Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells

Department of Cell and Developmental Biology, Institute of Molecular Genetics of the CAS, v. v. i., Videnska 1083, 142 20 Prague 4, Czech Republic
*
Author to whom correspondence should be addressed.
Genes 2018, 9(1), 20; https://doi.org/10.3390/genes9010020
Submission received: 30 November 2017
/
Revised: 22 December 2017
/
Accepted: 28 December 2017
/
Published: 8 January 2018
(This article belongs to the Special Issue Wnt Signaling in Stem Cells)
{kind=link}
{kind=link}
{kind=link}
Abstract
:In this review, we address aspects of Wnt, R-Spondin (RSPO) and Hippo signalling, in both healthy and transformed intestinal epithelium. In intestinal stem cells (ISCs), the Wnt pathway is essential for intestinal crypt formation and renewal, whereas RSPO-mediated signalling mainly affects ISC numbers. In human colorectal cancer (CRC), aberrant Wnt signalling is the driving mechanism initiating this type of neoplasia. The signalling role of the RSPO-binding transmembrane proteins, the leucine-rich-repeat-containing G-protein-coupled receptors (LGRs), is possibly more pleiotropic and not only limited to the enhancement of Wnt signalling. There is growing evidence for multiple crosstalk between Hippo and Wnt/β-catenin signalling. In the ON state, Hippo signalling results in serine/threonine phosphorylation of Yes-associated protein (YAP1) and tafazzin (TAZ), promoting formation of the β-catenin destruction complex. In contrast, YAP1 or TAZ dephosphorylation (and YAP1 methylation) results in β-catenin destruction complex deactivation and β-catenin nuclear localization. In the Hippo OFF state, YAP1 and TAZ are engaged with the nuclear β-catenin and participate in the β-catenin-dependent transcription program. Interestingly, YAP1/TAZ are dispensable for intestinal homeostasis; however, upon Wnt pathway hyperactivation, the proteins together with TEA domain (TEAD) transcription factors drive the transcriptional program essential for intestinal cell transformation. In addition, in many CRC cells, YAP1 phosphorylation by YES proto-oncogene 1 tyrosine kinase (YES1) leads to the formation of a transcriptional complex that includes YAP1, β-catenin and T-box 5 (TBX5) DNA-binding protein. YAP1/β-catenin/T-box 5-mediated transcription is necessary for CRC cell proliferation and survival. Interestingly, dishevelled (DVL) appears to be an important mediator involved in both Wnt and Hippo (YAP1/TAZ) signalling and some of the DVL functions were assigned to the nuclear DVL pool. Wnt ligands can trigger alternative signalling that directly involves some of the Hippo pathway components such as YAP1, TAZ and TEADs. By upregulating Wnt pathway agonists, the alternative Wnt signalling can inhibit the canonical Wnt pathway activity.
1. Introduction
In this review, we describe the molecular mechanisms and possible crosstalk of the signalling pathways that directly influence intestinal homeostasis and tumorigenesis. The role of Wnt/β-catenin signalling in both processes was documented by several major discoveries published in the 1990s. In 1996, reports from three laboratories showed that β-catenin, the main mediator of canonical Wnt signalling, interacts with the T-cell factor (TCF)/lymphoid enhancer factor (LEF) and converts these DNA binding proteins to transcriptional activators [1,2,3]. In 1997, several research teams reported that in cancer cells with inactive tumour suppressor adenomatous polyposis coli (APC) or harbouring stabilizing mutations in the Ctnnb1 gene (the gene encoding β-catenin), TCF/β-catenin-dependent transcription is constitutively active [4,5,6]. In 1998, these results were complemented by gene targeting in the mouse, showing that upon ablation of the β-catenin interacting partner TCF4, proliferative compartments in the small intestine are not formed [7]. In agreement with these observations were data showing that the growth of intestinal organoids depends on Wnt agonists R-Spondins (RSPOs) and organoids derived from APC-deficient intestinal tumours lost this dependency [8,9]. In 2011, de Lau and co-workers documented that the intestinal stem marker leucine-rich-repeat-containing G-protein-coupled receptor 5 (LGR5), whose expression is controlled by Wnt signalling, functions as a transmembrane RSPO receptor [9]. One year later, Koo and colleagues reported that RSPO/LGR signalling potentiates the surface expression of Wnt receptors frizzled (FZD) [10]. These findings seemingly completed our perception of the Wnt pathway as the major regulatory mechanism involved in intestinal epithelium renewal and transformation. The situation changed a few years ago when several laboratories documented that some effects previously attributed to hyperactive Wnt/β-catenin signalling are actually mediated by components of the Hippo pathway [11,12,13]. Additionally, Park and colleagues corroborated an alternative model of Wnt signalling that directly includes some effector proteins regulated by the Hippo pathway [14]. Moreover, several recent articles showed that besides RSPOs, LGR proteins associate with other ligands. Consequently, the mode of the LGR-mediated intracellular response is more complex than originally thought [15,16]. In summary, we attempted to recapitulate the published data related to possible interactions among the Wnt, Hippo and RSPO/LGR pathways. We also summarized some results obtained upon gene inactivation of individual RSPO ligands and LGR receptors in the mouse.
2. Intestinal Epithelium Architecture and Cellular Composition
The most distinct feature of stem cells is their self-renewal capacity and potency, i.e., the ability to differentiate into one or multiple types of differentiated cells. In contrast to embryonic stem cells that populate the early embryo and give rise to the entire organism, adult stem cells emerge during later developmental stages and their self-renewal and cell differentiation potential is limited to a particular organ or tissue [17]. Virtually all adult stem cells are found in a specific tissue microenvironment or compartment called the stem cell niche. The niche has specific physical and (bio)chemical properties and complex cellular composition that supports the stem cell growth. In addition, the niche keeps stem cells in the undifferentiated state, i.e., preserves their stemness and also determines stem cell numbers [18].
Intestinal epithelium—similarly to other tissues of the adult organism—is characterized by the presence of stem cells that ensure tissue homeostasis and regeneration upon tissue damage. With constituent cells renewing every 3–5 days, the intestinal epithelium represents the most dynamic cellular system in the mammalian body [19]. In the small intestine, the single-layer epithelium covers microscopic protuberances and invaginations called villi or crypts, respectively. The villi contain differentiated non-dividing cells. In contrast, proliferating cells including intestinal stem cells (ISCs) are localized to the crypts. The discovery indicating the crucial role of the Wnt signalling pathway in small intestinal crypts and ISC homeostasis has significantly influenced the stem cell field in the last twenty years [7]. Moreover, the fact that activation of the Wnt pathway is a typical feature of human intestinal tumours [4,5] enabled efficient combination of experiments using human tumour cells with the methodology utilizing gene targeting in the mouse.
The idea that gene expression triggered by aberrant Wnt signalling in human intestinal cancer cells mirrors the Wnt-dependent transcription program in the crypt cells led to the discovery of several ISC-specific markers [20,21]. Although the majority of identified Wnt target genes showed a broad expression pattern in the crypt cells, some of the genes displayed more localized expression. Notably mRNA encoding LGR5 was produced in crypt base columnar (CBC) cells, slender, actively dividing cells that were intermixed with Paneth cells at the bottom of the crypt [22]. Subsequent lineage tracing studies revealed asymmetric division of LGR5+ cells accompanied by their ability to differentiate into all cell lineages present in the intestinal epithelium. In the small intestine, these cell types include absorptive enterocytes, microfold (M) cells associated with the mucosal immune system and several populations of secretory cells. The latter are represented by mucus-producing goblet cells, various sorts of hormone-secreting enteroendocrine cells, tuft cells that secrete endogenous opioids but also contribute to immune type 2 responses against enteric parasite infections and Paneth cells characteristic by production of anti-bacterial enzymes and peptides [23]. Besides LGR5, additional ISC-specific markers have been identified such as achaete-scute complex homolog 2 (Ascl2), olfactomedin 4 (Olfm4) [24], SPARC-related modulator calcium binding 2 (Smoc2) [25] and Troy (alternative names—tumour necrosis factor receptor superfamily, member 19 (TNFRSF19) or TAJ) [26]. ISCs divide approximately once per day, generating highly proliferative progenitors called transit amplifying (TA) cells, localized in the crypt above the stem cell zone. Dividing cells move upwards from the crypt and, when they reach the crypt orifice, stop to divide and differentiate. Consequently, the differentiated cells on the villus continuously move and, once reaching the villus tip, are shed into the intestinal lumen. One exception from this scheme are Paneth cells. These long-lived cells do not migrate from the crypts but remain at the crypt base for 5–6 weeks. The morphology of the large intestine resembles the small intestine; however, it lacks villi and Paneth cells, the latter being substituted by the recently discovered deep crypt secretory (DCS) cells, marked by their expression of regenerating islet-derived family member 4 (Reg4) [27].
Before the stem cell identity of LGR5+ CBC cells had been confirmed, a number of studies proposed that intestinal homeostasis is maintained by slowly dividing cells localized four cell positions from the bottom of the crypt [28]. The “+4” cells, due to their quiescence-like state, displayed increased radiation resistance and were capable to repopulate the damaged epithelial layer [29]. However, despite remarkable effort, none of the markers assigned to these “reserved” ISCs proved to be specific [25,30]. However, there is growing evidence related to the high phenotypic plasticity of cells within the intestinal epithelium [31,32]. In the current view, rather than genuine ISCs, the +4 cells are considered to be non-dividing precursors of the secretory cell lineages that under stress conditions can dedifferentiate and replace the damaged or lost ISCs [33].
The ISC niche in the small intestine is composed of the extracellular matrix (ECM) and many cell types that include connective tissues cells (fibroblasts and myofibroblasts), endothelial and immune cells, cells of neuronal origin and smooth muscle and Paneth cells [34]. Interestingly, the tissue niche can be substituted by a “cocktail” of growth factors and signalling ligands that supports growth of isolated crypts or single LGR5+ ISCs in culture conditions. Upon embedding in Matrigel, the crypts (or single ISCs) generate three-dimensional structures called organoids or miniguts that resemble the organization, self-renewal and differentiation of the intestinal epithelium. The organoid medium has to be supplemented with the Wnt pathway agonist RSPO, the bone morphogenetic protein (BMP) pathway inhibitor noggin and epidermal growth factor (EGF). Essential factors, additional to RSPO, noggin and EGF, are supposedly produced by ISCs neighbouring Paneth cells. The Paneth cell-derived factors include Wnt3, the Notch ligand delta like 4 (Dll4) and tumour necrosis factor (TNF) [8,35]. Intriguingly, Paneth cell ablation does not impact ISCs in vivo; however, it has a deleterious effect on organoid growth [36]. The organoid cultures can be rescued by intestinal stromal cells, implying redundancy between Paneth cells and stroma as the Wnt source in the organism [37,38].
