
Identification of miRNAs Responsive to Botrytis cinerea in Herbaceous Peony (Paeonia lactiflora Pall.) by High-Throughput Sequencing

Abstract
:1. Introduction
2. Experimental Section
2.1. Plant Materials

2.2. Small RNA Library Construction and Sequencing
2.3. miRNA Identification and Corresponding Target Gene Prediction
2.4. Differentially Expressed miRNAs and Their Target Gene Annotation
2.5. Digital Gene Expression Analysis
2.6. Expression Analysis of Target Genes by qRT-PCR
3. Results
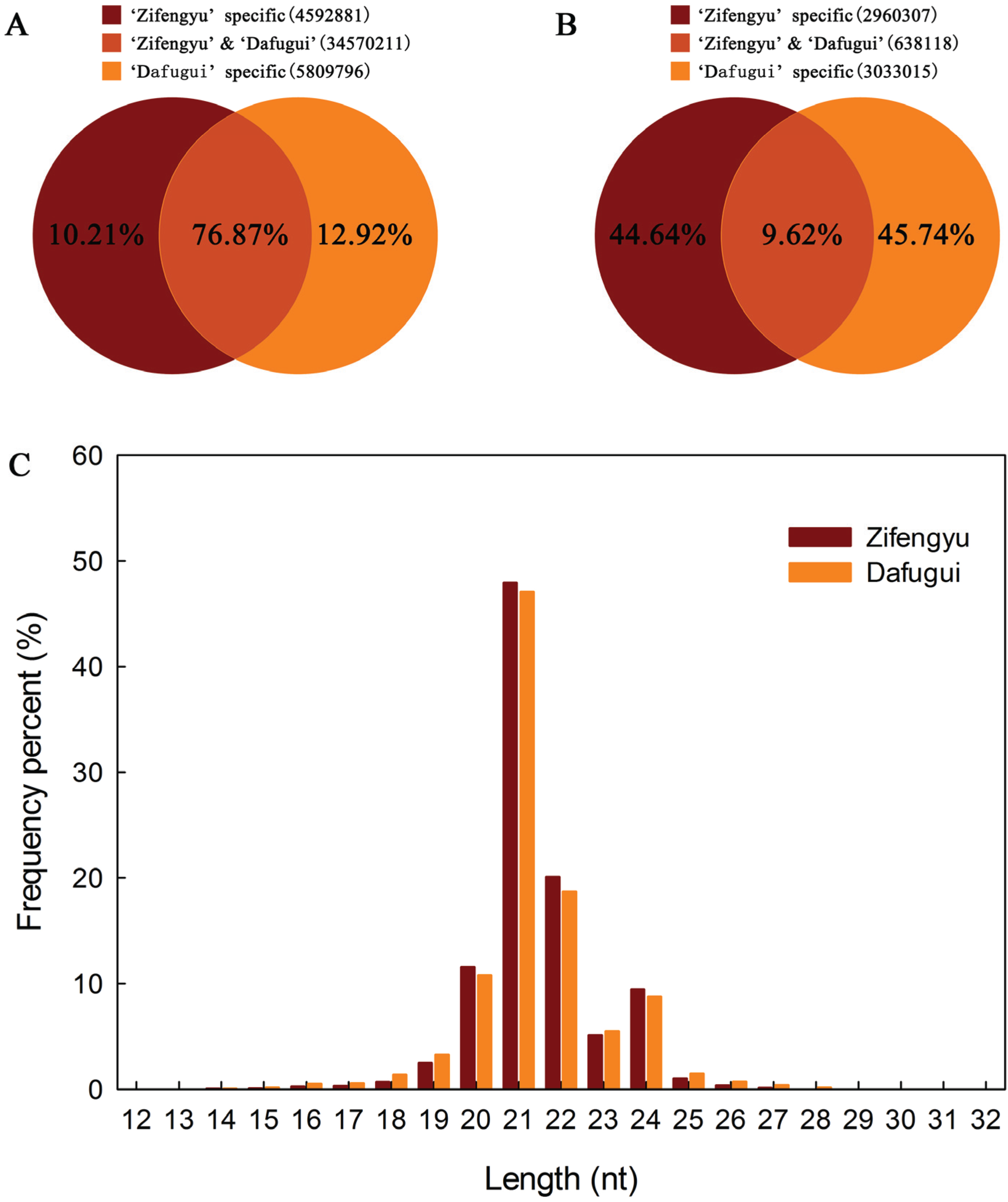
3.1. Sequence Analysis of sRNAs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Unique sRNAs | Total sRNAs | ||
|---|---|---|---|---|
| “Zifengyu” | “Dafugui” | “Zifengyu” | “Dafugui” | |
| rRNA | 60653 | 77278 | 1047570 | 1338595 |
| tRNA | 16257 | 19165 | 1180850 | 1206549 |
| snRNA | 4057 | 4310 | 16627 | 15686 |
| snoRNA | 1031 | 1033 | 3289 | 3180 |
| Unannotated | 3496291 | 3540283 | 19428764 | 17158589 |
| miRNA | 20136 | 29064 | 1843482 | 1729707 |
| Total | 3598425 | 3671133 | 23520582 | 21452306 |
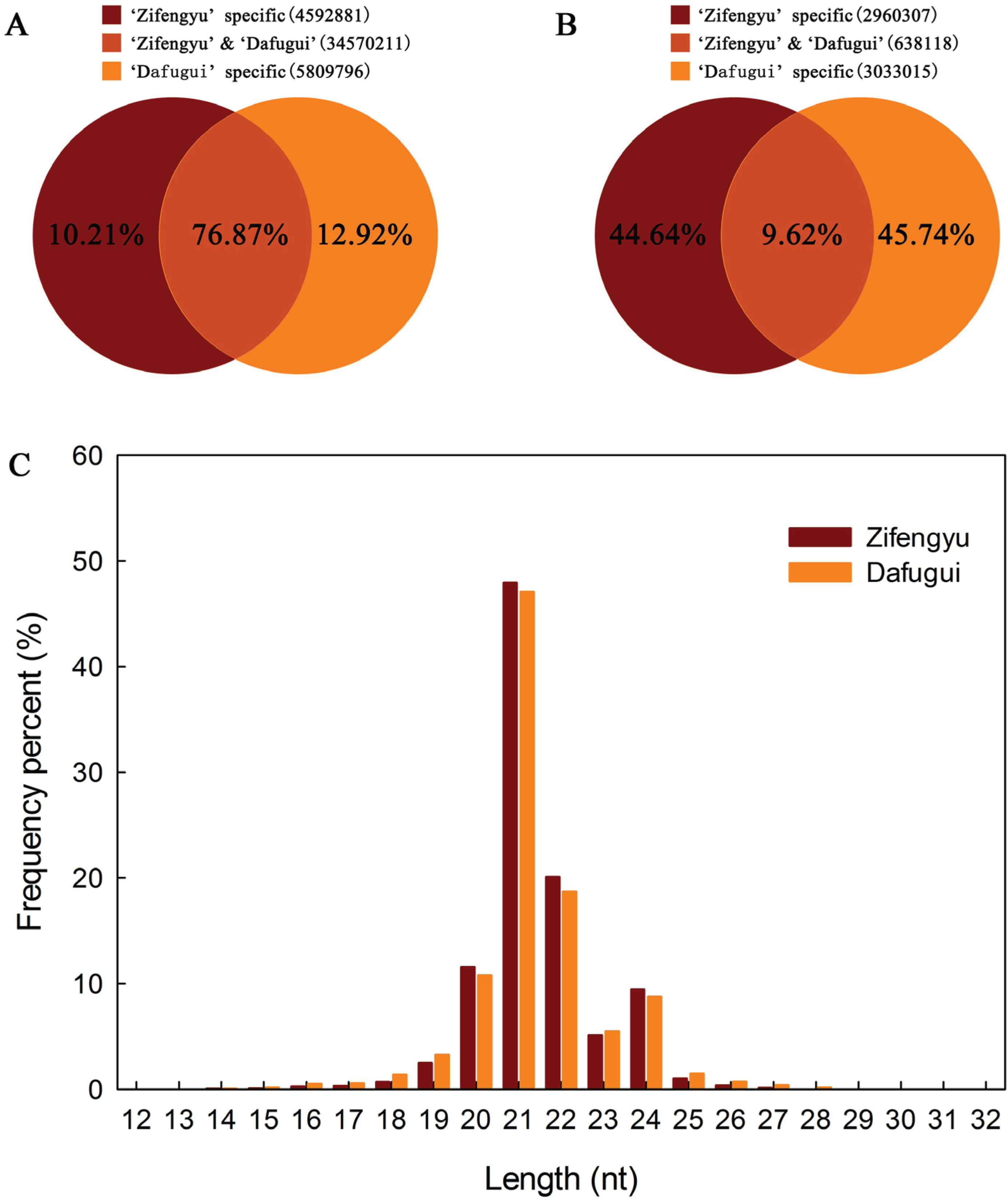
3.2. Identification of Conserved miRNAs
3.3. Identification of Novel miRNAs
| Name in miRBase | Sequence | Length (nt) | Start/End Precursor | Length of Precursor (nt) | Arm | MFE (kcal/mol) | Count |
|---|---|---|---|---|---|---|---|
| pla-MIR11601 | TGCTCTAAAAGATCGTAGTTC | 21 | 1–262 | 262 | 5' | −34.80 | 12 |
| pla-MIR11605 | TTGAGGCGGCATATTCTCAAT | 21 | 73–178 | 106 | 3' | −29.82 | 18 |
| pla-MIR11606 | TGGTGGACTCCAATTCGCATA | 21 | 1563–1694 | 132 | 3' | −49.60 | 1113 |
| pla-MIR11607 | GATCACTCGGTTGTCTGACACAC | 23 | 169–297 | 129 | 5' | −27.50 | 1258 |
| pla-MIR11608 | TCGCTTAGGGGTTGTTGAAGCGC | 23 | 119–222 | 104 | 5' | −38.60 | 29 |
| pla-MIR11609 | TAGCTTGGTGTGAGGTCAACTT | 22 | 132–309 | 178 | 5' | −43.50 | 11 |
