Reading and Language Disorders: The Importance of Both Quantity and Quality


{kind=link}
Abstract
:1. Introduction
2. Complex Traits, Complex Definitions
3. Changing and Heterogeneous Phenotypes
4. Monogenic Conditions Back in the Picture
5. Genetic Windows into Development
6. Classical Approaches
7. SLI and Dyslexia Linkage Loci
8. GWA Studies
9. GWA Studies of Speech and Language-Related Traits
10. GWA Study Design Factors
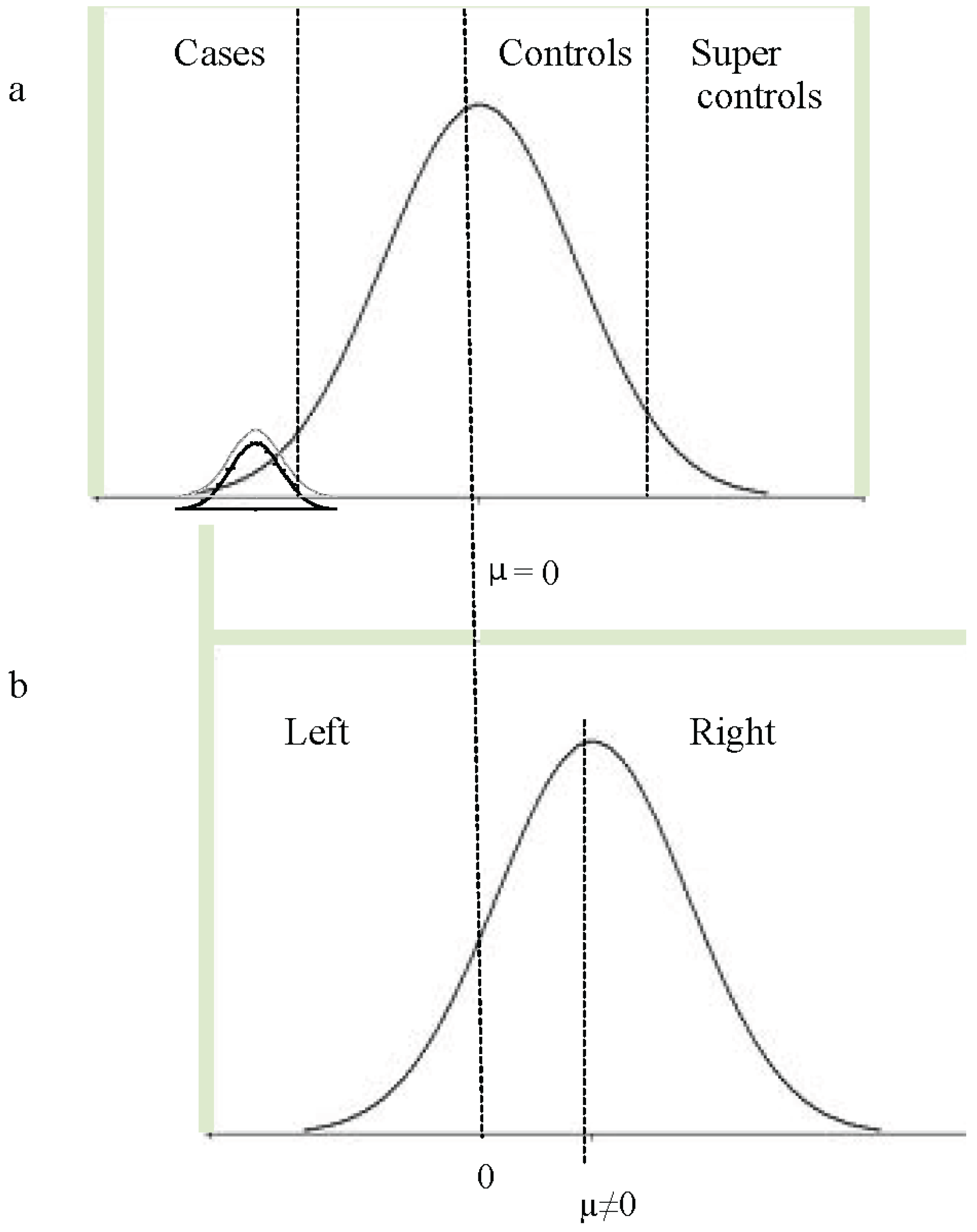
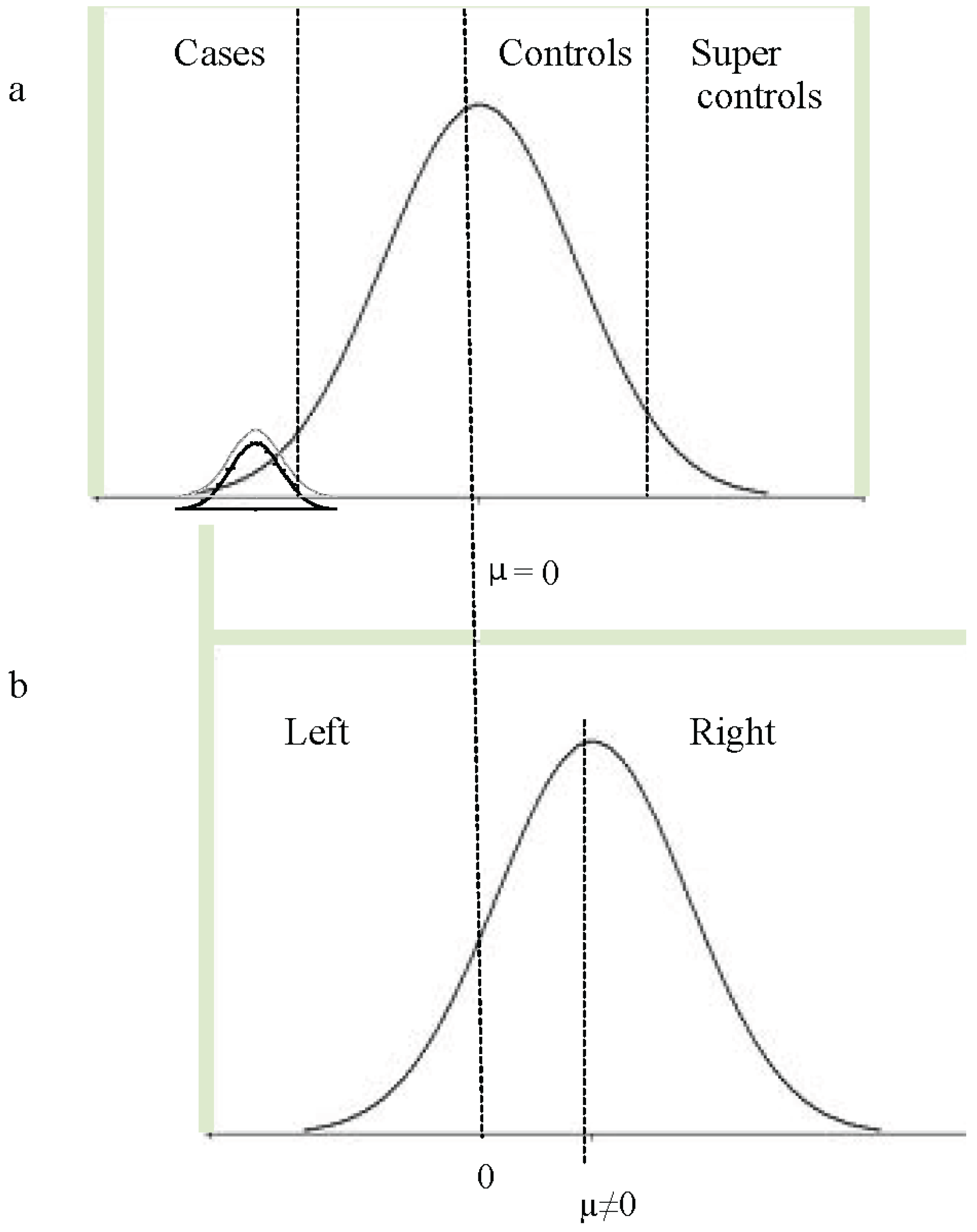
11. Dimensional and Categorical Models of Language Disorder
12. Cross-Linguistic Difficulties of Speech and Language Disorders
13. Mega-, Meta- and Mixed-GWA Studies
14. Quantitative vs. Qualitative, or Both
15. Filling the Gap
16. Difficulties of High-Throughput Sequencing Studies
17. Translational Relevance
18. Biology beyond the p-Values
19. Converging Evidence towards Biological Pathways
20. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harel, S.; Greenstein, Y.; Kramer, U.; Yifat, R.; Samuel, E.; Nevo, Y.; Leitner, Y.; Kutai, M.; Fattal, A.; Shinnar, S. Clinical characteristics of children referred to a child development center for evaluation of speech, language, and communication disorders. Pediatr. Neurol. 1996, 15, 305–311. [Google Scholar] [CrossRef]
- American-Psychiatric-Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed. (dsm-iv); American Psychiatric Association: Washington, DC, USA, 1994. [Google Scholar]
- Bishop, D.V. The role of genes in the etiology of specific language impairment. J. Commun. Disord. 2002, 35, 311–328. [Google Scholar] [CrossRef]
- Hayiou-Thomas, M.E. Genetic and environmental influences on early speech, language and literacy development. J. Commun. Disord. 2008, 41, 397–408. [Google Scholar] [CrossRef]
- DeFries, J.C.; Fulker, D.W.; LaBuda, M.C. Evidence for a genetic aetiology in reading disability of twins. Nature 1987, 329, 537–539. [Google Scholar]
- Bishop, D.V.M. Uncommon Understanding: Development and Disorders of Language Comprehension in Children; Psychology Press: Hove, UK, 1997. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Conti-Ramsden, G.; Botting, N. Characteristics of children attending language units in england: A national study of 7-year-olds. Int. J. Lang. Commun. Disord. 1999, 34, 359–366. [Google Scholar] [CrossRef]
- Snowling, M.J. Phonemic deficits in developmental dyslexia. Psychol. Res. 1981, 43, 219–234. [Google Scholar] [CrossRef]
- Ramus, F. Neurobiology of dyslexia: A reinterpretation of the data. Trends Neurosci. 2004, 27, 720–726. [Google Scholar] [CrossRef]
- Peterson, R.L.; Pennington, B.F. Developmental dyslexia. Lancet 2012, 379, 1997–2007. [Google Scholar] [CrossRef]
- Gathercole, S.E.; Baddeley, A.D. Phonological memory deficits in language disordered children: Is there a causal connection? J. Mem. Lang. 1990, 29, 336–360. [Google Scholar] [CrossRef]
- Tallal, P.; Piercy, M. Defects of non-verbal auditory perception in children with developmental aphasia. Nature 1973, 241, 468–469. [Google Scholar] [CrossRef]
- Gopnik, M. Feature-blind grammar and dysphagia. Nature 1990, 344, 715. [Google Scholar] [CrossRef]
- Bishop, D.V. The underlying nature of specific language impairment. J. Child Psychol. Psychiatry Allied Discip. 1992, 33, 3–66. [Google Scholar] [CrossRef]
- Abbeduto, L.; Brady, N.; Kover, S.T. Language development and fragile x syndrome: Profiles, syndrome-specificity, and within-syndrome differences. Ment. Retard. Dev. Disabil. Res. Rev. 2007, 13, 36–46. [Google Scholar] [CrossRef]
- Bishop, D.V.; Snowling, M.J. Developmental dyslexia and specific language impairment: Same or different? Psychol. Bull. 2004, 130, 858–886. [Google Scholar] [CrossRef]
- McArthur, G.M.; Hogben, J.H.; Edwards, V.T.; Heath, S.M.; Mengler, E.D. On the “specifics” of specific reading disability and specific language impairment. J. Child Psychol. Psychiatry Allied Discip. 2000, 41, 869–874. [Google Scholar] [CrossRef]
- Catts, H.W.; Adlof, S.M.; Hogan, T.P.; Weismer, S.E. Are specific language impairment and dyslexia distinct disorders? J. Speech Lang. Hear. Res. 2005, 48, 1378–1396. [Google Scholar] [CrossRef]
- Pieters, S.; de Block, K.; Scheiris, J.; Eyssen, M.; Desoete, A.; Deboutte, D.; van Waelvelde, H.; Roeyers, H. How common are motor problems in children with a developmental disorder: Rule or exception? Child Care Health Dev. 2012, 38, 139–145. [Google Scholar] [CrossRef]
