Characterization of the Genomic Architecture and Mutational Spectrum of a Small Cell Prostate Carcinoma


Abstract
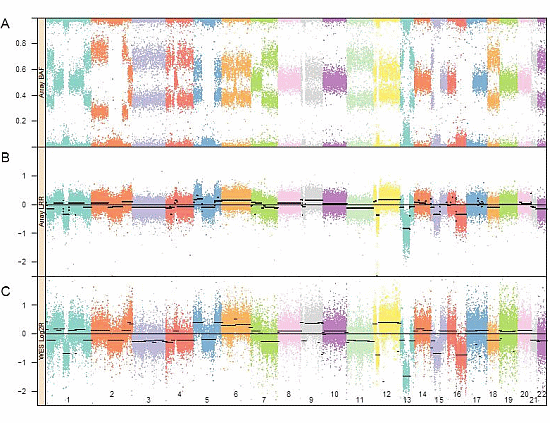
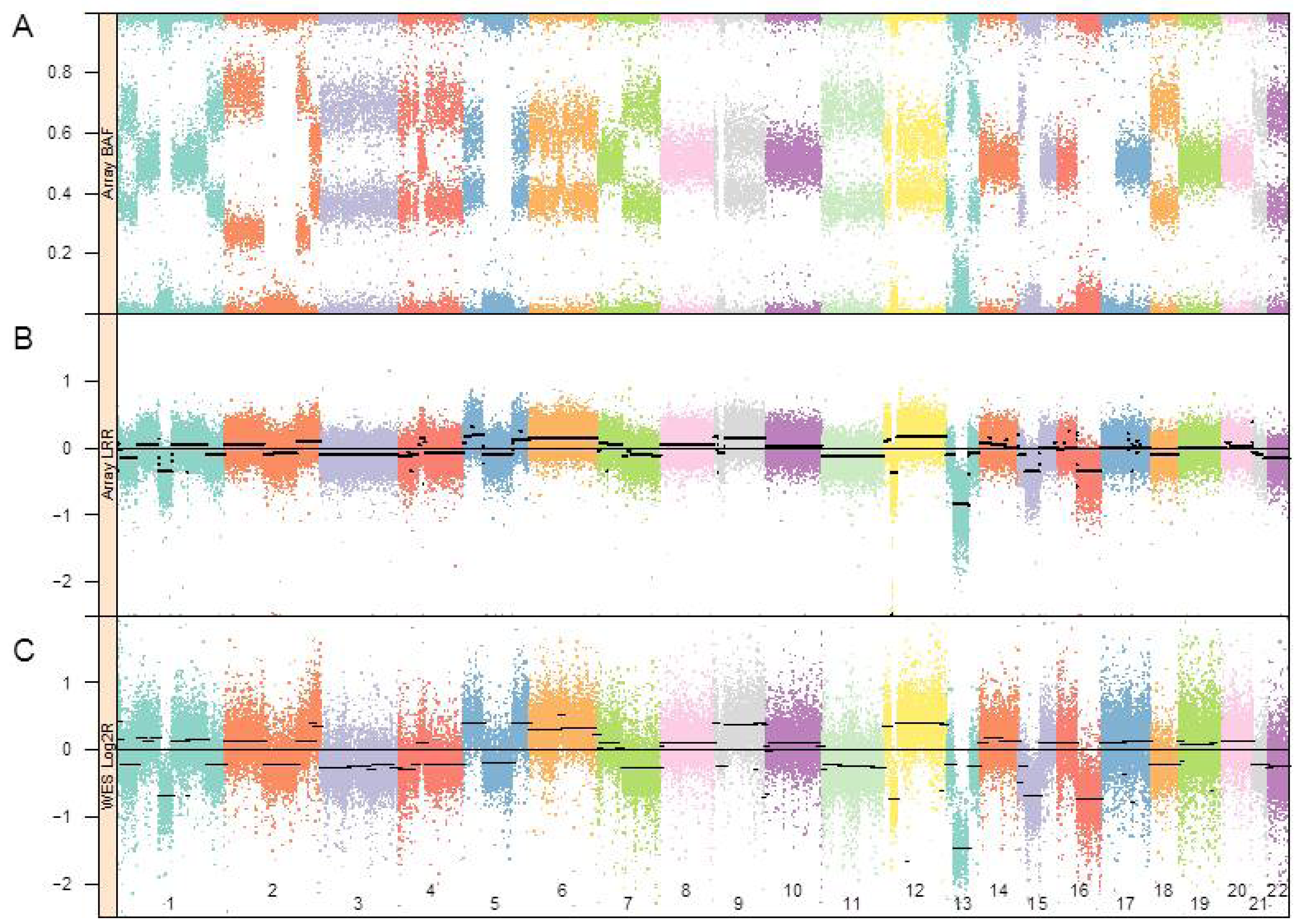
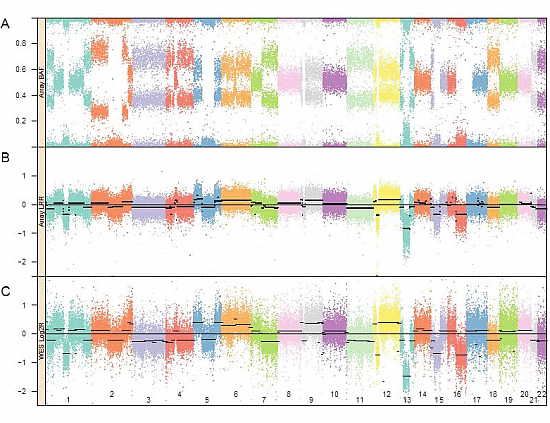
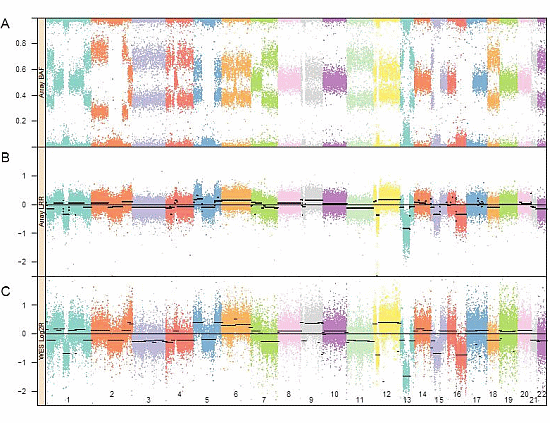
:
1. Introduction
2. Experimental
2.1. Sample
2.2. Genomic SNP Array and Analysis
2.3. Exome Capture and Sequencing
2.4. Sequence Interpretation
3. Results and Discussion
3.1. Genomic Landscape Detailed from Genotyping Data

3.2. Exome Variant Interpretation

{kind=link}
{kind=link}
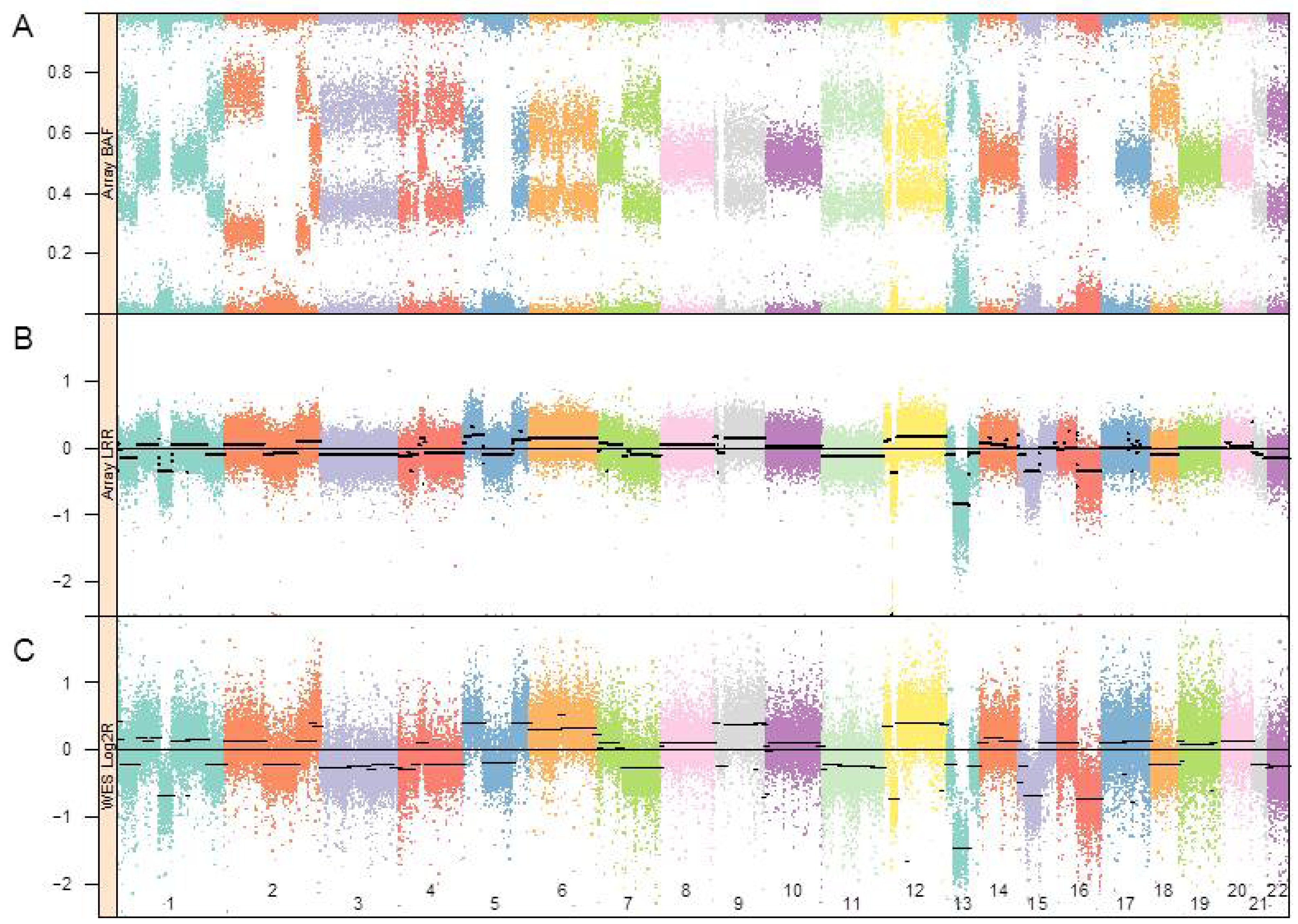{kind=link}
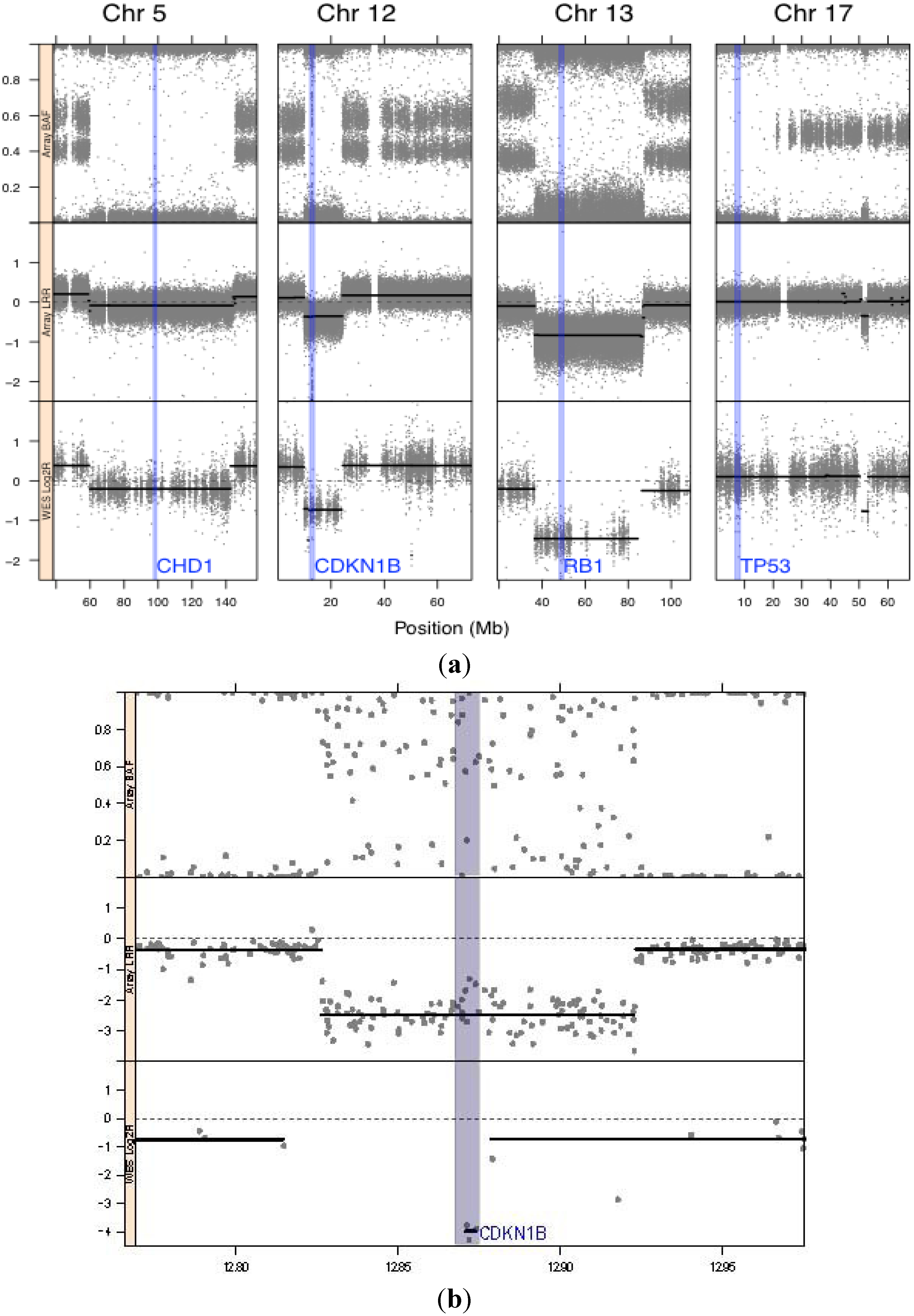{kind=link}
| Chr | Position | Gene | Driver | CN | EXC | LOH | Ref | SNV | % Var | Condel Score | Protein change | Ingenuity Assesment | Gene Description | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 17 | 7,578,394 | TP53 | HCD | 2.02 | 2.12 | YES | C | T | 0.97 | D(0.97–1.0) | H179R, H86R, H47R | Pathogenic | Tumor protein p53 | Damaging in all alternate translation products |
| 13 | 48,941,657 | RB1 | HCD | 1.12 | 0.72 | YES | G | T | 0.75 | STOP GAIN | E323* | Pathogenic | Retinoblastoma 1 | Premature termination codon is inferred damaging |
| 14 | 38,061,334 | FOXA1 | HCD | 2.11 | 2.25 | NO | G | T | 0.44 | D(1.0) | R219S, R186S | Likely Pathogenic | Forkhead box A1 | Damaging in two alternate translation products |