3. Wnt Signalling in the Intestine
Wnt signalling represents one of the key signalling mechanisms that regulates intestinal epithelium homeostasis. Nineteen mammalian secreted Wnt ligands have been identified, which contribute to a multitude of cellular processes such as proliferation, differentiation, cell-fate determination, migration and polarization [39]. Upon translation, all Wnt are acetylated in the endoplasmic reticulum (ER) by the acyl transferase porcupine (PORCN), which catalyses addition of palmitic and palmitoleic acid to the Wnt molecule [40,41]. Acylation is essential for Wnt passing through the secretory pathway and/or signalling properties of the Wnt ligand. Consequently, PORCN inhibition leads to reduction or absence of the Wnt ligand signalling capacity [42,43]. Wnt ligands are classified as canonical (e.g., Wnt1, Wnt3a and Wnt8a) and non-canonical (Wnt4, Wnt5a, Wnt11). Whereas the canonical ligands increase the stability of β-catenin as a key intracellular signalling molecule, non-canonical Wnts act β-catenin independently. The canonical Wnt/β-catenin pathway is triggered by interaction between the (canonical) Wnt ligand and the transmembrane receptor complex that is formed from the seven-pass transmembrane FZD and the co-receptor low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6). Among 10 FZDs encoded in the mouse genome, Fzd5 and Fzd7 mRNAs are highly enriched in the crypts and their expression declines towards the villus tip [44]. FZD7 appears to be the crucial Wnt signalling mediator in ISCs, since conditional deletion of the Fzd7 gene in LGR5+ cells leads to stem cells loss and impaired regeneration capacity after damage [45]. Additionally, increased Fzd7 expression levels were linked to progression of human intestinal cancer [46]. The Wnt-FZD-LRP5/6 interaction prevents activity of the β-catenin destruction complex that is composed of two scaffolding proteins, APC and AXIN and two serine/threonine kinases, casein kinase-1 (CK1) and glycogen synthetase kinase 3 (GSK3). In unstimulated cells, the complex binds β-catenin and sequentially phosphorylates the protein at serine 45 (CK1), threonine 41 (GSK3) and serines 37 and 33 (GSK3). Phosphorylated β-catenin molecules are polyubiquitinated by the E3 ligase β-transduction repeat-containing protein (β-TrCP) and degraded by the proteasome [47]. Upon Wnt stimulation, β-catenin accumulates in the cell cytoplasm and migrates to the nucleus, where it displaces the transcriptional groucho/transducin like enhancer of split (TLE) co-repressors from the nuclear high-mobility group (HMG) box containing DNA-binding factors TCF/LEF family ([48]). Beta-catenin activates transcription by recruitment of transcription co-activators and/or histone modifiers such as cyclic-AMP response element binding protein (CBP), ATP-dependent brahma related gene 1 (BRG1), p300 and B-cell lymphoma (BCL9) [49,50].
The contribution of Wnt/β-catenin signalling to intestinal self-renewal and tumorigenesis was established by gene targeting studies performed in the mouse and by analysis of human cancer cells. For example, disruption of the Tcf7l2 gene, encoding the β-catenin partner and Wnt signalling transcriptional regulator, caused complete loss of the intestinal crypt compartments accompanied by the absence of proliferating cells in the small intestine of new-born Tcf7l2−/− mice [7]. A similar phenotype was observed in transgenic animals overexpressing the secreted Wnt signalling inhibitor dickkopf-1 (Dkk1) [51]. In contrast, increased activation of the Wnt pathway represents a hallmark of the majority of intestinal adenomas or adenocarcinomas and the most frequent mutations found in sporadic tumours of the colon and rectum (colorectal cancer (CRC)) are mutational changes inactivating the APC tumour suppressor [52,53,54,55]. Consequently, in APC-deficient cells, the β-catenin destruction complex is not functional and the TCF4-β-catenin-dependent transcription is (aberrantly) activated [4]. In 1990, Moser and colleagues developed a mouse model of intestinal tumorigenesis based on random mutagenesis [56]. Two years later, Su and co-workers identified that the dominant mutation predisposing animals of this multiple intestinal neoplasia (Min) strain to numerous, mainly small intestinal tumours, inactivates the mouse Apc orthologue [57]. Correspondingly, conditional inactivation of Apc using the Cre/loxP system in LGR5+ cells initiates neoplastic growth in epithelia of the small intestine and colon [58]. Another example of genetic changes altering the Wnt pathway components and that have been identified in CRC, are missense mutations in the Ctnnb1 gene (encodes β-catenin) that stabilize the protein or mutations inactivating the additional component of the β-catenin destruction complex AXIN2 [5,59].
The Wnt/β-catenin pathway activity in the intestine generates a descending gradient along the crypt-villus axis that controls epithelial cell division, differentiation and migration. As already indicated, Paneth cells produce Wnt3, which stimulates the neighbouring cells; however, how the Wnt gradient is established and controlled remained unclear [35,44,60]. However, recent data obtained by Farin and colleagues using an epitope-tagged, functional Wnt3 knock-in allele, brought valuables insights into the Wnt ligand movement in the intestine. The ligand is distributed in a single-cell distance from its source (Paneth cell) to the neighbouring ISC. Then, the Wnt-FZD complex is passively distributed to daughter cells through mitosis. Subsequent cell divisions in the ISC and TA compartments results in dilution of surface-bound Wnt leading to the gradient formation [61].
The less-well characterized non-canonical Wnt pathways include Wnt/planar cell polarity (Wnt/PCP) and Wnt/calcium (Wnt/Ca2+) signalling. In Wnt/PCP signalling, the Wnt signal is transduced by a transmembrane receptor complex that involves the FZD receptor and the alternative co-receptors receptor tyrosine kinase-like orphan receptor 1/2 (ROR1/2), receptor-like tyrosine kinase (RYK) and protein tyrosine kinase 7 (PTK7). Wnt/PCP signalling activates the small GTPases RHO/RAS-related C3 botulinum toxin substrate 1 (RAC1) and results in cytoskeleton reorganization affecting cellular polarity and migration [62]. In Wnt/Ca2+ signalling, the Wnt FZD interaction leads to activation of phospholipase C (PLC) through heterotrimeric G proteins. Active PLC hydrolyses phospholipids to inositol 1,4,5-trisphosphate (IP3) and 1,2-diacyl glycerol. These molecules function as “second messengers” that induce multiple cellular responses such as Ca2+ release from the ER and protein kinase C (PKC) stimulation [63]. The role of non-canonical pathways in intestinal homeostasis has not been clarified yet. Wnt/PCP signalling possibly antagonizes the canonical Wnt/β-catenin pathway. Accordingly, increased production of Wnt5a is associated with improved prognosis in patients with intestinal tumours [64,65]. In contrast, Bakker and colleagues reported the opposite, i.e., correlation between Wnt5a expression and invasiveness of human and mouse colon cancer cells [66]. Thus, the exact role(s) of non-canonical pathways in intestinal homeostasis or tumour formation remains to be clarified.
4. R-Spondins/Leucine-Rich-Repeat-Containing G-Protein-Coupled Receptor Signalling
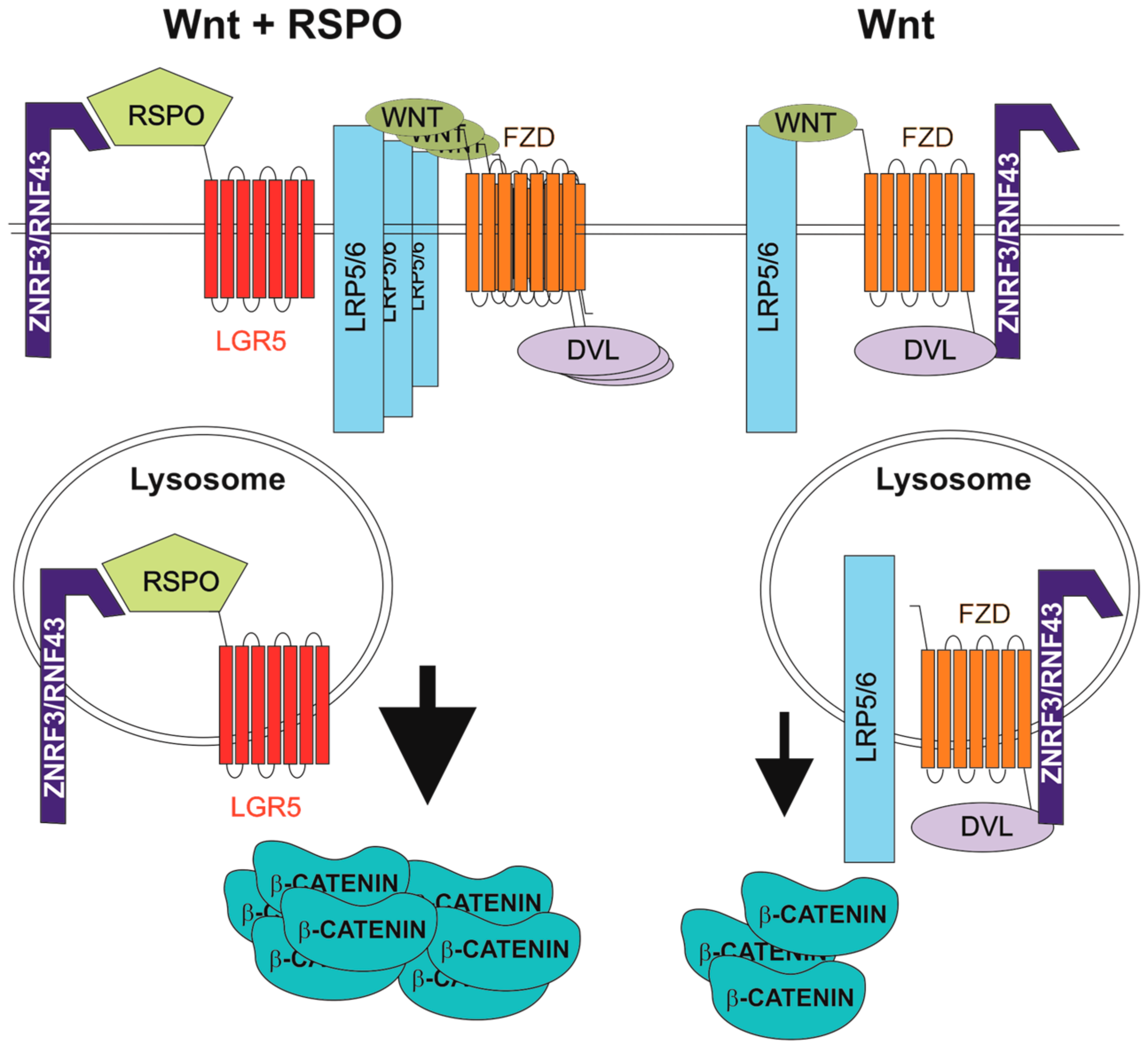