- Flapper, B.C.; Schoemaker, M.M. Developmental coordination disorder in children with specific language impairment: Co-morbidity and impact on quality of life. Res. Dev. Disabil. 2013, 34, 756–763. [Google Scholar] [CrossRef]
- Gooch, D.; Hulme, C.; Nash, H.M.; Snowling, M.J. Comorbidities in preschool children at family risk of dyslexia. J. Child Psychol. Psychiatry Allied Discip. 2013, 55, 237–246. [Google Scholar]
- Bloom, J.S.; Garcia-Barrera, M.A.; Miller, C.J.; Miller, S.R.; Hynd, G.W. Planum temporale morphology in children with developmental dyslexia. Neuropsychologia 2013, 51, 1684–1692. [Google Scholar] [CrossRef]
- Johnson, B.W.; McArthur, G.; Hautus, M.; Reid, M.; Brock, J.; Castles, A.; Crain, S. Lateralized auditory brain function in children with normal reading ability and in children with dyslexia. Neuropsychologia 2013, 51, 633–641. [Google Scholar] [CrossRef]
- Badcock, N.A.; Bishop, D.V.; Hardiman, M.J.; Barry, J.G.; Watkins, K.E. Co-localisation of abnormal brain structure and function in specific language impairment. Brain Lang. 2012, 120, 310–320. [Google Scholar] [CrossRef]
- Stoodley, C.J.; Fawcett, A.J.; Nicolson, R.I.; Stein, J.F. Impaired balancing ability in dyslexic children. Exp. Brain Res. 2005, 167, 370–380. [Google Scholar] [CrossRef]
- Budden, S. Clinical variability in early speech-language development in females with rett syndrome. Dev. Med. Child Neurol. 2012, 54, 392–393. [Google Scholar] [CrossRef]
- Roizen, N.J.; Antshel, K.M.; Fremont, W.; AbdulSabur, N.; Higgins, A.M.; Shprintzen, R.J.; Kates, W.R. 22q11.2ds deletion syndrome: Developmental milestones in infants and toddlers. J. Dev. Behav. Pediatr. 2007, 28, 119–124. [Google Scholar] [CrossRef]
- Starke, M.; Albertsson Wikland, K.; Moller, A. Parents’ descriptions of development and problems associated with infants with turner syndrome: A retrospective study. J. Paediatr. Child Health 2003, 39, 293–298. [Google Scholar] [CrossRef]
- Laws, G.; Bishop, D.V. Verbal deficits in down’s syndrome and specific language impairment: A comparison. Int. J. Lang. Commun. Disord. 2004, 39, 423–451. [Google Scholar] [CrossRef]
- Laws, G.; Bishop, D.V. A comparison of language abilities in adolescents with down syndrome and children with specific language impairment. J. Speech Lang. Hear. Res. 2003, 46, 1324–1339. [Google Scholar] [CrossRef]
- Simpson, N.H.; Addis, L.; Brandler, W.M.; Slonims, V.; Clark, A.; Watson, J.; Scerri, T.S.; Hennessy, E.R.; Bolton, P.F.; Conti-Ramsden, G.; et al. Increased prevalence of sex chromosome aneuploidies in specific language impairment and dyslexia. Dev. Med. Child Neurol. 2013. [Google Scholar] [CrossRef]
- Lai, C.S.; Fisher, S.E.; Hurst, J.A.; Vargha-Khadem, F.; Monaco, A.P. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature 2001, 413, 519–523. [Google Scholar] [CrossRef]
- Fisher, S.E.; Scharff, C. Foxp2 as a molecular window into speech and language. Trends Genet. 2009, 25, 166–177. [Google Scholar] [CrossRef]
- Kang, C.; Riazuddin, S.; Mundorff, J.; Krasnewich, D.; Friedman, P.; Mullikin, J.C.; Drayna, D. Mutations in the lysosomal enzyme-targeting pathway and persistent stuttering. N. Engl. J. Med. 2010, 362, 677–685. [Google Scholar] [CrossRef]
- Vernes, S.C.; Nicod, J.; Elahi, F.M.; Coventry, J.A.; Kenny, N.; Coupe, A.M.; Bird, L.E.; Davies, K.E.; Fisher, S.E. Functional genetic analysis of mutations implicated in a human speech and language disorder. Hum. Mol. Genet. 2006, 15, 3154–3167. [Google Scholar] [CrossRef]
- Vernes, S.C.; Newbury, D.F.; Abrahams, B.S.; Winchester, L.; Nicod, J.; Groszer, M.; Alarcon, M.; Oliver, P.L.; Davies, K.E.; Geschwind, D.H.; et al. A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 2008, 359, 2337–2345. [Google Scholar] [CrossRef]
- Alarcon, M.; Abrahams, B.S.; Stone, J.L.; Duvall, J.A.; Perederiy, J.V.; Bomar, J.M.; Sebat, J.; Wigler, M.; Martin, C.L.; Ledbetter, D.H.; et al. Linkage, association, and gene-expression analyses identify cntnap2 as an autism-susceptibility gene. Am. J. Hum. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef]
- Arking, D.E.; Cutler, D.J.; Brune, C.W.; Teslovich, T.M.; West, K.; Ikeda, M.; Rea, A.; Guy, M.; Lin, S.; Cook, E.H.; et al. A common genetic variant in the neurexin superfamily member cntnap2 increases familial risk of autism. Am. J. Hum. Genet. 2008, 82, 160–164. [Google Scholar] [CrossRef]