| 10 | 70,508,917 | CCAR1 | HCD | 2.05 | 2.13 | NO | G | A | 0.24 | N(0.02), D(0.81), D(0.83), N(0.02) | R269H, R258H, R284H, R89H | No Assessment | Cell division cycle and apoptosis regulator 1 | Probably damaging in two alternate translation products |
| 12 | 12.870 Mb– 12.875 Mb | CDKN1B | HCD | 0.36 | 0.13 | YES | None | Same as hg19 reference | DELETION | Not flagged | Cyclin-dependent kinase inhibitor 1B (p27/KIP1) | No mutations in coding regions |
| Chr | Position | Gene | CN | EXC | LOH | Ref | SNV | % Var | Condel Score | Protein change | Ingenuity Assesment | Gene Description | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 106,498,240 | NCK2 | 1.89 | 1.70 | YES | C | G | 1.00 | D(0.91) | P228R | Uncertain | NCK adaptor protein 2 | Promotes melanoma cell proliferation, migration and invasion |
| 2 | 107,041,278 | RGPD3 | 1.89 | 1.70 | YES | C | A | 0.90 | SIFT = Damaging | E1049* | Likely Pathogenic | RANBP2-like and GRIP domain containing 3 | Reported expression in testis and HeLa cells |
| 1 | 109,742,795–109,742,798 | KIAA1324, EIG21 | 1.57 | 1.24 | YES | G | 4 bp del | 0.85 | Frameshift | G829fs*10 | Uncertain | KIAA1324; Estrogen induced gene 121 | High expression is associated with shorter survival in ovarian cancer |