| pla-MIR11610 | ACAGATATGGTAGGGGGCACA | 21 | 10,914–10,989 | 76 | 3' | −36.70 | 222 |
| pla-MIR11611 | TAGGCAACCGTGGTAAAATGTC | 22 | 945–1045 | 101 | 3' | −48.10 | 6 |
| pla-MIR11612 | AAGACGGTCCAAAACGCCCAC | 21 | 151–283 | 133 | 3' | −63.20 | 490 |
| Name in miRBase | Sequence | Length (nt) | Start/End Precursor | Length of Precursor (nt) | Arm | MFE (kcal/mol) | Count |
|---|---|---|---|---|---|---|---|
| pla-MIR11598 | TCGTTCAAAGTAGGTTGTCAA | 21 | 113–349 | 237 | 5' | −45.60 | 16 |
| pla-MIR11599 | TTCAACCGTGGTAGATGTTAA | 21 | 95–306 | 212 | 5' | −92.70 | 13 |
| pla-MIR11600 | AACGTTCCTCGATTTCGCGAT | 21 | 471–546 | 76 | 3' | −24.90 | 6 |
| pla-MIR11601 | TGCTCTAAAAGATCGTAGTTC | 21 | 1–262 | 262 | 5' | −34.80 | 15 |
| pla-MIR11602 | TCTAACGGAACGCTATTGGATC | 22 | 6884–6998 | 115 | 3' | −22.10 | 8 |
| pla-MIR11603 | TTATAATTAGGTTGAGCGGAC | 21 | 40–313 | 274 | 5' | −62.00 | 2979 |
| pla-MIR11604 | TCCAGAGGGAGAACGTGGCGA | 21 | 328–406 | 79 | 3' | −26.90 | 9 |
| pla-MIR11605 | TTGAGGCGGCATATTCTCAAT | 21 | 73–178 | 106 | 3' | −29.82 | 41 |
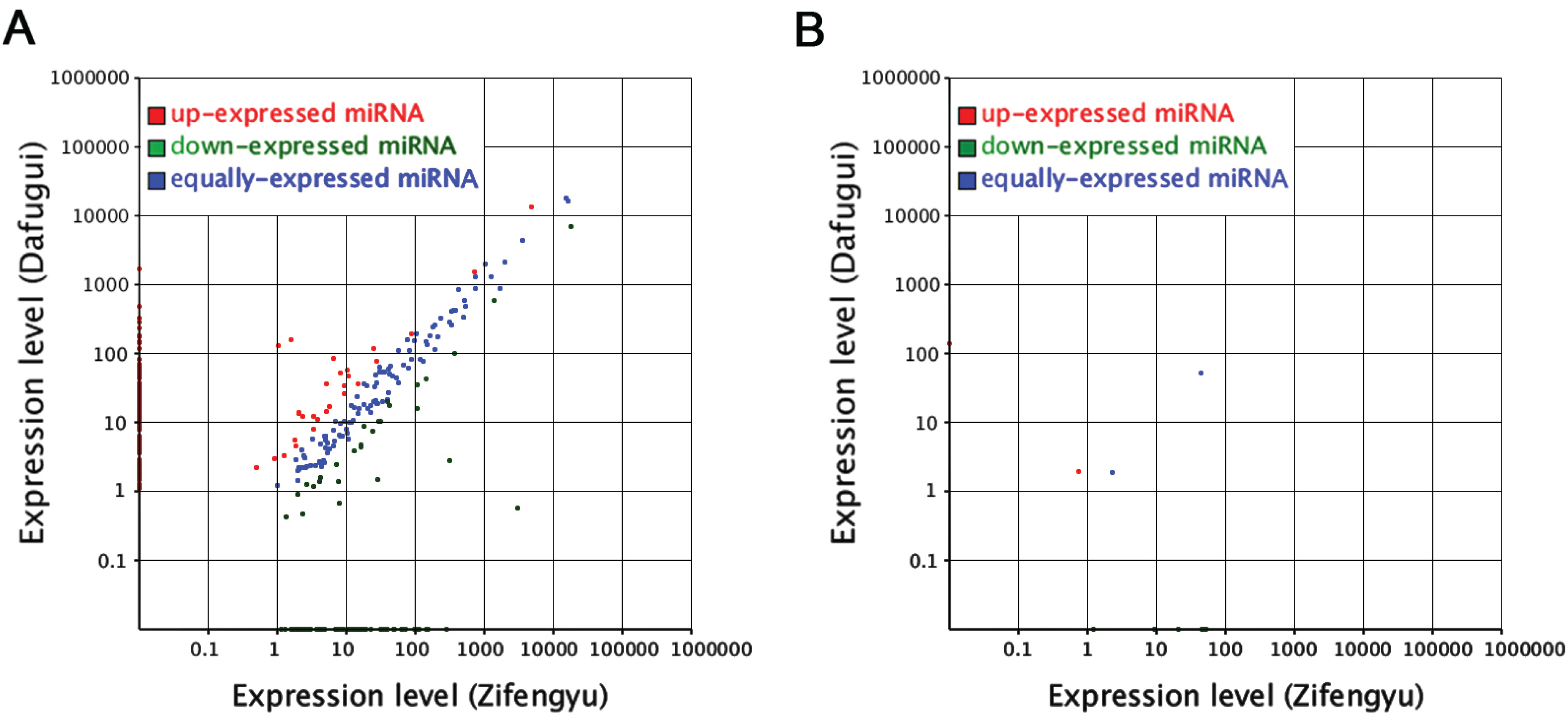
3.4. Comparative Analysis of miRNAs and Corresponding Target Genes between Two Libraries
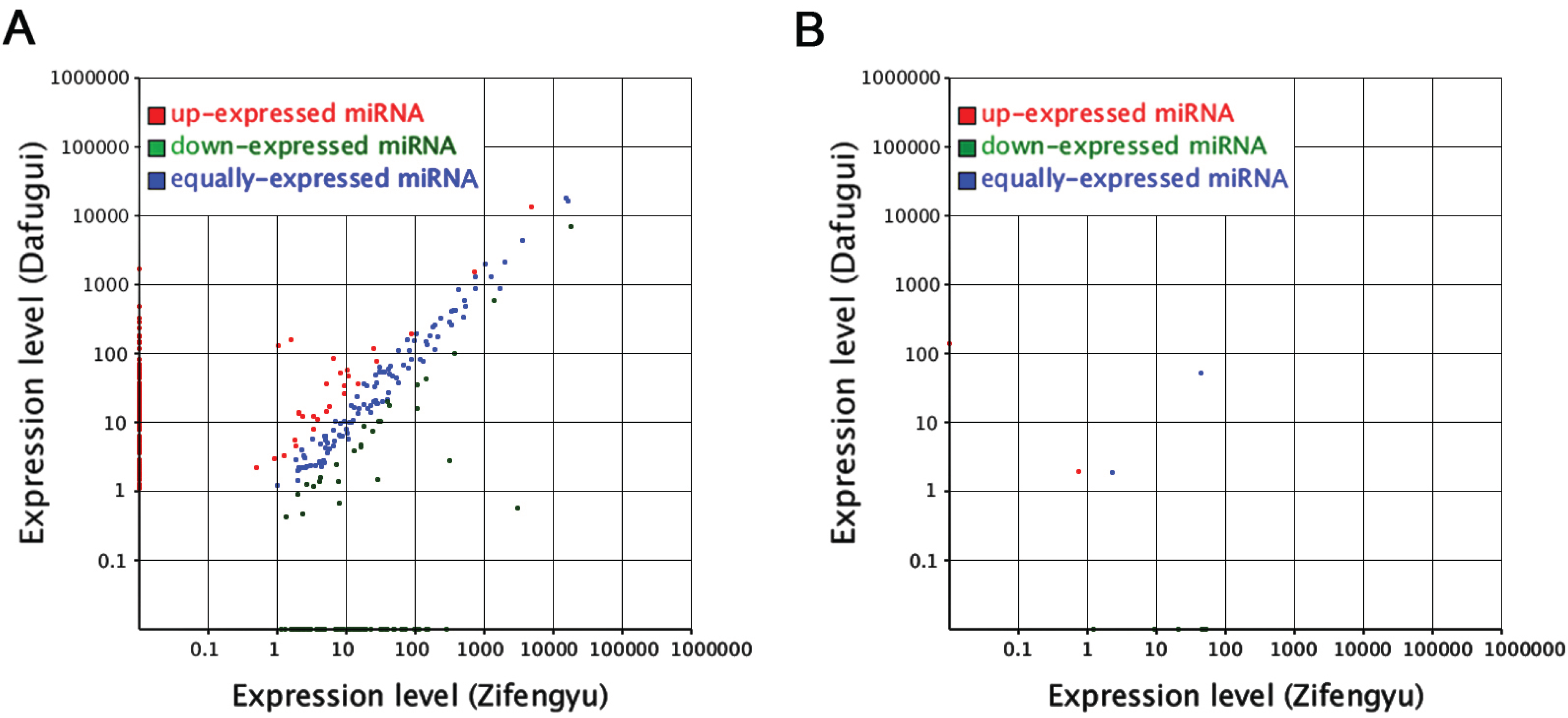
3.5. Analysis of the Candidate B. cinerea-Responsive miRNAs
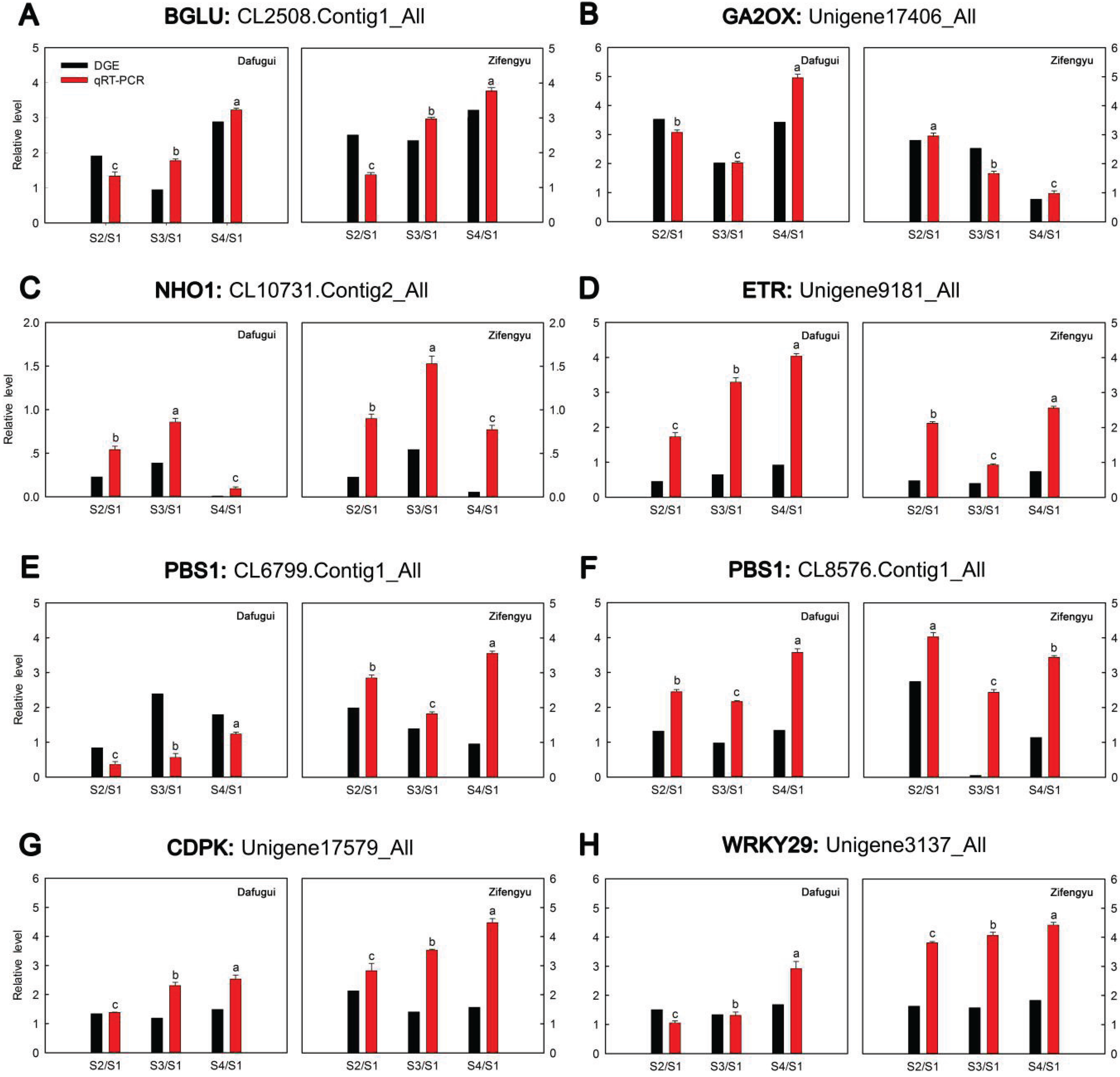
| miR-Name | Fold-Change (log2 Dafugui/Zifengyu) | Target Gene | Annotation | Expression Pattern of Target Gene (log2 Sn/S1) |
|---|---|---|---|---|
| miR5254 | 9.02815472 | CL2508.Contig1_All | beta-glucosidase |  |
| miR846 | 12.12015362 | Unigene17406_All | gibberellin 2-oxidase |  |
| miR165a-3p | 1.39849113 | CL10731.Contig2_All | nonhost1 |  |
| miR6432 | 8.82072147 | CL7438.Contig1_All | peroxin-10 |  |
| miR6450a | −11.19652845 | Unigene9181_All | ethylene receptor |  |