- Friedman, J.I.; Vrijenhoek, T.; Markx, S.; Janssen, I.M.; van der Vliet, W.A.; Faas, B.H.; Knoers, N.V.; Cahn, W.; Kahn, R.S.; Edelmann, L.; et al. Cntnap2 gene dosage variation is associated with schizophrenia and epilepsy. Mol. Psychiatry 2008, 13, 261–266. [Google Scholar] [CrossRef]
- Allison, D.B.; Thiel, B.; St Jean, P.; Elston, R.C.; Infante, M.C.; Schork, N.J. Multiple phenotype modeling in gene-mapping studies of quantitative traits: Power advantages. Am. J. Hum. Genet. 1998, 63, 1190–1201. [Google Scholar] [CrossRef]
- Whitehouse, A.J.; Bishop, D.V.; Ang, Q.W.; Pennell, C.E.; Fisher, S.E. Cntnap2 variants affect early language development in the general population. Genes Brain Behav. 2011, 10, 451–456. [Google Scholar] [CrossRef]
- Blair, D.R.; Lyttle, C.S.; Mortensen, J.M.; Bearden, C.F.; Jensen, A.B.; Khiabanian, H.; Melamed, R.; Rabadan, R.; Bernstam, E.V.; Brunak, S.; et al. A nondegenerate code of deleterious variants in mendelian loci contributes to complex disease risk. Cell 2013, 155, 70–80. [Google Scholar] [CrossRef]
- Tallal, P.; Ross, R.; Curtiss, S. Familial aggregation in specific language impairment. J. Speech Hear. Disord. 1989, 54, 167–173. [Google Scholar]
- Almasy, L.; Blangero, J. Human qtl linkage mapping. Genetica 2009, 136, 333–340. [Google Scholar] [CrossRef]
- Grigorenko, E.L.; Wood, F.B.; Meyer, M.S.; Hart, L.A.; Speed, W.C.; Shuster, A.; Pauls, D.L. Susceptibility loci for distinct components of developmental dyslexia on chromosomes 6 and 15. Am. J. Hum. Genet. 1997, 60, 27–39. [Google Scholar]
- Schulte-Korne, G.; Grimm, T.; Nothen, M.M.; Muller-Myhsok, B.; Cichon, S.; Vogt, I.R.; Propping, P.; Remschmidt, H. Evidence for linkage of spelling disability to chromosome 15. Am. J. Hum. Genet. 1998, 63, 279–282. [Google Scholar]
- Cardon, L.R.; Smith, S.D.; Fulker, D.W.; Kimberling, W.J.; Pennington, B.F.; DeFries, J.C. Quantitative trait locus for reading disability on chromosome 6. Science 1994, 266, 276–279. [Google Scholar]
- Fisher, S.E.; Marlow, A.J.; Lamb, J.; Maestrini, E.; Williams, D.F.; Richardson, A.J.; Weeks, D.E.; Stein, J.F.; Monaco, A.P. A quantitative-trait locus on chromosome 6p influences different aspects of developmental dyslexia. Am. J. Hum. Genet. 1999, 64, 146–156. [Google Scholar] [CrossRef]
- Gayan, J.; Smith, S.D.; Cherny, S.S.; Cardon, L.R.; Fulker, D.W.; Brower, A.M.; Olson, R.K.; Pennington, B.F.; DeFries, J.C. Quantitative-trait locus for specific language and reading deficits on chromosome 6p. Am. J. Hum. Genet. 1999, 64, 157–164. [Google Scholar] [CrossRef]
- Kaplan, D.E.; Gayan, J.; Ahn, J.; Won, T.W.; Pauls, D.; Olson, R.K.; DeFries, J.C.; Wood, F.; Pennington, B.F.; Page, G.P.; et al. Evidence for linkage and association with reading disability on 6p21.3–22. Am. J. Hum. Genet. 2002, 70, 1287–1298. [Google Scholar] [CrossRef]
- Fagerheim, T.; Raeymaekers, P.; Tonnessen, F.E.; Pedersen, M.; Tranebjaerg, L.; Lubs, H.A. A new gene (dyx3) for dyslexia is located on chromosome 2. J. Med. Genet. 1999, 36, 664–669. [Google Scholar]
- Francks, C.; Fisher, S.E.; Olson, R.K.; Pennington, B.F.; Smith, S.D.; DeFries, J.C.; Monaco, A.P. Fine mapping of the chromosome 2p12–16 dyslexia susceptibility locus: Quantitative association analysis and positional candidate genes sema4f and otx1. Psychiatr. Genet. 2002, 12, 35–41. [Google Scholar] [CrossRef]
- Hannula-Jouppi, K.; Kaminen-Ahola, N.; Taipale, M.; Eklund, R.; Nopola-Hemmi, J.; Kaariainen, H.; Kere, J. The axon guidance receptor gene robo1 is a candidate gene for developmental dyslexia. PLoS Genet. 2005, 1, e50. [Google Scholar]
- Nopola-Hemmi, J.; Myllyluoma, B.; Haltia, T.; Taipale, M.; Ollikainen, V.; Ahonen, T.; Voutilainen, A.; Kere, J.; Widen, E. A dominant gene for developmental dyslexia on chromosome 3. J. Med. Genet. 2001, 38, 658–664. [Google Scholar] [CrossRef]
- Fisher, S.E.; Francks, C.; Marlow, A.J.; MacPhie, I.L.; Newbury, D.F.; Cardon, L.R.; Ishikawa-Brush, Y.; Richardson, A.J.; Talcott, J.B.; Gayan, J.; et al. Independent genome-wide scans identify a chromosome 18 quantitative-trait locus influencing dyslexia. Nat. Genet. 2002, 30, 86–91. [Google Scholar] [CrossRef]
- Hsiung, G.Y.; Kaplan, B.J.; Petryshen, T.L.; Lu, S.; Field, L.L. A dyslexia susceptibility locus (dyx7) linked to dopamine d4 receptor (drd4) region on chromosome 11p15.5. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2004, 125B, 112–119. [Google Scholar] [CrossRef]