The human genome harbours four genes that encode related RSPO proteins. All RSPOs include a signal peptide, two furin-like cysteine-rich domains (FU1-CRD and FU2-CRD), a thrombospondin type 1 repeat domain and a basic amino acid-rich repeat domain [67]. Based on the physical interaction between RSPO1 and LRP6, RSPOs were first described as Wnt signalling agonists [68]. In 2011, three laboratories reported that RSPOs bind LGR5 (and its homologs LGR4 and LGR6) and that this interaction enhances the Wnt pathway output [9,69,70,71]. In the present model, RSPO-mediated potentiation of Wnt signalling depends on the cell surface clearance of the homologous transmembrane E3 ubiquitin ligases, ring finger 43 (RNF43) and zinc and ring finger 3 (ZNRF3). The ligases antagonize both canonical and non-canonical Wnt signalling through ubiquitination and degradation of Wnt receptors. In the presence of RSPO, a specific RSPO-LGR-RNF43/ZNRF3 complex is formed and the RNF43/ZNRF3 activity is suppressed [72]. Mechanistically, RNF43/ZNRF3 and LGR interact with the RSPO FU1-CRD and FU2-CRD domains, respectively. This leads to internalization of the ligases and their lysosomal degradation [73,74] (Figure 1). Recently, Loregger and co-workers determined another mode of RNF43 inhibitory action on Wnt signalling. RNF43 interacts with TCF4 and tethers the protein to the nuclear membrane. Since RNF43 functions even in cells with constitutive β-catenin activity, mimicking (or potentiation) of this specific RNF43 function might be used to treat CRC initiated by aberrant Wnt signalling [75]. However, based on an RNF43/ZNRF3 knockdown study, Carmon and colleagues suggested an alternative E3 ubiquitin ligase-independent, mechanism underlying the enhancement of Wnt pathway activity by RSPO-LGR signalling. RSPO1-LGR4 interacts with IQ motif containing GTPase activating protein 1 (IQGAP1) and the supramolecular complex recruits the cytoplasmic mediator of Wnt signalling, dishevelled (DVL). The complex promotes LRP5/6 phosphorylation, leading to increased canonical Wnt signalling [76]. This finding was later questioned by Jiang and colleagues, who analysed the results upon Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-mediated ZNRF3/RNF43 knockout in human cells. These authors highlighted the interaction between RSPO and RNF43/ZNRF3 as the main mechanism related to the “boost” of Wnt signalling. Moreover, they proposed a dual role for DVL in Wnt signalling regulation. In the absence of Wnt, DVL mediates interaction with FZD and ZNRF3/RNF43 and promotes receptor internalization. Upon ligand engagement, FZD is released from the interaction with DVL and RNF43/ZNRF3 stabilizing receptor expression on the cell surface [77].
In the intestine, RSPO1/2/3 mRNAs are predominantly produced in the stroma, whereas their expression in the epithelium is negligible [38]. The expression pattern corroborates the necessity to supplement organoid culture medium with exogenous RSPO to support organoid growth [8]. Interestingly, mice with conditional deletion of the Porcn gene in the intestinal epithelium showed no pathological changes but organoids derived from these mice failed to grow. Additionally, organoid viability was rescued by co-culture with cells producing RSPO1 (and Wnt3a). These findings support the idea that stromal production of RSPOs and Wnts is sufficient to support intestinal homeostasis [38]. Very recently, two articles brought an important insight related to Wnt and RSPO function in ISC self-renewal. The authors generated water-soluble Wnt agonists heterodimerizing the FZD–LRP5/LRP6 molecules and used them to activate Wnt signalling in experimental mice. Additionally, RSPOs were supplied as recombinant adenoviruses encoding RSPO1/2 (Ad-RSPO1/2). Moreover, these authors also achieved in vivo inhibition of Wnt/RSPO-mediated signalling by using adenoviral viruses producing FZD-CRD, Dkk1 (Ad-FZD-CRD, Ad-Dkk1), or RNF43/ZNRF3 and LGR5 soluble ectodomains (Ad-RNF43/ZNRF3-ECDs, Ad-LGR5-ECD). Intriguingly, contrary to the studies published previously [78], stimulation of the Wnt receptor did not induce crypt hyperplasia or increased ISC numbers. In contrast, infection with Ad-RSPOs provoked crypt (hyper)proliferation and increased production of LGR5+ cells. As expected, Wnt pathway inhibition (infection with Ad-FZD-CRD/Ad-Dkk1) caused crypt demise that was not rescued by RSPOs. Similarly, crypts were damaged upon RSPO signalling inhibition (upon infection with Ad-RNF43/ZNRF3-ECDs and Ad-LGR5-ECD); however, in this particular case the crypt formation was rescued by (simultaneous) Wnt signalling activation. In conclusion, these studies underlined the role of Wnt ligands in priming epithelial cell renewal via establishment of the ISC fate. RSPO signalling can induce LGR5+ cell amplification; however, the signalling is insufficient for formation (or maintenance) of ISCs when the Wnt signal is absent [79,80].
LGR5 and related LGR4 and LGR6 proteins represent the B subgroup of LGR transmembrane proteins, related to the G-protein-coupled receptors (GPCRs) of the hormone receptor class. All LGRs contain a large N-terminal extracellular domain; however, in contrast to LGR4/5/6, the other members of the LGR A and C subgroups bind hormones [81,82,83]. LGR4/5/6 proteins are highly homologous; nevertheless, their expression pattern appears to be only partially overlapping. As already described, LGR5 was identified as a Wnt signalling-regulated marker of ISCs [22]. Additionally, subsequent studies established LGR5 as a marker of stem cells in the hair follicle, stomach, mammary gland and many other mouse tissues [84,85,86,87,88,89]. Rather surprisingly, Lgr5 knockout mice develop normally but die shortly after birth because of ankyloglossia, a craniofacial defect that prevents new-borns from suckling. Otherwise, LGR5-deficient animals do not display any (remarkable) phenotypic abnormalities [90]. The other subgroup member, Lgr6, is expressed in the embryonic hair placode and in the area interconnecting adult hair follicles with sebaceous glands. The LGR6+ cells were identified as adult stem cells with the potential to generate all skin cell lineages. Remarkably, Lgr6 expression is Wnt signalling independent and Lgr6 knockouts do not manifest any apparent defects [91]. In contrast, LGR4 is expressed in proliferating cell compartments of many tissues including the bone, gastrointestinal tract, kidney, liver, mammary gland, pancreas and skin. Consequently, whole-body disruption of the Lgr4 gene is embryonic/perinatal lethal [92,93]. In the mouse crypt, conditional ablation of Lgr4 had damaging effects on ISC proliferation. Deficiency in both Lgr4 and Lgr5 genes worsened the phenotype and led to crypt demise [9]. Lgr4 gene expression is activated by BMP2 in osteoblastic cells [94] or by signal transducer and activator of transcription 3 (STAT3) signalling in osteosarcoma cells [95]. The latter observation is in agreement with the phenotype of Lgr4 null mice, which display delayed osteoblast differentiation [96]. It also corroborates the finding that the Wnt pathway regulates bone mass, skeletogenesis and maintenance of adult skeleton [97]. Interestingly, Luo and colleagues discovered that LGR4 functions as an alternative receptor for RANKL (also known as TNFSF11). In canonical signalling, RANKL binds its “classical” receptor RANK, inducing differentiation of osteoclasts and thereby bone resorption. In pathological conditions, the signalling might lead to osteoporosis [98]. In contrast, LGR4 produced on osteoclasts acts in the opposite manner and the RANKL-LGR4 pathway blocks osteoclastogenesis and bone resorption [16]. In agreement with these results, whole-genome sequencing and single-nucleotide polymorphism (SNP) chip analysis of more than 64 thousand Icelanders revealed the presence of a rare nonsense, i.e., inactivating, mutation in the Lgr4 gene that is linked to low bone mineral density [99]. Interestingly, a different gain-of-function LGR4 variant is linked with central obesity and a metabolic phenotype in young Chinese [100]. Complementary to this finding, ablation of mouse Lgr4 stimulates white-to-brown fat transition and promotes energy expenditure [101]. Currently, Deng and colleagues revealed that the BMP2 antagonist norrin interacts with LGR4 and that norrin-LGR4 potentiates the Wnt pathway similarly as the RSPO-LGR4 interaction [102]. Finally, Planas-Paz and colleagues showed that in the mouse liver, RSPO1-LGR4 signalling controls organ size and tissue regeneration. Nevertheless, the authors show that some of the effects mediated by RSPO1-LGR4 signalling are Wnt pathway independent [15]. In summary, all these results imply that the role of LGR4-mediated signalling is not limited to intestinal tissue and in addition, the receptor is possibly involved in multiple intracellular signalling mechanisms.
Similar to LGRs, RSPO functions do not (completely) overlap. Nevertheless, all RSPOs have the capacity to induce crypt cell proliferation and β-catenin activation [103]. RSPO1-deficient mice are viable; however, RSPO1 absence affects their ovary development [104]. Rspo2 gene knockout is associated with facial skeletal defects, limb loss and lung hyperplasia resulting in perinatal respiratory failure [105]. RSPO3 deficiency results in embryonic lethality, due to aberrant placental development and cardiovascular abnormalities [106,107]. Results related to Rspo4 gene targeting are not available yet; however, in humans, mutations affecting RSPO4 function are associated with anonychia, the absence of finger- and toenails [108].