| CL6799.Contig1_All | serine/threonine-protein kinase PBS1 |  | ||
| miR5558-3p | −2.35248612 | CL8576.Contig1_All | serine/threonine-protein kinase PBS1 |  |
| miR3897-3p | 7.50620839 | Unigene17579_All | calcium-dependent protein kinase |  |
| Unigene3137_All | WRKY transcription factor 29 |  |
3.6. qRT-PCR Analysis of Target Genes
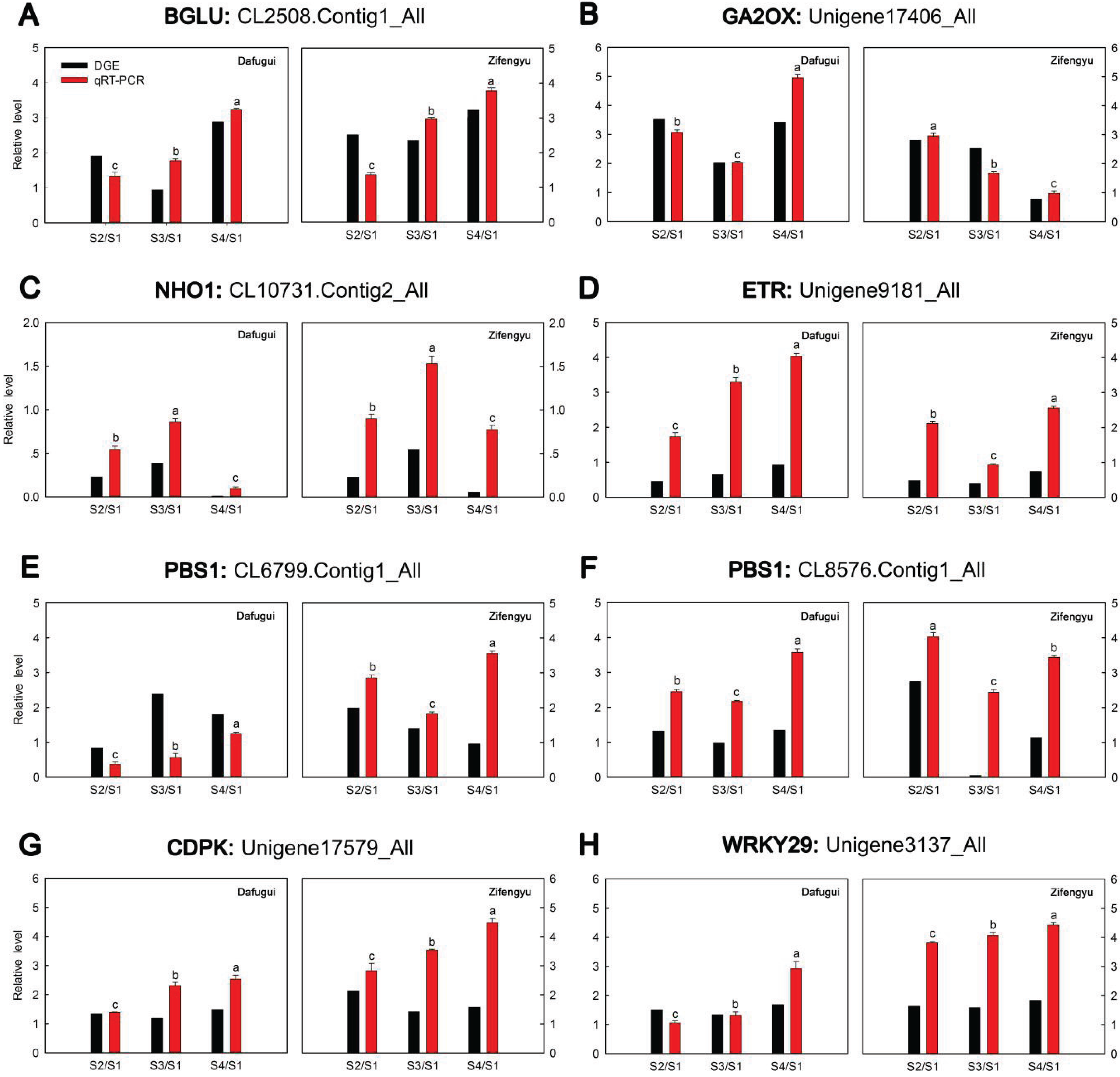
4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stevens, S.; Stevens, A.B.; Gast, K.L.B.; O’Mara, J.A.; Tisserat, N.A.; Bauernfeind, R. Commercial Specialty Cut Flower Production, Peonies; Cooperative Extension Service, Kansas State University: Manhattan, KS, USA, 1993. [Google Scholar]
- Wang, Y.; Jiang, W.; Wei, J.; Weng, M.; Han, J. Cultural implication and landscape application of Chinese herbaceous peony. Guangdong Agric. Sci. 2013, 20, 58–61. [Google Scholar]
- Lan, Y. Pathogenic identification and biological research on the gray-mold disease of peony. J. Nanjing For. Univ. 1987, 1, 8–14. [Google Scholar]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Two decades of miRNA biology: Lessons and challenges. RNA 2015, 21, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Zhang, B.H.; Pan, X.P.; Anderson, T.A. Identification of 188 conserved maize microRNAs and their targets. FEBS Lett. 2006, 580, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Guo, G.; Ni, Z.; Sunkar, R.; Du, J.; Zhu, J.K.; Sun, Q. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.Y.; Zhang, L.; Li, G.L.; Zhang, L.; Wang, X.J.; Cao, X.F.; Fang, X.; Chen, F. Identification of novel stress-regulated microRNAs from Oryza sativa L. Genomics 2010, 95, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Cai, W.J.; Wang, S.; Shan, C.M.; Wang, L.J.; Chen, X.Y. Temporal control of trichome distribution by microRNA156-targeted SPL genes in Arabidopsis thaliana. Plant Cell 2010, 22, 2322–2335. [Google Scholar] [CrossRef] [PubMed]