- Grigorenko, E.L.; Wood, F.B.; Meyer, M.S.; Pauls, J.E.; Hart, L.A.; Pauls, D.L. Linkage studies suggest a possible locus for developmental dyslexia on chromosome 1p. Am. J. Med. Genet. 2001, 105, 120–129. [Google Scholar] [CrossRef]
- Rabin, M.; Wen, X.L.; Hepburn, M.; Lubs, H.A.; Feldman, E.; Duara, R. Suggestive linkage of developmental dyslexia to chromosome 1p34-p36. Lancet 1993, 342, 178. [Google Scholar]
- De Kovel, C.G.; Franke, B.; Hol, F.A.; Lebrec, J.J.; Maassen, B.; Brunner, H.; Padberg, G.W.; Platko, J.; Pauls, D. Confirmation of dyslexia susceptibility loci on chromosomes 1p and 2p, but not 6p in a dutch sib-pair collection. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147, 294–300. [Google Scholar]
- Meng, H.; Smith, S.D.; Hager, K.; Held, M.; Liu, J.; Olson, R.K.; Pennington, B.F.; DeFries, J.C.; Gelernter, J.; O’Reilly-Pol, T.; et al. Dcdc2 is associated with reading disability and modulates neuronal development in the brain. Proc. Natl. Acad. Sci. USA 2005, 102, 17053–17058. [Google Scholar] [CrossRef]
- Francks, C.; Paracchini, S.; Smith, S.D.; Richardson, A.J.; Scerri, T.S.; Cardon, L.R.; Marlow, A.J.; MacPhie, I.L.; Walter, J.; Pennington, B.F.; et al. A 77-kilobase region of chromosome 6p22.2 is associated with dyslexia in families from the united kingdom and from the united states. Am. J. Hum. Genet. 2004, 75, 1046–1058. [Google Scholar] [CrossRef]
- Cope, N.; Harold, D.; Hill, G.; Moskvina, V.; Stevenson, J.; Holmans, P.; Owen, M.J.; O’Donovan, M.C.; Williams, J. Strong evidence that kiaa0319 on chromosome 6p is a susceptibility gene for developmental dyslexia. Am. J. Hum. Genet. 2005, 76, 581–591. [Google Scholar] [CrossRef]
- Paracchini, S.; Thomas, A.; Castro, S.; Lai, C.; Paramasivam, M.; Wang, Y.; Keating, B.J.; Taylor, J.M.; Hacking, D.F.; Scerri, T.; et al. The chromosome 6p22 haplotype associated with dyslexia reduces the expression of kiaa0319, a novel gene involved in neuronal migration. Hum. Mol. Genet. 2006, 15, 1659–1666. [Google Scholar] [CrossRef]
- Taipale, M.; Kaminen, N.; Nopola-Hemmi, J.; Haltia, T.; Myllyluoma, B.; Lyytinen, H.; Muller, K.; Kaaranen, M.; Lindsberg, P.J.; Hannula-Jouppi, K.; et al. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptide repeat domain protein dynamically regulated in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 11553–11558. [Google Scholar] [CrossRef]
- Bates, T.C.; Lind, P.A.; Luciano, M.; Montgomery, G.W.; Martin, N.G.; Wright, M.J. Dyslexia and dyx1c1: Deficits in reading and spelling associated with a missense mutation. Mol. Psychiatry 2009, 15, 1190–1196. [Google Scholar]
- Anthoni, H.; Zucchelli, M.; Matsson, H.; Muller-Myhsok, B.; Fransson, I.; Schumacher, J.; Massinen, S.; Onkamo, P.; Warnke, A.; Griesemann, H.; et al. A locus on 2p12 containing the co-regulated mrpl19 and c2orf3 genes is associated to dyslexia. Hum. Mol. Genet. 2007, 16, 667–677. [Google Scholar]
- Anthoni, H.; Sucheston, L.E.; Lewis, B.A.; Tapia-Paez, I.; Fan, X.; Zucchelli, M.; Taipale, M.; Stein, C.M.; Hokkanen, M.E.; Castren, E.; et al. The aromatase gene cyp19a1: Several genetic and functional lines of evidence supporting a role in reading, speech and language. Behav. Genet. 2012, 42, 509–527. [Google Scholar] [CrossRef]
- Scerri, T.S.; Schulte-Korne, G. Genetics of developmental dyslexia. Eur. Child Adolesc. Psychiatry 2010, 19, 179–197. [Google Scholar] [CrossRef]
- Currier, T.A.; Etchegaray, M.A.; Haight, J.L.; Galaburda, A.M.; Rosen, G.D. The effects of embryonic knockdown of the candidate dyslexia susceptibility gene homologue dyx1c1 on the distribution of gabaergic neurons in the cerebral cortex. Neuroscience 2011, 172, 535–546. [Google Scholar] [CrossRef]
- Tammimies, K.; Vitezic, M.; Matsson, H.; Le Guyader, S.; Burglin, T.R.; Ohman, T.; Stromblad, S.; Daub, C.O.; Nyman, T.A.; Kere, J.; et al. Molecular networks of dyx1c1 gene show connection to neuronal migration genes and cytoskeletal proteins. Biol. Psychiatry 2013, 73, 583–590. [Google Scholar] [CrossRef]
- Massinen, S.; Hokkanen, M.E.; Matsson, H.; Tammimies, K.; Tapia-Paez, I.; Dahlstrom-Heuser, V.; Kuja-Panula, J.; Burghoorn, J.; Jeppsson, K.E.; Swoboda, P.; et al. Increased expression of the dyslexia candidate gene dcdc2 affects length and signaling of primary cilia in neurons. PLoS One 2011, 6, e20580. [Google Scholar] [CrossRef]
