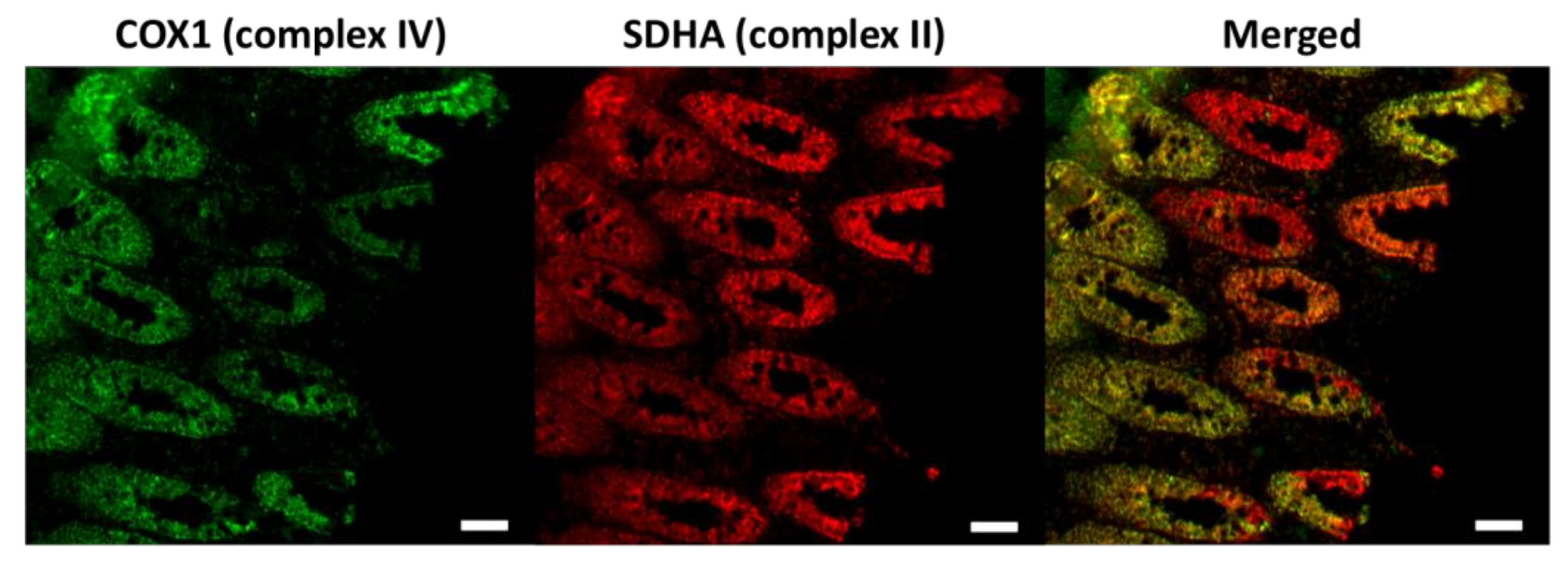
Figure 1.
Immunofluorescence images of the respiratory chain deficiency caused by the age-dependent accumulation of somatic mitochondrial DNA (mtDNA) mutations in the colon. Respiratory chain complex IV (marked by the COX1 antibody) is encoded by both mtDNA and nuclear DNA (nDNA), which is affected by the increased burden of mtDNA mutations. Complex II (labelled by the SDHA antibody) is entirely encoded by nDNA. The complex IV deficient colonic cells are indicated red in the merged picture. COX1, a key subunit of complex IV encoded by mtDNA. SDHA, one of the four nuclear encoded subunits of complex II. Scale bar: 50 µm.
Figure 1.
Immunofluorescence images of the respiratory chain deficiency caused by the age-dependent accumulation of somatic mitochondrial DNA (mtDNA) mutations in the colon. Respiratory chain complex IV (marked by the COX1 antibody) is encoded by both mtDNA and nuclear DNA (nDNA), which is affected by the increased burden of mtDNA mutations. Complex II (labelled by the SDHA antibody) is entirely encoded by nDNA. The complex IV deficient colonic cells are indicated red in the merged picture. COX1, a key subunit of complex IV encoded by mtDNA. SDHA, one of the four nuclear encoded subunits of complex II. Scale bar: 50 µm.