Aberrant RSPO/LGR signalling is frequently associated with CRC. Interestingly, in a fraction of CRCs harbouring intact Apc, Ctnnb1 or Axin2 genes, genetic rearrangements in the Rspo2 or Rpso3 locus were identified. The detected alterations generate fusions between Rspo2 and the eukaryotic translation initiation factor 3, subunit E (Eif3e) gene or between Rspo3 and the protein tyrosine phosphatase, receptor type, K (Ptprk) gene. Both fusions possibly lead to increased production of functional RSPO proteins resulting in aberrant Wnt signalling [109,110]. Furthermore, upregulation of LGR5 in colorectal tumours was associated with poor patient prognosis. This latter phenomenon is possibly associated with the involvement of LGR5+ tumour cells in relapse after therapy [111,112]. No direct association between LGR4 and CRC has yet been reported; however, increased LGR4 mRNA levels were observed in prostatic cancer [113]. Nevertheless, mutations inactivating LGR4 were associated with an increased incidence of biliary tract, gallbladder and skin cancer, suggesting a tumour suppressor role of LGR4 [99].
5. The Hippo Pathway
The Hippo signalling pathway was recently added to the list of signalling mechanisms involved in intestinal tumorigenesis. Importantly, several of the Hippo and Wnt pathway components are either shared between both pathways, or physically interact. The Hippo pathway was originally discovered in Drosophila melanogaster as an organ size regulatory system; however, its core components are evolutionally conserved [114,115]. The main modules identified in the mammalian Hippo pathway include serine/threonine STE20-like protein kinase 1 (MST1; alternative name STK4) and related MST2 (STK3), which are homologous to Drosophila Hippo, large tumour suppressor kinase 1/2 (LATS1/2) and scaffold proteins salvador family WW domain containing protein 1 (SAV1) and MOB Kinase Activator 1A/1B (MOB1A/1B). The pathway is stimulated when the cell receives growth inhibitory signals upon sensing high cell density and contact inhibition. The pathway regulates cellular localization of two proteins, Yes-associated protein (YAP1) and tafazzin (TAZ) [116,117]. Upon MST1/2 activation—via an extracellular stimulus and transmembrane signal relay mechanism that have not been clarified yet—MST1/2 form a complex with SAV1. Subsequently, the signal activates kinase LATS1/2 that interacts with MOB1A/1B. LATS1/2 phosphorylates YAP1 at serine 127 [118,119] and TAZ at serine 89 [120]. The phosphorylation enables binding of YAP1/TAZ to 14-3-3 proteins and prevents their transport to the nucleus. LATS-mediated phosphorylation of YAP1 at serine 381 primes subsequent phosphorylation at serine 384 and serine 387 executed by CK1. Phosphorylated YAP1 is recognized by β-TrCP, which leads to YAP ubiquitination and degradation [121].
In the Hippo signalling OFF state, YAP1 and TAZ mediators are translocated into the nucleus, where they bind members of the TEA domain transcription factor (TEAD) family [122] and promote transcription of pro-proliferative and anti-apoptotic genes. Alternatively, YAP1 and TAZ associate with runt-related transcription factor 2 (RUNX2) [123], SMAD [124] and AP-1 transcription complexes [125]. YAP1/TAZ can also function as transcription co-repressor after recruiting the nucleosome remodelling and deacetylase (NuRD) complex [126]. The absence of cell-cell contacts leads to RHO activation that, together with the actin cytoskeleton, promotes YAP1/TAZ nuclear translocation. This implies that cell contacts (or cell adhesion to the extracellular matrix) might be the impulse activating Hippo signalling. Another mechanism affecting YAP1/TAZ nuclear translocation is mediated by GPCRs that act via RHO-dependent LATS1/2 inactivation and YAP1/TAZ nuclear translocation [14].
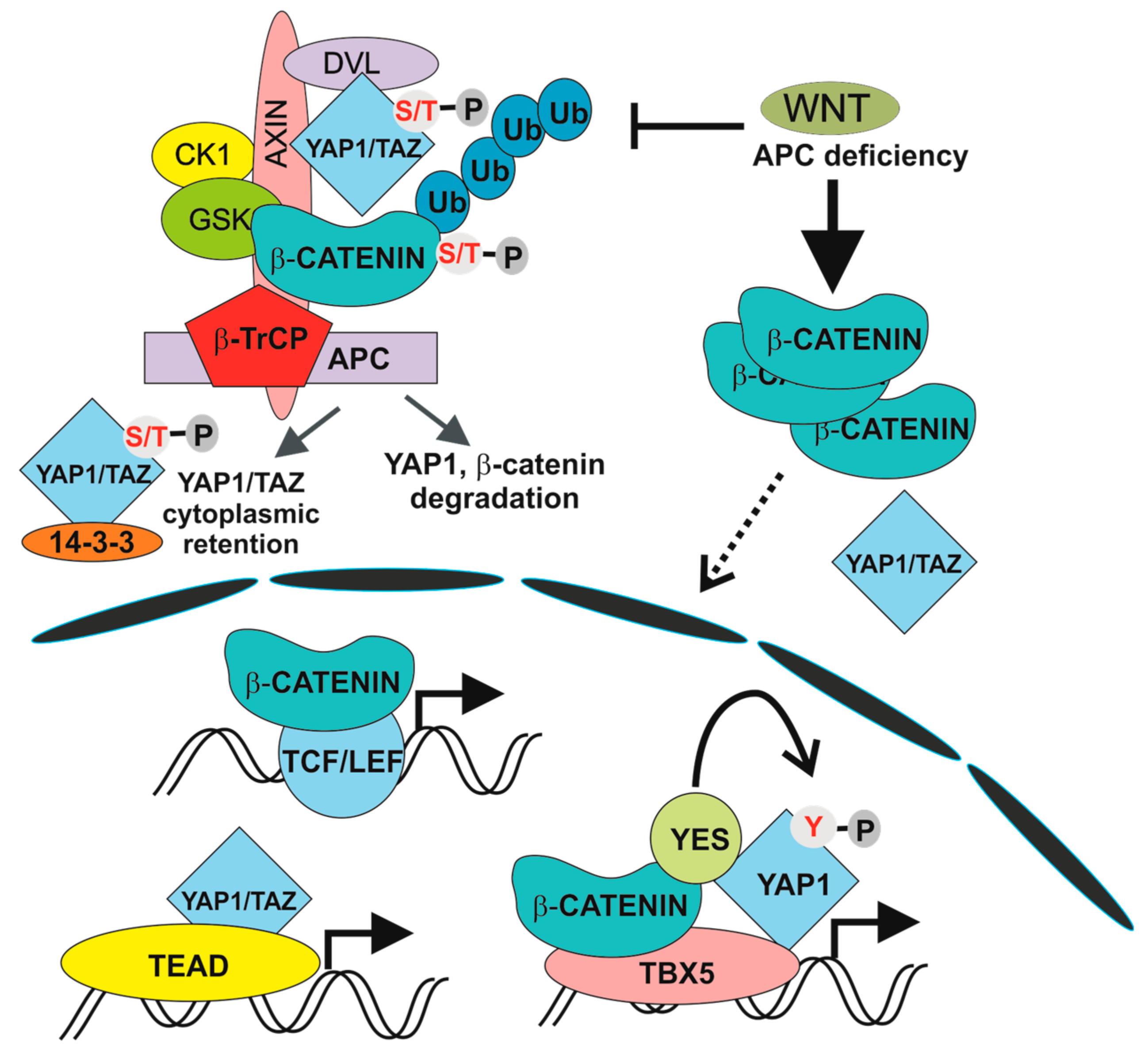
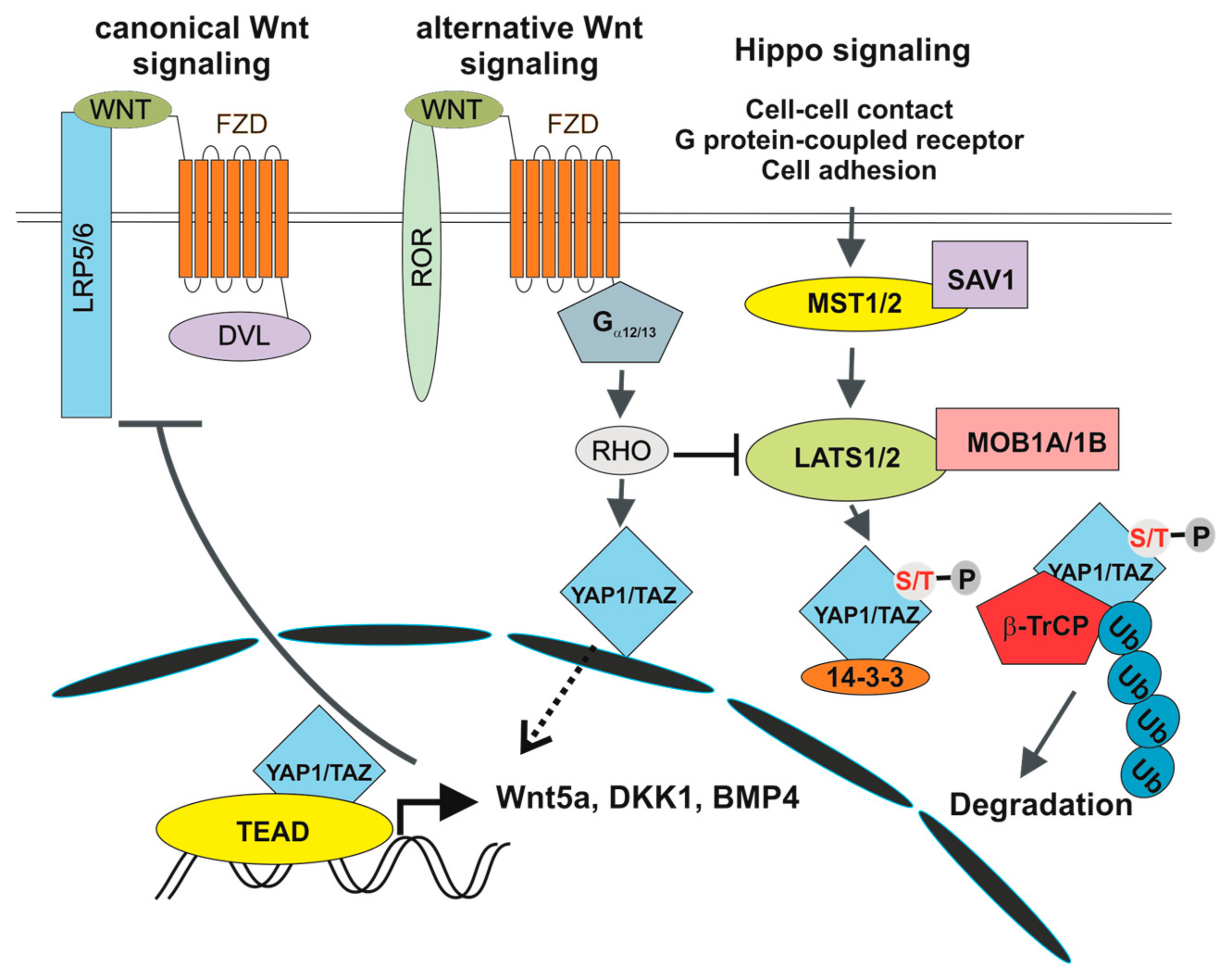
The involvement of the Hippo pathway in intestinal tumorigenesis was revealed by Camargo and co-workers. They showed that inducible YAP1 (over)expression in the mouse induced intestinal hyperplasia accompanied by the absence of mature Paneth and goblet cells. The observed phenotype, which was reversible and strictly dependent on YAP1 production, was not caused by cell dedifferentiation. Instead, differentiated cells were replaced by newly arising YAP1-expressing progenitors [11]. In addition, several studies associated deregulation of the Hippo pathway with poor prognosis of CRC patients [127,128]. There is growing evidence for multiple crosstalk between Hippo and Wnt/β-catenin signalling, although some results are controversial. Zhou and colleagues reported stimulation of Wnt/β-catenin signalling upon inactivation of MST1/2 kinases [128]. In addition, co-immunoprecipitation studies showed interaction between DVL and TAZ. Moreover, TAZ inhibited Wnt3a-induced DVL phosphorylation [129]. Similarly, Barry and colleagues reported functional interaction between YAP1 and DVL. YAP1 was detected in the cytoplasm of TA cells and in epithelial cells present on the villi, where it attenuated Wnt signalling by inhibiting DVL function. Moreover, overexpression of YAP1 mutant localized to the cytoplasm blocked crypt proliferation and induced intestinal degeneration. Additionally, YAP1 absence caused Wnt hypersensitivity during tissue regeneration that was connected with intestinal stem cell niche expansion. In their interpretation, the authors conclude that DVL nuclear translocation is the rescue mechanism in response to intestinal injury and that YAP1 controls this localization [130]. Additionally, direct interaction between β-catenin and phosphorylated YAP1/TAZ was reported to retain β-catenin cytoplasmic localization [131]. Intriguingly, Cai and colleagues documented interaction between SAV1, LATS and APC independently of β-catenin destruction complex formation. Consequently, APC deficiency leads to transcription of YAP1-specific target genes, including TAZ and β-catenin. In addition, ablation of the Yap1 or Taz gene abolished intestinal adenoma formation in APC-Min mice [132]. In contrast, the Piccolo group demonstrated direct regulation of TAZ and YAP1 activity by the β-catenin destruction complex. Upon Wnt pathway activation (and β-catenin destruction complex inactivation) YAP1 and TAZ are stabilized, translocate to the nucleus and together with TEAD transcription factors, drive expression of a specific set of target genes (Figure 2). The authors propose that a substantial portion of the genes regulated by aberrant Wnt signalling in CRC cells are in fact activated by YAP1/TAZ-TEAD complexes. The conclusion was supported by an intriguing observation showing that tumour formation in the intestine induced by conditional ablation of the Apc gene is completely suppressed by simultaneous inactivation of Yap1 and Taz [12,13]. Interestingly, individual and double deficiency of YAP1 and TAZ had no effect on crypt cell proliferation and intestinal architecture, indicating that YAP1/TAZ are dispensable for intestinal homeostasis [12]. A different model explaining the dependency of intestinal tumorigenesis on YAP1 was presented by Rosenbluh and colleagues. The authors performed a genome-scale loss-of-function screen to identify genes essential for cancer cells displaying active Wnt/β-catenin signalling. They found that proliferation and cell survival of the majority of CRC cells depends on the transcriptional activity of a protein complex that involves β-catenin, YAP1 and T-box 5 (TBX5) DNA-binding protein [133]. Interestingly, β-catenin transcriptional activity is influenced by the methylation status of YAP1. YAP1 methylated at lysine 494 by SET domain containing lysine methyltransferase 7 (SETD7) binds β-catenin and promotes its nuclear localization and transcription of Wnt target genes [134]. Besides that, YAP1 phosphorylation by YES proto-oncogene 1 tyrosine kinase (YES1) impacts on YAP1 transcriptional activity in CRC cells [133] (Figure 2). Finally, Park and colleagues corroborated an alternative model of Wnt signalling that directly includes some of the Hippo pathway components. In the “alternative” Wnt signalling pathway, the engagement of both types of canonical and noncanonical Wnt ligands with the receptor complex composed of FZD and ROR triggers a cascade of intracellular events that includes G12/G13 alpha subunits of heterotrimeric G proteins (Gα12/13), RHO GTPases, and LATS1/2 promoting YAP1/TAZ activation and TEAD-mediated transcription (Figure 3). In primary human bone-marrow stem cells, the pathway regulates osteogenic differentiation and cell migration. Intriguingly, some of the genes activated by the pathway encode inhibitors of the canonical Wnt pathway (e.g., DKK1) [14]. However, whether alternative Wnt signalling is a common signalling mechanism or is restricted to some specific cell types has yet to be defined.