- Adler, W.T.; Platt, M.P.; Mehlhorn, A.J.; Haight, J.L.; Currier, T.A.; Etchegaray, M.A.; Galaburda, A.M.; Rosen, G.D. Position of neocortical neurons transfected at different gestational ages with shrna targeted against candidate dyslexia susceptibility genes. PLoS One 2013, 8, e65179. [Google Scholar]
- Platt, M.P.; Adler, W.T.; Mehlhorn, A.J.; Johnson, G.C.; Wright, K.A.; Choi, R.T.; Tsang, W.H.; Poon, M.W.; Yeung, S.Y.; Waye, M.M.; et al. Embryonic disruption of the candidate dyslexia susceptibility gene homolog kiaa0319-like results in neuronal migration disorders. Neuroscience 2013, 248C, 585–593. [Google Scholar]
- Wang, Y.; Yin, X.; Rosen, G.; Gabel, L.; Guadiana, S.M.; Sarkisian, M.R.; Galaburda, A.M.; Loturco, J.J. Dcdc2 knockout mice display exacerbated developmental disruptions following knockdown of doublecortin. Neuroscience 2011, 190, 398–408. [Google Scholar] [CrossRef]
- Szalkowski, C.E.; Fiondella, C.G.; Galaburda, A.M.; Rosen, G.D.; Loturco, J.J.; Fitch, R.H. Neocortical disruption and behavioral impairments in rats following in utero rnai of candidate dyslexia risk gene kiaa0319. Int. J. Dev. Neurosci. 2012, 30, 293–302. [Google Scholar] [CrossRef]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. Dyx1c1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef]
- Chandrasekar, G.; Vesterlund, L.; Hultenby, K.; Tapia-Paez, I.; Kere, J. The zebrafish orthologue of the dyslexia candidate gene dyx1c1 is essential for cilia growth and function. PLoS One 2013, 8, e63123. [Google Scholar]
- Ivliev, A.E.; 't Hoen, P.A.C.; van Roon-Mom, W.M.; Peters, D.J.M.; Sergeeva, M.G. Exploring the transcriptome of ciliated cells using in silico dissection of human tissues. PLoS One 2012, 7, e35618. [Google Scholar]
- Villanueva, P.; Newbury, D.F.; Jara, L.; de Barbieri, Z.; Mirza, G.; Palomino, H.M.; Fernandez, M.A.; Cazier, J.B.; Monaco, A.P.; Palomino, H. Genome-wide analysis of genetic susceptibility to language impairment in an isolated chilean population. Eur. J. Hum. Genet. 2011, 19, 687–695. [Google Scholar] [CrossRef]
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Smith, B.J.; Vieland, V.J.; Tallal, P.; Brzustowicz, L.M. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum. Hered. 2004, 57, 10–20. [Google Scholar] [CrossRef]
- Bartlett, C.W.; Flax, J.F.; Logue, M.W.; Vieland, V.J.; Bassett, A.S.; Tallal, P.; Brzustowicz, L.M. A major susceptibility locus for specific language impairment is located on 13q21. Am. J. Hum. Genet. 2002, 71, 45–55. [Google Scholar]
- SLI Consortium (SLIC). Highly significant linkage to the sli1 locus in an expanded sample of individuals affected by specific language impairment. Am. J. Hum. Genet. 2004, 74, 1225–1238. [Google Scholar]
- SLI Consortium. A genomewide scan identifies two novel loci involved in specific language impairment. Am. J. Hum. Genet. 2002, 70, 384–398. [Google Scholar] [CrossRef]
- Monaco, A.P. Multivariate linkage analysis of specific language impairment (sli). Ann. Hum. Genet. 2007, 71, 660–673. [Google Scholar] [CrossRef]
- Falcaro, M.; Pickles, A.; Newbury, D.F.; Addis, L.; Banfield, E.; Fisher, S.E.; Monaco, A.P.; Simkin, Z.; Conti-Ramsden, G. Genetic and phenotypic effects of phonological short-term memory and grammatical morphology in specific language impairment. Genes Brain Behav. 2008, 7, 393–402. [Google Scholar] [CrossRef]
- Newbury, D.F.; Winchester, L.; Addis, L.; Paracchini, S.; Buckingham, L.L.; Clark, A.; Cohen, W.; Cowie, H.; Dworzynski, K.; Everitt, A.; et al. Cmip and atp2c2 modulate phonological short-term memory in language impairment. Am. J. Hum. Genet. 2009, 85, 264–272. [Google Scholar]
- Ceroni, F.; Simpson, N.H.; Francks, C.; Baird, G.; Conti-Ramsden, G.; Clark, A.; Bolton, P.F.; Hennessy, E.R.; Donnelly, P.; Bentley, D.R.; et al. Homozygous microdeletion of exon 5 in znf277 in a girl with specific language impairment. Eur. J. Hum. Genet. 2014. [Google Scholar] [CrossRef]
- Nudel, R.; Simpson, N.H.; Baird, G.; O’Hare, A.; Conti-Ramsden, G.; Bolton, P.F.; Hennessy, E.R.; Consortium, S.L.I.; Monaco, A.P.; Knight, J.C.; et al. Associations of hla alleles with specific language impairment. J. Neurodev. Disord. 2014. [Google Scholar] [CrossRef]
- Zollner, S.; Pritchard, J.K. Overcoming the winner’s curse: Estimating penetrance parameters from case-control data. Am. J. Hum. Genet. 2007, 80, 605–615. [Google Scholar] [CrossRef]
- The International Hapmap Project. Available online: http://www.hapmap.org/ (accessed on 31 March 2014).