- Carlsbecker, A.; Lee, J.Y.; Roberts, C.J.; Dettmer, J.; Lehesranta, S.; Zhou, J.; Lindgren, O.; Moreno-Risueno, M.A.; Vatén, A.; Thitamadee, S.; et al. Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 2010, 465, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.S.; Li, J.Y.; Stahle, M.L.; Dubroué, A.; Gubler, F.; Millar, A.A. Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc. Natl. Acad. Sci. USA 2007, 104, 16371–16376. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, Y.C.; Wang, C.Y.; Luo, Y.C.; Huang, Q.J.; Chen, S.Y.; Zhou, H.; Qu, L.H.; Chen, Y.Q. Expression analysis of phytohormone regulated microRNAs in rice, implying their regulation roles in plant hormone signaling. FEBS Lett. 2009, 583, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Carnavale Bottino, M.; Rosario, S.; Grativol, C.; Thiebaut, F.; Rojas, C.A.; Farrineli, L.; Hemerly, A.S.; Ferreira, P.C. High-throughput sequencing of small RNA transcriptome reveals salt stress regulated microRNAs in sugarcane. PLoS ONE 2014, 8, e59423. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Mineta, K.; Hirai, M.Y.; Suzuki, Y.; Kanaya, S.; Takahashi, H.; Onouchi, H.; Yamaguchi, J.; Naito, S. Changes in mRNA stability associated with cold stress in Arabidopsis cells. Plant Cell Physiol. 2013, 54, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wang, J.; Cheng, H.; Wang, X.; Yu, D. Detection and evolutionary analysis of soybean miRNAs responsive to soybean mosaic virus. Planta 2013, 237, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.B.; Wu, F.L. Characterization of miRNAs associated with Botrytis cinerea infection of tomato leaves. BMC Plant Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.B.; Wu, F.L.; Xiao, L.; Liang, G.W.; Zhen, Y.X.; Guo, Z.K.; Guo, A.G. Microarray-based analysis of tomato miRNA regulated by Botrytis cinerea. J. Plant Growth Regul. 2012, 31, 38–46. [Google Scholar] [CrossRef]