Acknowledgments
We thank S. Takacova and T. Epp for critically reading the manuscript. The experimental work related to the topic of this review is supported by the Ministry of Education, Youth and Sports of the Czech Republic (grant number LO1419) and the institutional grant (RVO 68378050).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destree, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Huber, O.; Korn, R.; McLaughlin, J.; Ohsugi, M.; Herrmann, B.G.; Kemler, R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 1996, 59, 3–10. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; vanWichen, D.; deWeger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signalling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single LGR5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as mediator of Wnt signalling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt signalling activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Marti, P.; et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, Z.; Ma, Y.; Yue, Z.; Lin, H.; Qu, G.; Huang, J.; Dai, W.; Li, C.; Zheng, C.; et al. Lgr4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat. Med. 2016, 22, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Stem cells. What is an adult stem cell? Science 2015, 350, 1319–1320. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Wagers, A.J. No place like home: Anatomy and function of the stem cell niche. Nat. Rev. Mol. Cell Biol. 2008, 9, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Sancho, E.; Verweij, C.; de Lau, W.; Oving, I.; Hurlstone, A.; van der Horn, K.; Batlle, E.; Coudreuse, D.; Haramis, A.P.; et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [Google Scholar] [CrossRef]
- Van der Flier, L.G.; Sabates-Bellver, J.; Oving, I.; Haegebarth, A.; De Palo, M.; Anti, M.; Van Gijn, M.E.; Suijkerbuijk, S.; Van de Wetering, M.; Marra, G.; et al. The intestinal Wnt/TCF signature. Gastroenterology 2007, 132, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene LGR5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; Haegebarth, A.; Stange, D.E.; van de Wetering, M.; Clevers, H. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology 2009, 137, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Stange, D.E.; Schepers, A.G.; van de Wetering, M.; Koo, B.K.; Itzkovitz, S.; Volckmann, R.; Kung, K.S.; Koster, J.; Radulescu, S.; et al. The LGR5 intestinal stem cell signature: Robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012, 31, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Fafilek, B.; Krausova, M.; Vojtechova, M.; Pospichalova, V.; Tumova, L.; Sloncova, E.; Huranova, M.; Stancikova, J.; Hlavata, A.; Svec, J.; et al. Troy, a tumour necrosis factor receptor family member, interacts with LGR5 to inhibit Wnt signalling in intestinal stem cells. Gastroenterology 2013, 144, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sachs, N.; Wiebrands, K.; Ellenbroek, S.I.; Fumagalli, A.; Lyubimova, A.; Begthel, H.; van den Born, M.; van Es, J.H.; Karthaus, W.R.; et al. Reg4+ deep crypt secretory cells function as epithelial niche for LGR5+ stem cells in colon. Proc. Natl. Acad. Sci. USA 2016, 113, E5399–E5407. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S.; Booth, C.; Pritchard, D.M. The intestinal epithelial stem cell: The mucosal governor. Int. J. Exp. Pathol. 1997, 78, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; de Sauvage, F.J. A reserve stem cell population in small intestine renders LGR5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Itzkovitz, S.; Lyubimova, A.; Blat, I.C.; Maynard, M.; van Es, J.; Lees, J.; Jacks, T.; Clevers, H.; van Oudenaarden, A. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat. Cell Biol. 2012, 14, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asfaha, S.; Hayakawa, Y.; Muley, A.; Stokes, S.; Graham, T.A.; Ericksen, R.E.; Westphalen, C.B.; von Burstin, J.; Mastracci, T.L.; Worthley, D.L.; et al. Krt19+/LGR5− cells are radioresistant cancer-initiating stem cells in the colon and intestine. Cell Stem Cell 2015, 16, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; de Sauvage, F.; van Es, J.H.; et al. Replacement of lost LGR5-positive stem cells through plasticity of their enterocyte-lineage daughters. Cell Stem Cell 2016, 18, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Buczacki, S.J.; Zecchini, H.I.; Nicholson, A.M.; Russell, R.; Vermeulen, L.; Kemp, R.; Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing LGR5. Nature 2013, 495, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Meran, L.; Baulies, A.; Li, V.S.W. Intestinal stem cell niche: The extracellular matrix and cellular components. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for LGR5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Van Es, J.H.; Clevers, H. Redundant sources of wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 2012, 143, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Escudero, S.; Shivdasani, R.A. Intact function of LGR5 receptor-expressing intestinal stem cells in the absence of Paneth cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3932–3937. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, Z.; Greicius, G.; Madan, B.; Biechele, S.; Zhong, Z.; Zaribafzadeh, H.; Edison; Aliyev, J.; Wu, Y.; Bunte, R.; et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development 2014, 141, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, R.; Nusse, R. Towards an integrated view of Wnt signalling in development. Development 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R., 3rd; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Takada, R.; Satomi, Y.; Kurata, T.; Ueno, N.; Norioka, S.; Kondoh, H.; Takao, T.; Takada, S. Monounsaturated fatty acid modification of Wnt protein: Its role in Wnt secretion. Dev. Cell 2006, 11, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Doubravska, L.; Krausova, M.; Gradl, D.; Vojtechova, M.; Tumova, L.; Lukas, J.; Valenta, T.; Pospichalova, V.; Fafilek, B.; Plachy, J.; et al. Fatty acid modification of Wnt1 and Wnt3a at serine is prerequisite for lipidation at cysteine and is essential for Wnt signalling. Cell. Signal. 2011, 23, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Langton, P.F.; Kakugawa, S.; Vincent, J.P. Making, exporting and modulating Wnts. Trends Cell Biol. 2016, 26, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Gregorieff, A.; Pinto, D.; Begthel, H.; Destree, O.; Kielman, M.; Clevers, H. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology 2005, 129, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, D.J.; Phesse, T.J.; Barker, N.; Schwab, R.H.; Amin, N.; Malaterre, J.; Stange, D.E.; Nowell, C.J.; Currie, S.A.; Saw, J.T.; et al. Frizzled7 functions as a Wnt receptor in intestinal epithelial LGR5+ stem cells. Stem Cell Rep. 2015, 4, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ueno, K.; Hazama, S.; Mitomori, S.; Nishioka, M.; Suehiro, Y.; Hirata, H.; Oka, M.; Imai, K.; Dahiya, R.; Hinoda, Y. Down-regulation of frizzled-7 expression decreases survival, invasion and metastatic capabilities of colon cancer cells. Br. J. Cancer 2009, 101, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Hrckulak, D.; Kolar, M.; Strnad, H.; Korinek, V. TCF/LEF transcription factors: An update from the internet resources. Cancers 2016, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Tortelote, G.G.; Reis, R.R.; de Almeida Mendes, F.; Abreu, J.G. Complexity of the Wnt/beta-catenin pathway: Searching for an activation model. Cell. Signal. 