- 1000 Genomes: A Deep Catalogue of Human Genetic Variation. Available online: http://www.1000genomes.org/ (accessed on 31 March 2014).
- Visscher, P.M.; Brown, M.A.; McCarthy, M.I.; Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 2012, 90, 7–24. [Google Scholar] [CrossRef]
- Meaburn, E.L.; Harlaar, N.; Craig, I.W.; Schalkwyk, L.C.; Plomin, R. Quantitative trait locus association scan of early reading disability and ability using pooled DNA and 100k snp microarrays in a sample of 5760 children. Mol. Psychiatry 2008, 13, 729–740. [Google Scholar] [CrossRef]
- Roeske, D.; Ludwig, K.U.; Neuhoff, N.; Becker, J.; Bartling, J.; Bruder, J.; Brockschmidt, F.F.; Warnke, A.; Remschmidt, H.; Hoffmann, P.; et al. First genome-wide association scan on neurophysiological endophenotypes points to trans-regulation effects on slc2a3 in dyslexic children. Mol. Psychiatry 2011, 16, 97–107. [Google Scholar]
- Risch, N.; Merikangas, K. The future of genetic studies of complex human diseases. Science 1996, 273, 1516–1517. [Google Scholar] [CrossRef]
- Field, L.L.; Shumansky, K.; Ryan, J.; Truong, D.; Swiergala, E.; Kaplan, B.J. Dense-map genome scan for dyslexia supports loci at 4q13, 16p12, 17q22; suggests novel locus at 7q36. Genes Brain Behav. 2013, 12, 56–69. [Google Scholar] [CrossRef]
- Luciano, M.; Evans, D.M.; Hansell, N.K.; Medland, S.E.; Montgomery, G.W.; Martin, N.G.; Wright, M.J.; Bates, T.C. A genome-wide association study for reading and language abilities in two population cohorts. Genes Brain Behav. 2013, 12, 645–652. [Google Scholar] [CrossRef]
- Eicher, J.D.; Powers, N.R.; Miller, L.L.; Akshoomoff, N.; Amaral, D.G.; Bloss, C.S.; Libiger, O.; Schork, N.J.; Darst, B.F.; Casey, B.J.; et al. Genome-wide association study of shared components of reading disability and language impairment. Genes Brain Behav. 2013, 12, 792–801. [Google Scholar] [CrossRef]
- Mott, R.; Yuan, W.; Kaisaki, P.; Gan, X.; Cleak, J.; Edwards, A.; Baud, A.; Flint, J. The architecture of parent-of-origin effects in mice. Cell 2014, 156, 332–342. [Google Scholar] [CrossRef]
- Nudel, R.; Simpson, N.H.; Baird, G.; O’Hare, A.; Conti-Ramsden, G.; Bolton, P.F.; Hennessy, E.R.; SLIC; Ring, S.M.; Davey Smith, G.; et al. Genome-wide association analyses of child genotype effects and parent-of-origin effects in specific language impairment (sli). Genes Brain Behav. 2014. [Google Scholar] [CrossRef]
- McCarthy, M.I.; Hirschhorn, J.N. Genome-wide association studies: Potential next steps on a genetic journey. Hum. Mol. Genet. 2008, 17, R156–R165. [Google Scholar] [CrossRef]
- Leonard, L. Is specific language impairment a useful construct? In Cambridge Monographs and Texts in Applied Psycholinguistics; Rosenberg, S., Ed.; Cambridge University Press: Cambridge, UK, 1987; pp. 1–39. [Google Scholar]
- Rescorla, L.; Ratner, N.B. Phonetic profiles of toddlers with specific expressive language impairment (sli-e). J. Speech Hear. Res. 1996, 39, 153–165. [Google Scholar]
- Dollaghan, C.A. Taxometric analyses of specific language impairment in 3- and 4-year-old children. J. Speech Lang. Hear. Res. 2004, 47, 464–475. [Google Scholar] [CrossRef]
- Dollaghan, C.A. Taxometric analyses of specific language impairment in 6-year-old children. J. Speech Lang. Hear. Res. 2011, 54, 1361–1371. [Google Scholar] [CrossRef]
- Leonard, L.B. Specific langauage impairment across languages. Child Dev. Perspect. 2014, 8, 1–5. [Google Scholar] [CrossRef]
- Bishop, D.V. Is specific language impairment a valid diagnostic category? Genetic and psycholinguistic evidence. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1994, 346, 105–111. [Google Scholar] [CrossRef]
- Hayiou-Thomas, M.E.; Oliver, B.; Plomin, R. Genetic influences on specific versus nonspecific language impairment in 4-year-old twins. J. Learn. Disabil. 2005, 38, 222–232. [Google Scholar] [CrossRef]
- Bishop, D.V.; Hayiou-Thomas, M.E. Heritability of specific language impairment depends on diagnostic criteria. Genes Brain Behav. 2008, 7, 365–372. [Google Scholar] [CrossRef]
- Eley, T.C.; Bishop, D.V.; Dale, P.S.; Oliver, B.; Petrill, S.A.; Price, T.S.; Purcell, S.; Saudino, K.J.; Simonoff, E.; Stevenson, J.; et al. Genetic and environmental origins of verbal and performance components of cognitive delay in 2-year-olds. Dev. Psychol. 1999, 35, 1122–1131. [Google Scholar] [CrossRef]
- Paracchini, S.; Steer, C.D.; Buckingham, L.L.; Morris, A.P.; Ring, S.; Scerri, T.; Stein, J.; Pembrey, M.E.; Ragoussis, J.; Golding, J.; et al. Association of the kiaa0319 dyslexia susceptibility gene with reading skills in the general population. Am. J. Psychiatry 2008, 165, 1576–1584. [Google Scholar] [CrossRef]
- Scerri, T.S.; Morris, A.P.; Buckingham, L.L.; Newbury, D.F.; Miller, L.L.; Monaco, A.P.; Bishop, D.V.; Paracchini, S. Dcdc2, kiaa0319 and cmip are associated with reading-related traits. Biol. Psychiatry 2011, 70, 237–245. [Google Scholar] [CrossRef]