| 2 | 102,407,183 | MAP4K4 | 1.89 | 1.70 | YES | G | T | 0.49 | D(0.88) or N(0.02) | G42V, G4V | Likely Pathogenic | Mitogen-activated protein kinase kinase kinase kinase 4 | Often overexpressed in cancer and has roles in various cancer processes |
| 19 | 50,247,621 | TSKS | 2.00 | 2.14 | NO | C | T | 0.45 | N(0.05) | E410K | Likely Pathogenic | Testis-specific kinase substrate | Low expression in some embryonal carcinoma lines |
| 15 | 88,678,358 | NTRK3 | 2.02 | 2.14 | NO | C | T | 0.41 | D(0.72–0.87) | G295D, G393D | Likely Pathogenic | Neurotrophic tyrosine kinase, receptor, type 3 | Potential tumor suppressor, often fused with ETV6 in thyroid cancer |
| 16 | 7,759,062 | RBFOX1 | 2.10 | 2.14 | NO | G | A | 0.40 | D(0.91–1.0) | G307R, G334R, G355R, G339R, G377R | Uncertain | RNA binding protein, fox-1 homolog (C. elegans) 1 | Related gene RBFOX2 is a Candidate Driver (CD) |
| 19 | 38,865,389 | PSMD8 | 2.00 | 2.10 | NO | C | T | 0.38 | N(0.00) | R50C | Uncertain | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 8 | Upregulated in a choriocarcinoma cell line |
| 12 | 31,254,871 | DDX11 | 2.25 | 2.61 | NO | C | G | 0.34 | N(0.37) | H693Q, H317Q | Likely Pathogenic | DEAD/H box helicase 11 | Associated with small-cell carcinoma |
| 1 | 36,290,920 | AGO4 | 1.83 | 1.71 | NO | G | A | 0.24 | N(0.00) | M105V | Uncertain | Argonaute RISC catalytic component 4 | Down-regulated in hepatocellular cancer |