- Zhao, D.Q.; Zhou, C.H.; Tao, J. Carotenoid accumulation and carotenogenic genes expression during two types of persimmon fruit (Diospyros kaki L.) development. Plant Mol. Biol. Rep. 2011, 29, 646–654. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.X.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weigel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.J.; Hao, Z.J.; Meng, J.S.; Liu, D.; Wei, M.R.; Tao, J. Digital gene expression analysis to screen disease resistance-relevant genes from leaves of herbaceous peony (Paeonia lactiflora Pall.) infected by Botrytis cinerea. PLoS ONE 2015, 10, e0133305. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.Q.; Jiang, Y.; Ning, C.L.; Meng, J.S.; Lin, S.S.; Ding, W. Transcriptome sequencing of a chimaera reveals coordinated expression of anthocyanin biosynthetic genes mediating yellow formation in herbaceous peony (Paeonia lactiflora Pall.). BMC Genomics 2014, 15, 689. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Tao, J.; Han, C.; Ge, J. Actin as an alternative internal control gene for gene expression analysis in herbaceous peony (Paeonia lactiflora Pall.). Afr. J. Agric. Res. 2012, 7, 2153–2159. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 36, 1101–1108. [Google Scholar] [CrossRef]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006, 38, S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Chiou, T.J.; Lin, S.I.; Aung, K.; Zhu, J.K. A miRNA involved in phosphate starvation response in Arabidopsis. Curr. Biol. 2005, 15, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Wang, L.; Chen, S.; Jiang, J.; Guan, Z.; Li, P.; Chen, F. Identification of nitrogen starvation-responsive microRNAs in Chrysanthemum nankingense. Plant Physiol. Biochem. 