- Newbury, D.F.; Paracchini, S.; Scerri, T.S.; Winchester, L.; Addis, L.; Richardson, A.J.; Walter, J.; Stein, J.F.; Talcott, J.B.; Monaco, A.P. Investigation of dyslexia and sli risk variants in reading- and language-impaired subjects. Behav. Genet. 2011, 41, 90–104. [Google Scholar] [CrossRef]
- Landerl, K.; Ramus, F.; Moll, K.; Lyytinen, H.; Leppanen, P.H.; Lohvansuu, K.; O’Donovan, M.; Williams, J.; Bartling, J.; Bruder, J.; et al. Predictors of developmental dyslexia in european orthographies with varying complexity. J. Child Psychol. Psychiatry Allied Discip. 2013, 54, 686–694. [Google Scholar] [CrossRef]
- Becker, J.; Czamara, D.; Scerri, T.S.; Ramus, F.; Csepe, V.; Talcott, J.B.; Stein, J.; Morris, A.; Ludwig, K.U.; Hoffmann, P.; et al. Genetic analysis of dyslexia candidate genes in the european cross-linguistic neurodys cohort. Eur. J. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Schumacher, J.; Anthoni, H.; Dahdouh, F.; Konig, I.R.; Hillmer, A.M.; Kluck, N.; Manthey, M.; Plume, E.; Warnke, A.; Remschmidt, H.; et al. Strong genetic evidence of dcdc2 as a susceptibility gene for dyslexia. Am. J. Hum. Genet. 2006, 78, 52–62. [Google Scholar] [CrossRef]
- Bedore, L.M.; Leonard, L.B. Grammatical morphology deficits in spanish-speaking children with specific language impairment. J. Speech Lang. Hear. Res. 2001, 44, 905–924. [Google Scholar] [CrossRef]
- Dispaldro, M.; Leonard, L.B.; Deevy, P. Clinical markers in italian-speaking children with and without specific language impairment: A study of non-word and real word repetition as predictors of grammatical ability. Int. J. Lang. Commun. Disord. 2013, 48, 554–564. [Google Scholar] [CrossRef]
- Paradis, J.; Crago, M.; Genesee, F.; Rice, M. French-english bilingual children with sli: How do they compare with their monolingual peers? J. Speech Lang. Hear. Res. 2003, 46, 113–127. [Google Scholar] [CrossRef]
- Do, R.; Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Gao, C.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat. Genet. 2013, 45, 1345–1352. [Google Scholar] [CrossRef]
- Global Lipids Genetics, C.; Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef]
- Magi, R.; Manning, S.; Yousseif, A.; Pucci, A.; Santini, F.; Karra, E.; Querci, G.; Pelosini, C.; McCarthy, M.I.; Lindgren, C.M.; et al. Contribution of 32 GWAS-identified common variants to severe obesity in european adults referred for bariatric surgery. PLoS One 2013, 8, e70735. [Google Scholar] [CrossRef]
- Berndt, S.I.; Gustafsson, S.; Magi, R.; Ganna, A.; Wheeler, E.; Feitosa, M.F.; Justice, A.E.; Monda, K.L.; Croteau-Chonka, D.C.; Day, F.R.; et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat. Genet. 2013, 45, 501–512. [Google Scholar] [CrossRef]
- Yang, J.; Loos, R.J.; Powell, J.E.; Medland, S.E.; Speliotes, E.K.; Chasman, D.I.; Rose, L.M.; Thorleifsson, G.; Steinthorsdottir, V.; Magi, R.; et al. Fto genotype is associated with phenotypic variability of body mass index. Nature 2012, 490, 267–272. [Google Scholar] [CrossRef]
- Lango, H.; Consortium, U.K.T.D.G.; Palmer, C.N.; Morris, A.D.; Zeggini, E.; Hattersley, A.T.; McCarthy, M.I.; Frayling, T.M.; Weedon, M.N. Assessing the combined impact of 18 common genetic variants of modest effect sizes on type 2 diabetes risk. Diabetes 2008, 57, 3129–3135. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium; Genetic Risk Outcome of Psychosis (GROUP) Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium; Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide snps. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef]
- Scerri, T.S.; Brandler, W.M.; Paracchini, S.; Morris, A.P.; Ring, S.M.; Talcott, J.B.; Stein, J.; Monaco, A.P. Pcsk6 is associated with handedness in individuals with dyslexia. Hum. Mol. Genet. 2011, 20, 608–614. [Google Scholar] [CrossRef]
- Brandler, W.M.; Morris, A.P.; Evans, D.M.; Scerri, T.S.; Kemp, J.P.; Timpson, N.J.; St Pourcain, B.; Smith, G.D.; Ring, S.M.; Stein, J.; et al. Common variants in left/right asymmetry genes and pathways are associated with relative hand skill. PLoS Genet. 2013, 9, e1003751. [Google Scholar] [CrossRef]
- Hamada, H.; Meno, C.; Watanabe, D.; Saijoh, Y. Establishment of vertebrate left-right asymmetry. Nat. Rev. Genet. 2002, 3, 103–113. [Google Scholar]
- Ludwig, K.U.; Samann, P.; Alexander, M.; Becker, J.; Bruder, J.; Moll, K.; Spieler, D.; Czisch, M.; Warnke, A.; Docherty, S.J.; et al. A common variant in myosin-18b contributes to mathematical abilities in children with dyslexia and intraparietal sulcus variability in adults. Transl. Psychiatry 2013, 3, e229. [Google Scholar] [CrossRef]
- Medland, S.E.; Lindgren, C.M.; Magi, R.; Neale, B.M.; Albrecht, E.; Esko, T.; Evans, D.M.; Hottenga, J.J.; Ikram, M.A.; Mangino, M.; et al. Meta-Analysis of GWAS for Handedness: Results from the ENGAGE consortium. Available online: http://www.ashg.org/2009meeting/abstracts/fulltext/f21141.htm/ (accessed on 31 March 2014).