| 12 | 31,250,875 | DDX11 | 2.25 | 2.61 | NO | G | C | 0.14 | D(0.49) | A607P | Pathogenic | DEAD/H box helicase 11 | Expressed at high levels in melanoma |
| 19 | 4,689,651 | DPP9 | 2.00 | 2.09 | NO | G | T | 0.12 | D(0.50) | S560R | Uncertain | Dipeptidyl-peptidase 9 | Expressed in breast and ovarian cancers |
| 10 | 81,921,760 | ANXA11 | 2.05 | 2.13 | NO | G | A | 0.04 | D(0.85–0.95) | R338C, R371C, R4C | Not flagged | Annexin A11 | May enhance metastasis and invasion; related gene ANXA6 is a Candidate Driver (CD) |
| 9 | 100,843,284 | TRIM14 | 2.23 | 2.59 | NO | C | T | 0.03 | D(0.82–0.94) | R264W | Not flagged | Tripartite motif containing 14 | Related gene TRIM7 is a High Confidence Driver (HCD) |
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aparicio, A.; Logothetis, C.J.; Maity, S.N. Understanding the lethal variant of prostate cancer: Power of examining extremes. Cancer Discov. 2011, 1, 466–468. [Google Scholar] [CrossRef]
- Spiess, P.E.; Pettaway, C.A.; Vakar-Lopez, F.; Kassouf, W.; Wang, X.; Busby, J.E.; Do, K.A.; Davuluri, R.; Tannir, N.M. Treatment outcomes of small cell carcinoma of the prostate: A single-center study. Cancer 2007, 110, 1729–1737. [Google Scholar] [CrossRef]