2017, 40, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Davis, C.R.; Wang, H.T.; Chu, P.; Lee, M.; Yuan, J.; Nusse, R.; Kuo, C.J. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc. Natl. Acad. Sci. USA 2004, 101, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Nishisho, I.; Kinzler, K.W.; Vogelstein, B.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Nagase, H. Mutations of the adenomatous polyposis coli gene in familial polyposis coli patients and sporadic colorectal tumors. Princess Takamatsu Symp. 1991, 22, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Bright-Thomas, R.M.; Hargest, R. APC, beta-catenin and hTCF-4; an unholy trinity in the genesis of colorectal cancer. Eur. J. Surg. Oncol. 2003, 29, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, S.M.; Petty, E.M.; Stoffel, E.M.; Fearon, E.R. An AXIN2 mutant allele associated with predisposition to colorectal neoplasia has context-dependent effects on AXIN2 protein function. Neoplasia 2015, 17, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.S.; Dismuke, A.D.; Powell, A.E.; Carroll, K.H.; Wong, M.H. Wnt-reporter expression pattern in the mouse intestine during homeostasis. BMC Gastroenterol. 2008, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Jordens, I.; Mosa, M.H.; Basak, O.; Korving, J.; Tauriello, D.V.; de Punder, K.; Angers, S.; Peters, P.J.; Maurice, M.M.; et al. Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 2016, 530, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Mlodzik, M. Planar cell polarity signaling: From fly development to human disease. Annu. Rev. Genet. 2008, 42, 517–540. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Mehdawi, L.M.; Prasad, C.P.; Ehrnstrom, R.; Andersson, T.; Sjolander, A. Non-canonical WNT5A signaling up-regulates the expression of the tumor suppressor 15-PGDH and induces differentiation of colon cancer cells. Mol. Oncol. 2016, 10, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Medegan, B.; Braun, D.P. Wnt9A induction linked to suppression of human colorectal cancer cell proliferation. Int. J. Mol. Sci. 2016, 17, 495. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.R.; Das, A.M.; Helvensteijn, W.; Franken, P.F.; Swagemakers, S.; van der Valk, M.A.; ten Hagen, T.L.; Kuipers, E.J.; van Veelen, W.; Smits, R. Wnt5a promotes human colon cancer cell migration and invasion but does not augment intestinal tumorigenesis in Apc1638N mice. Carcinogenesis 2013, 34, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.K.; Lee, J.S. Cellular signaling and biological functions of R-spondins. Cell. Signal. 2012, 24, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Yokota, C.; Semenov, M.V.; Doble, B.; Woodgett, J.; He, X. R-spondin1 is a high affinity ligand for LRP6 and induces LRP6 phosphorylation and beta-catenin signaling. J. Biol. Chem. 2007, 282, 15903–15911. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Ruffner, H.; Sprunger, J.; Charlat, O.; Leighton-Davies, J.; Grosshans, B.; Salathe, A.; Zietzling, S.; Beck, V.; Therier, M.; Isken, A.; et al. R-spondin potentiates Wnt/beta-catenin signaling through orphan receptors LGR4 and LGR5. PLoS ONE 2012, 7, e40976. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/LGR5/RNF43 module: Regulator of Wnt signal strength. Genes Dev. 2014, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zamponi, R.; Charlat, O.; Ramones, M.; Swalley, S.; Jiang, X.; Rivera, D.; Tschantz, W.; Lu, B.; Quinn, L.; et al. Interaction with both ZNRF3 and LGR4 is required for the signalling activity of R-spondin. EMBO Rep. 2013, 14, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Zebisch, M.; Xu, Y.; Krastev, C.; MacDonald, B.T.; Chen, M.; Gilbert, R.J.; He, X.; Jones, E.Y. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 2013, 4, 2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loregger, A.; Grandl, M.; Mejias-Luque, R.; Allgauer, M.; Degenhart, K.; Haselmann, V.; Oikonomou, C.; Hatzis, P.; Janssen, K.P.; Nitsche, U.; et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated beta-catenin by sequestering TCF4 to the nuclear membrane. Sci. Signal. 2015, 8, ra90. [Google Scholar] [CrossRef] [PubMed]
- Carmon, K.S.; Gong, X.; Yi, J.; Thomas, A.; Liu, Q. RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E1221–E1229. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Charlat, O.; Zamponi, R.; Yang, Y.; Cong, F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell 2015, 58, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of APC in vivo immediately perturbs Wnt signaling, differentiation and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.S.; Janda, C.Y.; Chang, J.; Zheng, G.X.Y.; Larkin, K.A.; Luca, V.C.; Chia, L.A.; Mah, A.T.; Han, A.; Terry, J.M.; et al. Non-equivalence of Wnt and R-spondin ligands during LGR5+ intestinal stem-cell self-renewal. Nature 2017, 545, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Janda, C.Y.; Dang, L.T.; You, C.; Chang, J.; de Lau, W.; Zhong, Z.A.; Yan, K.S.; Marecic, O.; Siepe, D.; Li, X.; et al. Surrogate Wnt agonists that phenocopy canonical Wnt and beta-catenin signalling. Nature 2017, 545, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Wu, F.J.; Kudo, M.; Hsiao, C.L.; Hsueh, A.J.; Luo, C.W. A naturally occurring LGR4 splice variant encodes a soluble antagonist useful for demonstrating the gonadal roles of LGR4 in mammals. PLoS ONE 2014, 9, e106804. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.Y.; Liang, S.G.; Hsueh, A.J. Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine-rich repeats and a G protein-coupled, seven-transmembrane region. Mol. Endocrinol. 1998, 12, 1830–1845. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.; Wang, R.; Bailey, W.; Xie, G.; Chen, F.; Caskey, C.T.; Liu, Q. Identification and cloning of an orphan G protein-coupled receptor of the glycoprotein hormone receptor subfamily. Biochem. Biophys. Res. Commun. 1998, 247, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Jaks, V.; Barker, N.; Kasper, M.; van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgard, R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 2008, 40, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Brenot, A.; Lawson, D.A.; Linnemann, J.R.; Van Kappel, E.C.; Wong, K.C.; de Sauvage, F.; Klein, O.D.; Werb, Z. Lgr5-expressing cells are sufficient and necessary for postnatal mammary gland organogenesis. Cell Rep. 2013, 3, 70–78. [Google Scholar] [CrossRef] [PubMed]
- de Visser, K.E.; Ciampricotti, M.; Michalak, E.M.; Tan, D.W.; Speksnijder, E.N.; Hau, C.S.; Clevers, H.; Barker, N.; Jonkers, J. Developmental stage-specific contribution of LGR5+ cells to basal and luminal epithelial lineages in the postnatal mammary gland. J. Pathol. 2012, 228, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.K.; Li, Y.; Redding, K.M.; Iwatsuki, K.; Margolskee, R.F.; Jiang, P. Lgr5-EGFP marks taste bud stem/progenitor cells in posterior tongue. Stem Cells 2013, 31, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Rookmaaker, M.B.; Kujala, P.; Ng, A.; Leushacke, M.; Snippert, H.; van de Wetering, M.; Tan, S.; Van Es, J.H.; Huch, M.; et al. Lgr5+ve stem/progenitor cells contribute to nephron formation during kidney development. Cell Rep. 2012, 2, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Mazerbourg, S.; Bouley, D.M.; Luo, C.W.; Kawamura, K.; Kuwabara, Y.; Baribault, H.; Tian, H.; Hsueh, A.J. Neonatal lethality of LGR5 null mice is associated with ankyloglossia and gastrointestinal distension. Mol. Cell. Biol. 2004, 24, 9736–9743. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; van Es, J.H.; Barker, N.; van de Wetering, M.; van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 2010, 327, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.A.; Mitchell, K.J.; Goodrich, L.V.; Lu, X.; Pinson, K.; Scherz, P.; Skarnes, W.C.; Tessier-Lavigne, M. Defining brain wiring patterns and mechanisms through gene trapping in mice. Nature 2001, 410, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Mazerbourg, S.; Bouley, D.M.; Sudo, S.; Klein, C.A.; Zhang, J.V.; Kawamura, K.; Goodrich, L.V.; Rayburn, H.; Tessier-Lavigne, M.; Hsueh, A.J. Leucine-rich repeat-containing, G protein-coupled receptor 4 null mice exhibit intrauterine growth retardation associated with embryonic and perinatal lethality. Mol. Endocrinol. 2004, 18, 2241–2254. [Google Scholar] [CrossRef] [PubMed]