2015, 91, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, L.; Liu, X.; Cui, D.; Chen, T.; Zhang, H.; Jiang, C.; Xu, C.; Li, P.; Li, S.; et al. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE 2013, 8, e55107. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Wang, F.; Xu, Z.S.; Jiang, Q.; Ma, J.; Tan, G.F.; Xiong, A.S. High throughput sequencing of two celery cultivars small RNAs identifies microRNAs involved in temperature stress response. BMC Genomics 2014. [Google Scholar] [CrossRef]
- Ding, Y.F.; Zhu, C. The role of microRNAs in copper and cadmium homeostasis. Biochem. Biophys. Res. Commun. 2009, 386, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Gal-On, A.; Kaplan, I.; Palukaitis, P. Differential effects of satellite RNA on the accumulation of cucumber mosaic virus RNAs and their encoded proteins in tobacco vs. zucchini squash with two strains of CMV helper virus. Virology 1995, 208, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Diermann, N.; Matoušek, J.; Junge, M.; Riesner, D.; Steger, G. Characterization of plant miRNAs and small RNAs derived from potato spindle tuber viroid (PSTVd) in infected tomato. Biol. Chem. 2010, 391, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.P.; Xiao, Y.N.; Zhao, J.R. Digital gene expression analysis of early root infection resistance to Sporisorium reilianum f. sp. zeae in maize. Mol. Genet. Genomics 2013, 288, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jiang, H.Z.; Zhu, X.Y.; Wang, W.N.; He, X.H.; Shi, Y.Z. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genomics 2013. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Pan, X.; Wilson, I.W.; Li, F.; Liu, M.; Teng, W.; Zhang, B. High throughput sequencing technology reveals that the taxoid elicitor methyl jasmonate regulates microRNA expression in Chinese yew (Taxus chinensis). Gene 2009, 436, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, I.A.; Fossdal, C.G.; Johnsen, Ø. MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010, 187, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, C.; Zhang, C.; Korir, N.K.; Yu, H.; Ma, Z.; Fang, J. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 2010. [Google Scholar] [CrossRef] [PubMed]
- Donaire, L.; Pedrola, L.; Rosa Rde, L.; Llave, C. High-throughput sequencing of RNA silencing-associated small RNAs in olive (Olea europaea L.). PLoS ONE 2011, 6, e27916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Li, G.; Mi, S.; Li, S.; Hannon, G.J.; Wang, X.J.; Qi, Y. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 2007, 21, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Cang, J.; Yu, J.; Lu, B.W.; Liu, L.J.; Wang, J.F. Solexa sequencing and bioinformatics analysis of small RNA in winter wheat. Chin. Bull. Bot. 2014, 49, 8–18. [Google Scholar]
- De Cremer, K.; Mathys, J.; Vos, C.; Froenicke, L.; Michelmore, R.W.; Cammue, B.P.; de Coninck, B. RNAseq-based transcriptome analysis of Lactuca sativa infected by the fungal necrotroph Botrytis cinerea. Plant Cell Environ. 2013, 36, 1992–2007. [Google Scholar] [PubMed]
- Smith, J.E.; Mengesha, B.; Tang, H.; Mengiste, T.; Bluhm, B.H. Resistance to Botrytis cinerea in Solanum lycopersicoides involves widespread transcriptional reprogramming. BMC Genomics 2014. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Anduro, G.; Ceniceros-Ojeda, E.A.; Casados-Vazquez, L.E. Genome-wide analysis of the beta-glucosidase gene family in maize (Zea mays L. var B73). Plant Mol. Biol. 2011, 77, 159–183. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.J.; Cho, K.C.; Hwang, O.J.; Choi, Y.S.; Shin, A.Y.; Hwang, I.; Kim, J.I. Overexpression of an Arabidopsis β-glucosidase gene enhances drought resistance with dwarf phenotype in creeping bentgrass. Plant Cell Rep. 2012, 31, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Tang, X.Y.; Zhou, J.M. Arabidopsis NHO1 is required for general resistance against Pseudomonas bacteria. Plant Cell 2001, 13, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Xing, T.; Wang, X.J.; Malik, K.; Miki, B.L. Ectopic expression of an Arabidopsis Calmodulin-like domain protein kinase-enhanced NADPH oxidase activity and oxidative burst in tomato protoplasts. Mol. Plant Microbe Interact. 2001, 14, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Meyerowitz, E.M. Ethylene responses are negatively regulated by a receptor gene family in Arabidopsis thaliana. Cell 1998, 94, 261–271. [Google Scholar] [CrossRef]
- Zhu, J.F.; Li, W.F.; Yang, W.H. Identification of microRNAs in Caragana intermedia by high-throughput sequencing and expression analysis of 12 microRNAs and their targets under salt stress. Plant Cell Rep. 2013, 32, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Gong, S.; Hao, Z.; Tao, J. Identification of miRNAs Responsive to Botrytis cinerea in Herbaceous Peony (Paeonia lactiflora Pall.) by High-Throughput Sequencing. Genes 2015, 6, 918-934. https://doi.org/10.3390/genes6030918
Zhao D, Gong S, Hao Z, Tao J. Identification of miRNAs Responsive to Botrytis cinerea in Herbaceous Peony (Paeonia lactiflora Pall.) by High-Throughput Sequencing. Genes. 2015; 6(3):918-934. https://doi.org/10.3390/genes6030918
Chicago/Turabian StyleZhao, Daqiu, Saijie Gong, Zhaojun Hao, and Jun Tao. 2015. "Identification of miRNAs Responsive to Botrytis cinerea in Herbaceous Peony (Paeonia lactiflora Pall.) by High-Throughput Sequencing" Genes 6, no. 3: 918-934. https://doi.org/10.3390/genes6030918