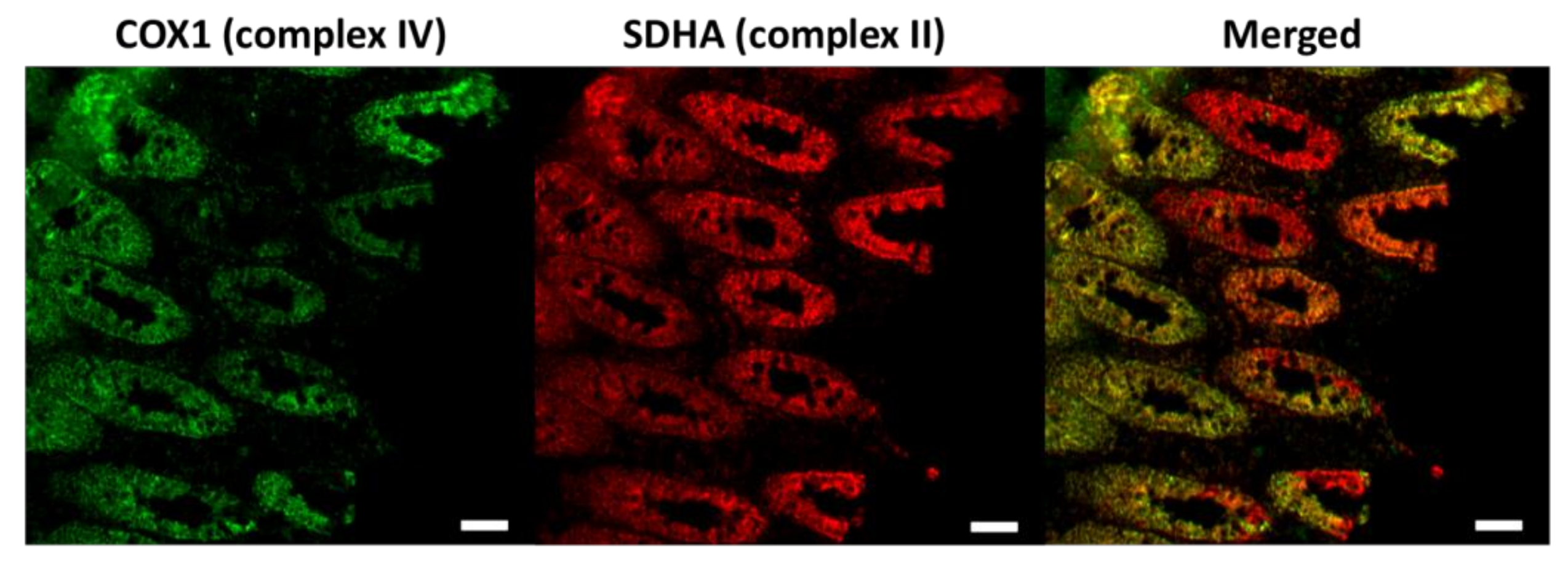
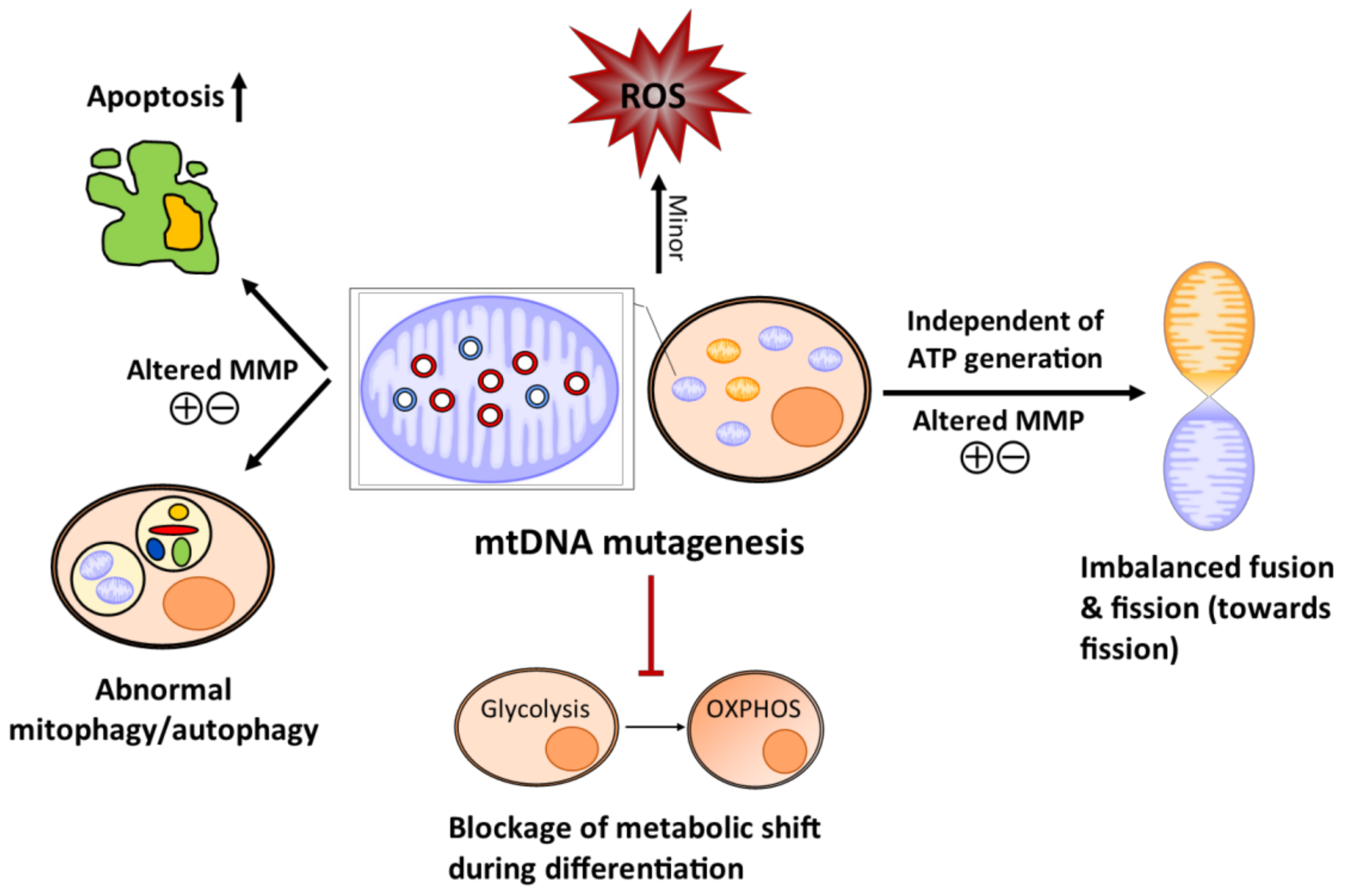
Figure 2.
Schematic diagram of how the mitochondrial abnormalities caused by the age-dependent accumulation of somatic mtDNA mutations affect stem cell homeostasis in the mtDNA mutator mice. Mitochondrial DNA mutagenesis causes minor changes in reactive oxygen species (ROS)/redox level, which may alter stem cell identity (quiescence and regeneration). The somatic mtDNA mutation increases apoptosis and also shifts the level of mitophagy/autophagy in the stem cell population possibly through the loss of the mitochondrial membrane potential (MMP), which may eventually engender stem cell/progenitor depletion and accelerate stem cell senescence. Mitochondrial DNA mutagenesis imbalances mitochondrial dynamics towards fission independent of ATP production, which can affect stem cell self-renewal and differentiation. The amassing of somatic mtDNA mutations prevents stem cells from converting glycolysis to oxidative phosphorylation (OXPHOS) as they differentiate, resulting in cell death and failure to produce progenies. Normal mitochondria are coloured orange and dysfunctional mitochondria are in blue. Mutated mtDNA are red and the normal mtDNA are blue.
Figure 2.
Schematic diagram of how the mitochondrial abnormalities caused by the age-dependent accumulation of somatic mtDNA mutations affect stem cell homeostasis in the mtDNA mutator mice. Mitochondrial DNA mutagenesis causes minor changes in reactive oxygen