- Pawaputanon Na Mahasarakham, C.; Ezura, Y.; Kawasaki, M.; Smriti, A.; Moriya, S.; Yamada, T.; Izu, Y.; Nifuji, A.; Nishimori, K.; Izumi, Y.; et al. BMP-2 enhances Lgr4 gene expression in osteoblastic cells. J. Cell. Physiol. 2016, 231, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, W.; Guo, C.A.; Han, N.; Pan, J.F.; Fei, T.; Yan, Z.Q. Stat3 upregulates leucine-rich repeat-containing G protein-coupled receptor 4 expression in osteosarcoma cells. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhou, W.; Zhou, X.; Li, D.; Weng, J.; Yi, Z.; Cho, S.G.; Li, C.; Yi, T.; Wu, X.; et al. Regulation of bone formation and remodeling by G-protein-coupled receptor 48. Development 2009, 136, 2747–2756. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, A.; Norozi, F.; Shahrabi, S.; Shahjahani, M.; Saki, N. Wnt/beta-catenin signaling in bone marrow niche. Cell Tissue Res. 2016, 363, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Boyle, W.J.; Simonet, W.S.; Kostenuik, P.J.; Dougall, W.C.; Sullivan, J.K.; San Martin, J.; Dansey, R. Bench to bedside: Elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat. Rev. Drug Discov. 2012, 11, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Styrkarsdottir, U.; Thorleifsson, G.; Sulem, P.; Gudbjartsson, D.F.; Sigurdsson, A.; Jonasdottir, A.; Oddsson, A.; Helgason, A.; Magnusson, O.T.; Walters, G.B.; et al. Nonsense mutation in the LGR4 gene is associated with several human diseases and other traits. Nature 2013, 497, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ning, T.; Shi, J.; Chen, M.; Ding, L.; Huang, Y.; Kauderer, S.; Xu, M.; Cui, B.; Bi, Y.; et al. Association of a gain-of-function variant in LGR4 with central obesity. Obesity (Silver Spring) 2017, 25, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, R.; Wang, F.; Hong, J.; Li, X.; Chen, M.; Ke, Y.; Zhang, X.; Ma, Q.; Wang, R.; et al. Ablation of LGR4 promotes energy expenditure by driving white-to-brown fat switch. Nat. Cell Biol. 2013, 15, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Reddy, P.; Cheng, Y.; Luo, C.W.; Hsiao, C.L.; Hsueh, A.J. Multi-functional norrin is a ligand for the LGR4 receptor. J. Cell Sci. 2013, 126, 2060–2068. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Zhao, J.; Andarmani, S.; Kakitani, M.; Oshima, T.; Binnerts, M.E.; Abo, A.; Tomizuka, K.; Funk, W.D. R-spondin proteins: A novel link to beta-catenin activation. Cell Cycle 2006, 5, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Tomizuka, K.; Horikoshi, K.; Kitada, R.; Sugawara, Y.; Iba, Y.; Kojima, A.; Yoshitome, A.; Yamawaki, K.; Amagai, M.; Inoue, A.; et al. R-spondin1 plays an essential role in ovarian development through positively regulating Wnt-4 signaling. Hum. Mol. Genet. 2008, 17, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Yamada, W.; Nagao, K.; Horikoshi, K.; Fujikura, A.; Ikeda, E.; Inagaki, Y.; Kakitani, M.; Tomizuka, K.; Miyazaki, H.; Suda, T.; et al. Craniofacial malformation in R-spondin2 knockout mice. Biochem. Biophys. Res. Commun. 2009, 381, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Mieda, M.; Ikeda, T.; Hamada, Y.; Nakamura, H.; Okamoto, H. R-spondin3 is required for mouse placental development. Dev. Biol. 2007, 301, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Kazanskaya, O.; Ohkawara, B.; Heroult, M.; Wu, W.; Maltry, N.; Augustin, H.G.; Niehrs, C. The Wnt signaling regulator R-spondin 3 promotes angioblast and vascular development. Development 2008, 135, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Bruchle, N.O.; Frank, J.; Frank, V.; Senderek, J.; Akar, A.; Koc, E.; Rigopoulos, D.; van Steensel, M.; Zerres, K.; Bergmann, C. RSPO4 is the major gene in autosomal-recessive anonychia and mutations cluster in the furin-like cysteine-rich domains of the Wnt signaling ligand R-spondin 4. J. Investig. Dermatol. 2008, 128, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Schatoff, E.M.; Murphy, C.; Zafra, M.P.; Wilkinson, J.E.; Elemento, O.; Dow, L.E. R-spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat. Commun. 2017, 8, 15945. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.Y.; Lee, T.L.; Hao, S.S.; Mahooti, S.; Baird, S.M.; Donoghue, D.J.; Haas, M. Visualization of early prostatic adenocarcinoma as a stem cell disease. Oncotarget 2016, 7, 76159–76168. [Google Scholar] [CrossRef] [PubMed]
- Merlos-Suarez, A.; Barriga, F.M.; Jung, P.; Iglesias, M.; Cespedes, M.V.; Rossell, D.; Sevillano, M.; Hernando-Momblona, X.; da Silva-Diz, V.; Munoz, P.; et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 2011, 8, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Yue, J.; Wang, J.; Zhang, L.; Fan, R.; Zhang, H.; Zhang, Q. GPCR48/LGR4 promotes tumorigenesis of prostate cancer via PI3K/Akt signaling pathway. Med. Oncol. 2015, 32, 49. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The Hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Hariharan, I.K. The Hippo pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011288. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Hong, W. The emerging role of the Hippo pathway in cell contact inhibition, organ size control and cancer development in mammals. Cancer Cell 2008, 13, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Konsavage, W.M., Jr.; Yochum, G.S. Intersection of Hippo/YAP and Wnt/beta-catenin signaling pathways. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Marignani, P.A.; Sarbassova, D.; Yagi, R.; Hall, R.A.; Donowitz, M.; Hisaminato, A.; Fujiwara, T.; Ito, Y.; Cantley, L.C.; et al. Taz: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000, 19, 6778–6791. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. Tead mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.K.; Sullivan, A.J.; Medina, R.; Ito, Y.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 2004, 23, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the Hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; McNeill, H.; et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 2010, 18, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imajo, M.; Miyatake, K.; Iimura, A.; Miyamoto, A.; Nishida, E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signalling. EMBO J. 2012, 31, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Maitra, A.; Anders, R.A.; Taketo, M.M.; Pan, D. Beta-catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015, 29, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Beta-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Oudhoff, M.J.; Braam, M.J.S.; Freeman, S.A.; Wong, D.; Rattray, D.G.; Wang, J.; Antignano, F.; Snyder, K.; Refaeli, I.; Hughes, M.R.; et al. SETD7 controls intestinal regeneration and tumorigenesis by regulating Wnt/beta-catenin and Hippo/YAP signaling. Dev. Cell 2016, 37, 47–57. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Enhancement of Wnt signalling by R-spondin (RSPO) ligands. DVL, dishevelled; FZD, frizzled; LGR5, leucine-rich-repeat-containing G-protein-coupled receptor 5; LRP5/6, low-density lipoprotein receptor-related protein 5 or 6; RNF43, ring finger 43; ZNRF3, zinc and ring finger 3.
Figure 1.
Enhancement of Wnt signalling by R-spondin (RSPO) ligands. DVL, dishevelled; FZD, frizzled; LGR5, leucine-rich-repeat-containing G-protein-coupled receptor 5; LRP5/6, low-density lipoprotein receptor-related protein 5 or 6; RNF43, ring finger 43; ZNRF3, zinc and ring finger 3.
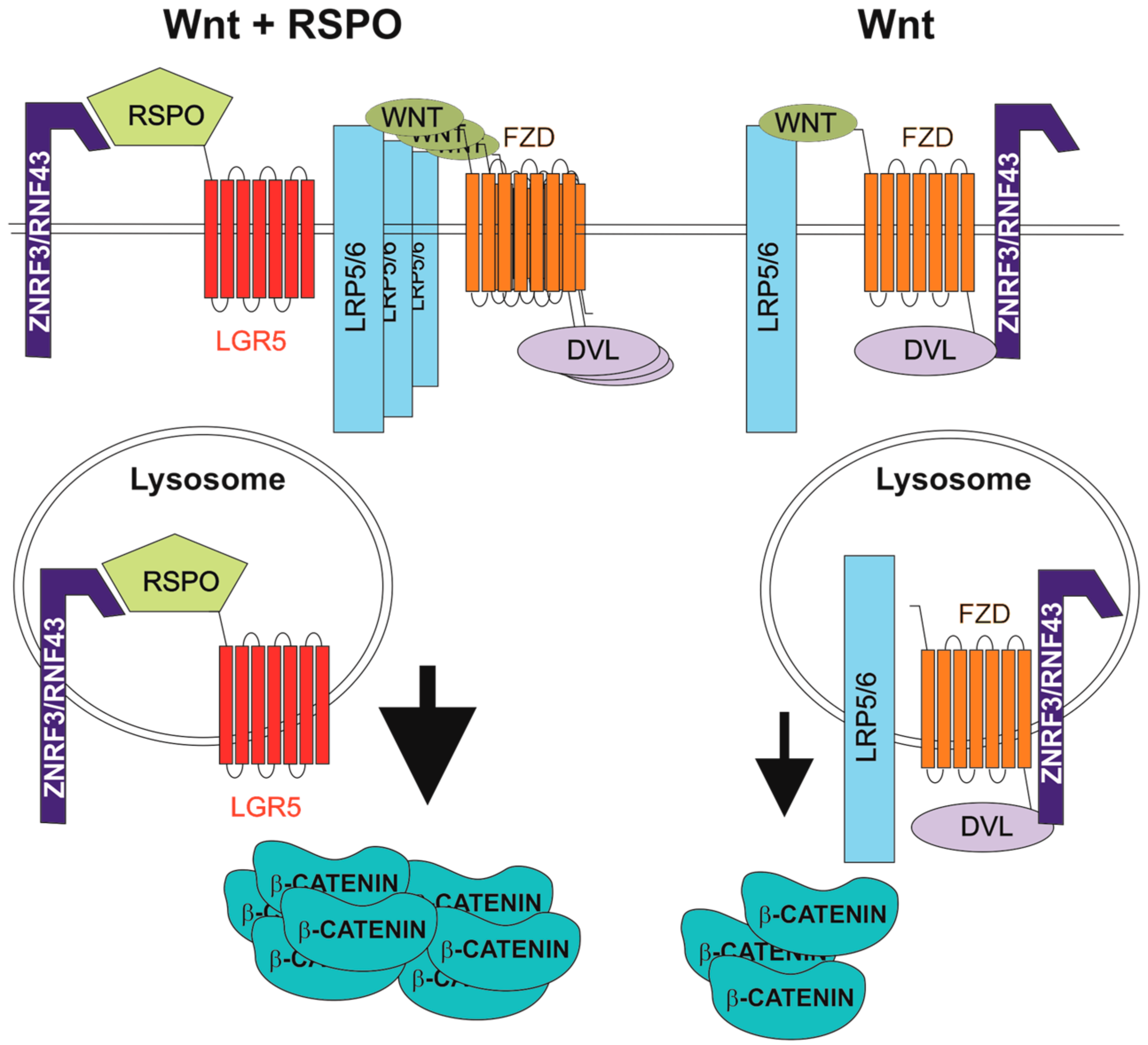
Figure 2.