- Wang, W.; Epstein, J.I. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am. J. Surg. Pathol. 2008, 32, 65–71. [Google Scholar] [CrossRef]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Meulenbeld, H.J.; Bleuse, J.P.; Vinci, E.M.; Raymond, E.; Vitali, G.; Santoro, A.; Dogliotti, L.; Berardi, R.; Cappuzzo, F.; Tagawa, S.T.; et al. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int. 2013, 111, 44–52. [Google Scholar] [CrossRef]
- Tzelepi, V.; Zhang, J.; Lu, J.F.; Kleb, B.; Wu, G.; Wan, X.; Hoang, A.; Efstathiou, E.; Sircar, K.; Navone, N.M.; et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin. Cancer Res. 2012, 18, 666–677. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Imamura, Y.; Sakamoto, S.; Endo, T.; Utsumi, T.; Fuse, M.; Suyama, T.; Kawamura, K.; Imamoto, T.; Yano, K.; Uzawa, K.; et al. FOXA1 promotes tumor progression in prostate cancer via the insulin-like growth factor binding protein 3 pathway. PLoS One 2012, 7, e42456. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Foye, A.; Wagle, N.; Kim, W.; Carter, S.L.; McKenna, A.; Simko, J.P.; Garraway, L.A.; Febbo, P.G. Successful whole-exome sequencing from a prostate cancer bone metastasis biopsy. Prostate Cancer Prostatic Dis. 2014, 17, 23–27. [Google Scholar] [CrossRef]
- Genetic Resources Core Facility. Available online: http://grcf.jhmi.edu/ (accessed on 2 January 2014).