species (ROS)/redox level, which may alter stem cell identity (quiescence and regeneration). The somatic mtDNA mutation increases apoptosis and also shifts the level of mitophagy/autophagy in the stem cell population possibly through the loss of the mitochondrial membrane potential (MMP), which may eventually engender stem cell/progenitor depletion and accelerate stem cell senescence. Mitochondrial DNA mutagenesis imbalances mitochondrial dynamics towards fission independent of ATP production, which can affect stem cell self-renewal and differentiation. The amassing of somatic mtDNA mutations prevents stem cells from converting glycolysis to oxidative phosphorylation (OXPHOS) as they differentiate, resulting in cell death and failure to produce progenies. Normal mitochondria are coloured orange and dysfunctional mitochondria are in blue. Mutated mtDNA are red and the normal mtDNA are blue.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Su, T.; Turnbull, D.M.; Greaves, L.C. Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes 2018, 9, 182. https://doi.org/10.3390/genes9040182
AMA Style
Su T, Turnbull DM, Greaves LC. Roles of Mitochondrial DNA Mutations in Stem Cell Ageing. Genes. 2018; 9(4):182. https://doi.org/10.3390/genes9040182
Chicago/Turabian StyleSu, Tianhong, Doug M. Turnbull, and Laura C. Greaves. 2018. "Roles of Mitochondrial DNA Mutations in Stem Cell Ageing" Genes 9, no. 4: 182. https://doi.org/10.3390/genes9040182
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.