Transcription complexes induced by physiological and aberrant Wnt pathway activation. β-TrCP, β-transduction repeat-containing protein; APC, adenomatous polyposis coli, CK1, casein kinase-1, GSK3, glycogen synthetase kinase 3; S/T-P, the phosphorylated serine or threonine residue; TAZ, tafazzin; TEAD, TEA domain transcription factors; TBX5, T-box 5; TCF/LEF, T-cell factor /lymphoid enhancer factor; Ub, ubiquitin; Y-P, the phosphorylated tyrosine residue; YAP1, Yes-associated protein; YES; YES proto-oncogene 1.
Figure 2.
Transcription complexes induced by physiological and aberrant Wnt pathway activation. β-TrCP, β-transduction repeat-containing protein; APC, adenomatous polyposis coli, CK1, casein kinase-1, GSK3, glycogen synthetase kinase 3; S/T-P, the phosphorylated serine or threonine residue; TAZ, tafazzin; TEAD, TEA domain transcription factors; TBX5, T-box 5; TCF/LEF, T-cell factor /lymphoid enhancer factor; Ub, ubiquitin; Y-P, the phosphorylated tyrosine residue; YAP1, Yes-associated protein; YES; YES proto-oncogene 1.
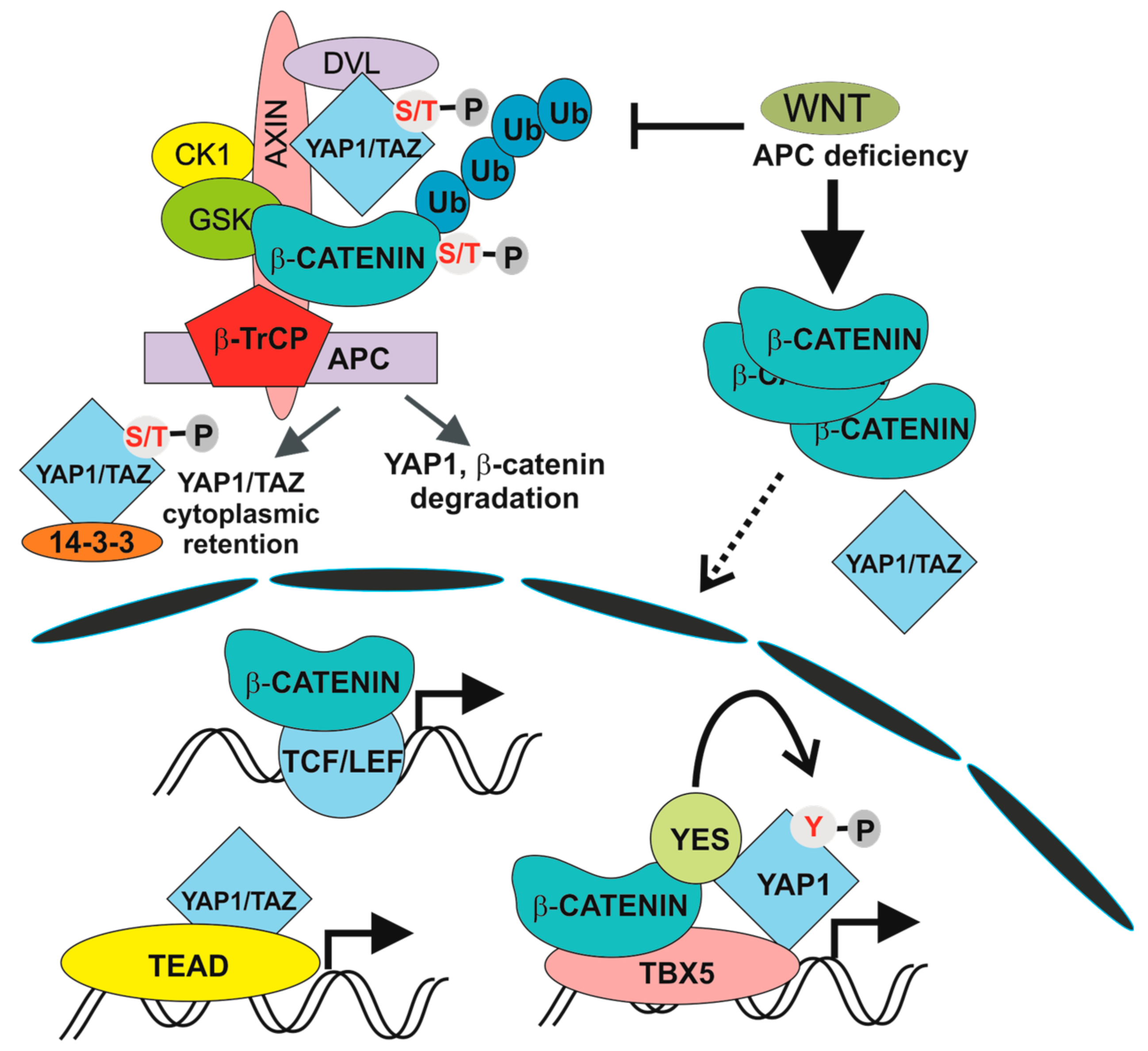
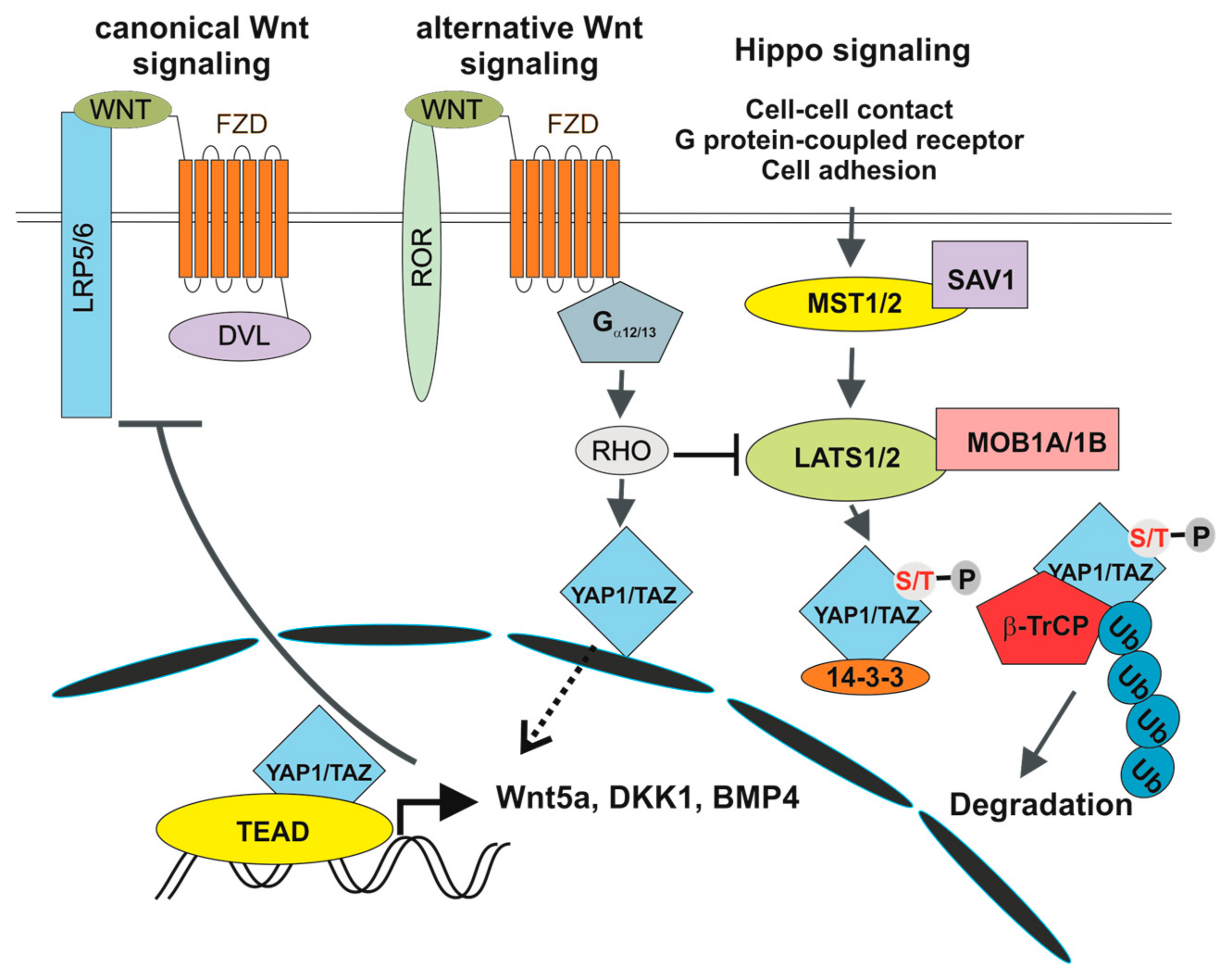
Figure 3.
Crosstalk between canonical and alternative Wnt signalling and the Hippo pathway. 14-3-3, 14-3-3 protein; BMP4, bone morphogenetic protein 4; DKK1, dickkopf-1; Gα12/13, G12/G13 alpha subunits of heterotrimeric G proteins; LATS1/2, large tumour suppressor kinase 1/2; MOB1A/1B, MOB Kinase Activator 1A/1B; MST1, STE20-like protein kinase 1; RHO, RHO GTPase; ROR1/2, receptor tyrosine kinase-like orphan receptor 1/2; SAV1, salvador family WW domain containing protein 1.
Figure 3.
Crosstalk between canonical and alternative Wnt signalling and the Hippo pathway. 14-3-3, 14-3-3 protein; BMP4, bone morphogenetic protein 4; DKK1, dickkopf-1; Gα12/13, G12/G13 alpha subunits of heterotrimeric G proteins; LATS1/2, large tumour suppressor kinase 1/2; MOB1A/1B, MOB Kinase Activator 1A/1B; MST1, STE20-like protein kinase 1; RHO, RHO GTPase; ROR1/2, receptor tyrosine kinase-like orphan receptor 1/2; SAV1, salvador family WW domain containing protein 1.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kriz, V.; Korinek, V. Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells. Genes 2018, 9, 20. https://doi.org/10.3390/genes9010020
AMA Style
Kriz V, Korinek V. Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells. Genes. 2018; 9(1):20. https://doi.org/10.3390/genes9010020
Chicago/Turabian StyleKriz, Vitezslav, and Vladimir Korinek. 2018. "Wnt, RSPO and Hippo Signalling in the Intestine and Intestinal Stem Cells" Genes 9, no. 1: 20. https://doi.org/10.3390/genes9010020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.