- Eriksson, N.; Macpherson, J.M.; Tung, J.Y.; Hon, L.S.; Naughton, B.; Saxonov, S.; Avey, L.; Wojcicki, A.; Pe’er, I.; Mountain, J. Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet. 2010, 6, e1000993. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in asd. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar]
- Vissers, L.E.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M.; et al. A de novo paradigm for mental retardation. Nat. Genet. 2011, 42, 1109–1112. [Google Scholar]
- Toma, C.; Torrico, B.; Hervas, A.; Valdes-Mas, R.; Tristan-Noguero, A.; Padillo, V.; Maristany, M.; Salgado, M.; Arenas, C.; Puente, X.S.; et al. Exome sequencing in multiplex autism families suggests a major role for heterozygous truncating mutations. Mol. Psychiatry 2013. [Google Scholar] [CrossRef]
- Yu, T.W.; Chahrour, M.H.; Coulter, M.E.; Jiralerspong, S.; Okamura-Ikeda, K.; Ataman, B.; Schmitz-Abe, K.; Harmin, D.A.; Adli, M.; Malik, A.N.; et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron 2013, 77, 259–273. [Google Scholar] [CrossRef]
- Worthey, E.A.; Raca, G.; Laffin, J.J.; Wilk, B.M.; Harris, J.M.; Jakielski, K.J.; Dimmock, D.P.; Strand, E.A.; Shriberg, L.D. Whole-exome sequencing supports genetic heterogeneity in childhood apraxia of speech. J. Neurodev. Disord. 2013, 5, 29. [Google Scholar] [CrossRef]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Coe, B.P.; Parikh, S.; Friedman, N.; Goldstein, A.; Filipink, R.A.; McConnell, J.S.; Angle, B.; Meschino, W.S.; et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012, 367, 1321–1331. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef]
- Leblond, C.S.; Jutta, H.; Delorme, R.; Proepper, C.; Betancur, C.; Huguet, G.; Konyukh, M.; Chaste, P.; Ey, E.; Rastam, M.; et al. Genetic and functional analyses of shank2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012, 8, e1002521. [Google Scholar] [CrossRef]
- Newbury, D.F.; Mari, F.; Sadighi Akha, E.; Macdermot, K.D.; Canitano, R.; Monaco, A.P.; Taylor, J.C.; Renieri, A.; Fisher, S.E.; Knight, S.J. Dual copy number variants involving 16p11 and 6q22 in a case of childhood apraxia of speech and pervasive developmental disorder. Eur. J. Hum. Genet. 2013, 21, 361–365. [Google Scholar] [CrossRef]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Lappalainen, T.; Sammeth, M.; Friedlander, M.R.; 't Hoen, P.A.; Monlong, J.; Rivas, M.A.; Gonzalez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef]
- Gratten, J.; Visscher, P.M.; Mowry, B.J.; Wray, N.R. Interpreting the role of de novo protein-coding mutations in neuropsychiatric disease. Nat. Genet. 2013, 45, 234–238. [Google Scholar]
- Laederich, M.B.; Horton, W.A. Fgfr3 targeting strategies for achondroplasia. Expert Rev. Mol. Med. 2012, 14, e11. [Google Scholar] [CrossRef]
- Foldynova-Trantirkova, S.; Wilcox, W.R.; Krejci, P. Sixteen years and counting: The current understanding of fibroblast growth factor receptor 3 (fgfr3) signaling in skeletal dysplasias. Hum. Mutat. 2012, 33, 29–41. [Google Scholar] [CrossRef]
- Eglinton, E.; Annett, M. Handedness and dyslexia: A meta-analysis. Percept. Mot. Skills 1994, 79, 1611–1616. [Google Scholar] [CrossRef]
- Bishop, D.V. How to increase your chances of obtaining a significant association between handedness and disorder. J. Clin. Exp. Neuropsychol. 1990, 12, 812–816. [Google Scholar] [CrossRef]
- Dragovic, M.; Hammond, G. Handedness in schizophrenia: A quantitative review of evidence. Acta Psychiatr. Scand. 2005, 111, 410–419. [Google Scholar] [CrossRef]
- Bishop, D.V. Cerebral asymmetry and language development: Cause, correlate, or consequence? Science 2013. [Google Scholar] [CrossRef]
- Paracchini, S.; Scerri, T.; Monaco, A.P. The genetic lexicon of dyslexia. Annu. Rev. Genomics Hum. Genet. 2007, 8, 57–79. [Google Scholar] [CrossRef]
- Brandler, W.M.; Paracchini, S. The genetic relationship between handedness and neurodevelopmental disorders. Trends Mol. Med. 2014, 20, 83–90. [Google Scholar]
- Velayos-Baeza, A.; Toma, C.; da Roza, S.; Paracchini, S.; Monaco, A.P. Alternative splicing in the dyslexia-associated gene kiaa0319. Mamm. Genome 2007, 18, 627–634. [Google Scholar] [CrossRef]
- Field, S.; Riley, K.L.; Grimes, D.T.; Hilton, H.; Simon, M.; Powles-Glover, N.; Siggers, P.; Bogani, D.; Greenfield, A.; Norris, D.P. Pkd1l1 establishes left-right asymmetry and physically interacts with pkd2. Development 2011, 138, 1131–1142. [Google Scholar] [CrossRef]
- Lee, J.E.; Gleeson, J.G. Cilia in the nervous system: Linking cilia function and neurodevelopmental disorders. Curr. Opin. Neurol. 2011, 24, 98–105. [Google Scholar] [CrossRef]
- Sun, T.; Walsh, C.A. Molecular approaches to brain asymmetry and handedness. Nat. Rev. Neurosci. 2006, 7, 655–662. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Newbury, D.F.; Monaco, A.P.; Paracchini, S. Reading and Language Disorders: The Importance of Both Quantity and Quality. Genes 2014, 5, 285-309. https://doi.org/10.3390/genes5020285
Newbury DF, Monaco AP, Paracchini S. Reading and Language Disorders: The Importance of Both Quantity and Quality. Genes. 2014; 5(2):285-309. https://doi.org/10.3390/genes5020285
Chicago/Turabian StyleNewbury, Dianne F., Anthony P. Monaco, and Silvia Paracchini. 2014. "Reading and Language Disorders: The Importance of Both Quantity and Quality" Genes 5, no. 2: 285-309. https://doi.org/10.3390/genes5020285
APA StyleNewbury, D. F., Monaco, A. P., & Paracchini, S. (2014). Reading and Language Disorders: The Importance of Both Quantity and Quality. Genes, 5(2), 285-309. https://doi.org/10.3390/genes5020285