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef]
- Magi, A.; Tattini, L.; Cifola, I.; D’Aurizio, R.; Benelli, M.; Mangano, E.; Battaglia, C.; Bonora, E.; Kurg, A.; Seri, M.; et al. EXCAVATOR: Detecting copy number variants from whole-exome sequencing data. Genome Biol. 2013. [Google Scholar] [CrossRef]
- Barnhart, M.G.S.; Hetrick, K.; Goldstein, J.; Marosy, D.; Mohr, D.; Craig, B.; Watkins, L., Jr.; Doheny, K. CIDRSeqSuite 2.0: An automated analysis pipeline for next-generation sequencing. In Proceedings of the presented at the 61st annual meeting of the American society for human genetics, Montreal, QC, Canada, 12 October 2011.
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Picard. Available online: http://picard.sourceforge.net/ (accessed on 2 January 2014).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 11, 1–33. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Scripps Genome Adviser. Available online: http://genomics.scripps.edu/ADVISER/ (accessed on 2 January 2014).
- Saunders, C.T.; Wong, W.S.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef]
- Christoforides, A.; Carpten, J.D.; Weiss, G.J.; Demeure, M.J.; von Hoff, D.D.; Craig, D.W. Identification of somatic mutations in cancer through Bayesian-based analysis of sequenced genome pairs. BMC Genomics 2013. [Google Scholar] [CrossRef]
- IntOgen: Interactive Onco Genomics Mutations Server. Available online: http://www.intogen.org/mutations/ (accessed 4 January 2014).
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef]
- Condel: CONsensus DELeteriousness Score of Missense SNVs Server. Available online: http://bg.upf.edu/condel/analysis/ (accessed 4 January 2014).
- Gonzalez-Perez, A.; Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef]
- Petty, E.; Pillus, L. Balancing chromatin remodeling and histone modifications in transcription. Trends Genet. 2013, 29, 621–629. [Google Scholar] [CrossRef]
- Cuesta, R.; Martinez-Sanchez, A.; Gebauer, F. miR-181a regulates cap-dependent translation of p27(kip1) mRNA in myeloid cells. Mol. Cell. Biol. 2009, 29, 2841–2851. [Google Scholar] [CrossRef]
- Fero, M.L.; Randel, E.; Gurley, K.E.; Roberts, J.M.; Kemp, C.J. The murine gene p27Kip1 is haplo-insufficient for tumor suppression. Nature 1998, 396, 177–180. [Google Scholar] [CrossRef]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Clark, K.L.; Halay, E.D.; Lai, E.; Burley, S.K. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 1993, 364, 412–420. [Google Scholar] [CrossRef]
- Burkhardt, L.; Fuchs, S.; Krohn, A.; Masser, S.; Mader, M.; Kluth, M.; Bachmann, F.; Huland, H.; Steuber, T.; Graefen, M.; et al. CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res. 2013, 73, 2795–2805. [Google Scholar] [CrossRef]
- Quayle, S.N.; Lee, J.Y.; Cheung, L.W.; Ding, L.; Wiedemeyer, R.; Dewan, R.W.; Huang-Hobbs, E.; Zhuang, L.; Wilson, R.K.; Ligon, K.L.; et al. Somatic mutations of PIK3R1 promote gliomagenesis. PLoS One 2012, 7, e49466. [Google Scholar] [CrossRef]
- Labelle-Cote, M.; Dusseault, J.; Ismail, S.; Picard-Cloutier, A.; Siegel, P.M.; Larose, L. Nck2 promotes human melanoma cell proliferation, migration and invasion in vitro and primary melanoma-derived tumor growth in vivo. BMC Cancer 2011. [Google Scholar] [CrossRef]
- Ciccarelli, F.D.; von Mering, C.; Suyama, M.; Harrington, E.D.; Izaurralde, E.; Bork, P. Complex genomic rearrangements lead to novel primate gene function. Genome Res. 2005, 15, 343–351. [Google Scholar] [CrossRef]
- Schlumbrecht, M.P.; Xie, S.S.; Shipley, G.L.; Urbauer, D.L.; Broaddus, R.R. Molecular clustering based on ERalpha and EIG121 predicts survival in high-grade serous carcinoma of the ovary/peritoneum. Mod. Pathol. 2011, 24, 453–462. [Google Scholar] [CrossRef]
- Deng, L.; Feng, J.; Broaddus, R.R. The novel estrogen-induced gene EIG121 regulates autophagy and promotes cell survival under stress. Cell Death Dis. 2010. [Google Scholar] [CrossRef]
- Qiu, M.H.; Qian, Y.M.; Zhao, X.L.; Wang, S.M.; Feng, X.J.; Chen, X.F.; Zhang, S.H. Expression and prognostic significance of MAP4K4 in lung adenocarcinoma. Pathol. Res. Pract. 2012, 208, 541–548. [Google Scholar] [CrossRef]
- Venables, J.P.; Brosseau, J.P.; Gadea, G.; Klinck, R.; Prinos, P.; Beaulieu, J.F.; Lapointe, E.; Durand, M.; Thibault, P.; Tremblay, K.; et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol. Cell. Biol. 2013, 33, 396–405. [Google Scholar] [CrossRef]
- Zhou, D.; Yang, L.; Zheng, L.; Ge, W.; Li, D.; Zhang, Y.; Hu, X.; Gao, Z.; Xu, J.; Huang, Y.; et al. Exome capture sequencing of adenoma reveals genetic alterations in multiple cellular pathways at the early stage of colorectal tumorigenesis. PLoS One 2013, 8, e53310. [Google Scholar]
- Scorilas, A.; Yousef, G.M.; Jung, K.; Rajpert-De Meyts, E.; Carsten, S.; Diamandis, E.P. Identification and characterization of a novel human testis-specific kinase substrate gene which is downregulated in testicular tumors. Biochem. Biophys. Res. Commun. 2001, 285, 400–408. [Google Scholar] [CrossRef]
- Luo, Y.; Kaz, A.M.; Kanngurn, S.; Welsch, P.; Morris, S.M.; Wang, J.; Lutterbaugh, J.D.; Markowitz, S.D.; Grady, W.M. NTRK3 is a potential tumor suppressor gene commonly inactivated by epigenetic mechanisms in colorectal cancer. PLoS Genet. 2013, 9, e1003552. [Google Scholar] [CrossRef]
- Kralik, J.M.; Kranewitter, W.; Boesmueller, H.; Marschon, R.; Tschurtschenthaler, G.; Rumpold, H.; Wiesinger, K.; Erdel, M.; Petzer, A.L.; Webersinke, G. Characterization of a newly identified ETV6-NTRK3 fusion transcript in acute myeloid leukemia. Diagn. Pathol. 2011. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Banno, K.; Shimizu, T.; Ueki, A.; Tsuji, K.; Masuda, K.; Kisu, I.; Nomura, H.; Tominaga, E.; Nagano, O.; et al. Gene expression profile of a newly established choriocarcinoma cell line, iC3-1, compared to existing choriocarcinoma cell lines and normal placenta. Placenta 2013, 34, 110–118. [Google Scholar] [CrossRef]
- Kitagawa, N.; Ojima, H.; Shirakihara, T.; Shimizu, H.; Kokubu, A.; Urushidate, T.; Totoki, Y.; Kosuge, T.; Miyagawa, S.; Shibata, T. Downregulation of the microRNA biogenesis components and its association with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2013, 104, 543–551. [Google Scholar] [CrossRef]
- Bhattacharya, C.; Wang, X.; Becker, D. The DEAD/DEAH box helicase, DDX11, is essential for the survival of advanced melanomas. Mol. Cancer 2012. [Google Scholar] [CrossRef]
- Wilson, C.H.; Abbott, C.A. Expression profiling of dipeptidyl peptidase 8 and 9 in breast and ovarian carcinoma cell lines. Int. J. Oncol. 2012, 41, 919–932. [Google Scholar]
- Wang, J.; Guo, C.; Liu, S.; Qi, H.; Yin, Y.; Liang, R.; Sun, M.Z.; Greenaway, F.T. Annexin A11 in disease. Clin. Chim. Acta 2014, 431, 164–168. [Google Scholar] [CrossRef]
- Prensner, J.R.; Iyer, M.K.; Sahu, A.; Asangani, I.A.; Cao, Q.; Patel, L.; Vergara, I.A.; Davicioni, E.; Erho, N.; Ghadessi, M.; et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat. Genet. 2013, 45, 1392–1398. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Jin, C.; Yang, J.C.; Tanasa, B.; Li, W.; Merkurjev, D.; Ohgi, K.A.; Meng, D.; Zhang, J.; et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature 2013, 500, 598–602. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef]
- Comstock, C.E.; Augello, M.A.; Schiewer, M.J.; Karch, J.; Burd, C.J.; Ertel, A.; Knudsen, E.S.; Jessen, W.J.; Aronow, B.J.; Knudsen, K.E. Cyclin D1 is a selective modifier of androgen-dependent signaling and androgen receptor function. J. Biol. Chem. 2011, 286, 8117–8127. [Google Scholar] [CrossRef]
- Xu, Y.; Shao, Q.S.; Yao, H.B.; Jin, Y.; Ma, Y.Y.; Jia, L.H. Up-expression of FOXC1 correlates with poor prognosis in gastric cancer patients. Histopathology 2013. [Google Scholar] [CrossRef]
- Wei, L.X.; Zhou, R.S.; Xu, H.F.; Wang, J.Y.; Yuan, M.H. High expression of FOXC1 is associated with poor clinical outcome in non-small cell lung cancer patients. Tumour Biol. 2013, 34, 941–946. [Google Scholar] [CrossRef]
- Sizemore, S.T.; Keri, R.A. The forkhead box transcription factor FOXC1 promotes breast cancer invasion by inducing matrix metalloprotease 7 (MMP7) expression. J. Biol. Chem. 2012, 287, 24631–24640. [Google Scholar] [CrossRef]
- Youngren, K.K.; Coveney, D.; Peng, X.; Bhattacharya, C.; Schmidt, L.S.; Nickerson, M.L.; Lamb, B.T.; Deng, J.M.; Behringer, R.R.; Capel, B.; et al. The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumors. Nature 2005, 435, 360–364. [Google Scholar] [CrossRef]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl. Med. 2012. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Scott, A.F.; Mohr, D.W.; Ling, H.; Scharpf, R.B.; Zhang, P.; Liptak, G.S. Characterization of the Genomic Architecture and Mutational Spectrum of a Small Cell Prostate Carcinoma. Genes 2014, 5, 366-384. https://doi.org/10.3390/genes5020366
Scott AF, Mohr DW, Ling H, Scharpf RB, Zhang P, Liptak GS. Characterization of the Genomic Architecture and Mutational Spectrum of a Small Cell Prostate Carcinoma. Genes. 2014; 5(2):366-384. https://doi.org/10.3390/genes5020366
Chicago/Turabian StyleScott, Alan F., David W. Mohr, Hua Ling, Robert B. Scharpf, Peng Zhang, and Gregory S. Liptak. 2014. "Characterization of the Genomic Architecture and Mutational Spectrum of a Small Cell Prostate Carcinoma" Genes 5, no. 2: 366-384. https://doi.org/10.3390/genes5020366
APA StyleScott, A. F., Mohr, D. W., Ling, H., Scharpf, R. B., Zhang, P., & Liptak, G. S. (2014). Characterization of the Genomic Architecture and Mutational Spectrum of a Small Cell Prostate Carcinoma. Genes, 5(2), 366-384. https://doi.org/10.3390/genes5020366