Genome-Wide Sequencing Reveals Two Major Sub-Lineages in the Genetically Monomorphic Pathogen Xanthomonas Campestris Pathovar Musacearum


Abstract
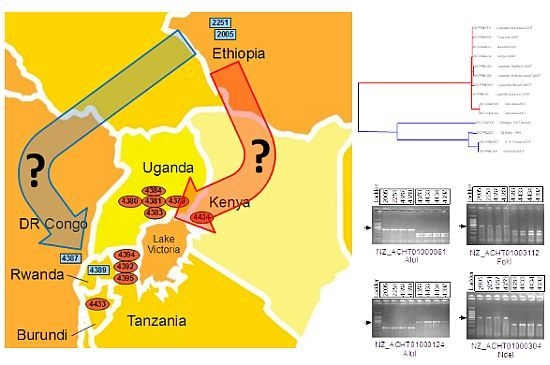
:
1. Introduction
2. Results and Discussion
2.1. Genome Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Source and Date of Isolation | Coverage | SRA Accession | |
|---|---|---|---|---|
| Xcm NCPPB2005 | Ethiopia 1967 | 72× | SRR489154.7 | |
| Xcm NCPPB2251 | Ethiopia 1969 | 13× | SRR494492.2 | |
| Xcm NCPPB4379 | Uganda (Kayunga) 2007 | 102× | SRR494484.2 | |
| Xcm NCPPB4380 | Uganda (Kiboga) 2007 | 113× | SRR494485.2 | |
| Xcm NCPPB4381 | Uganda (Luwero) 2007 | 56× | SRR020203.3 | |
| Xcm NCPPB4383 | Uganda (Wakiso) 2007 | 11× | SRR494493.2 | |
| Xcm NCPPB4384 | Uganda (Nakaongola) 2007 | 55× | SRR494488.2 | |
| Xcm NCPPB4387 | D. R. Congo (Kivu province) 2007 | 13× | SRR494494.1 | |
| Xcm NCPPB4389 | Rwanda (Gisenyi province) 2007 | 16× | SRR494495.2 | |
| Xcm NCPPB4392 | Tanzania (Muleba district, Kagera region) 2007 | 72× | SRR494498.3 | |
| Xcm NCPPB4394 | Tanzania (Muleba district, Kagera region) 2007 | 92× | SRR494489.1 | |
| Xcm NCPPB4395 | Tanzania (Muleba district, Kagera region) 2007 | 117× | SRR494490.2 | |
| Xcm NCPPB4433 | Burundi 2008 | 13× | SRR494496.1 | |
| Xcm NCPPB4434 | Kenya (Teso district) 2008 | 15× | SRR494497.1 | |
| Xvv NCPPB206 | South Africa 1948 | 70× | SRR494500.3 | |
| Xvv NCPPB702 | Zimbabwe 1959 | 35× | SRR020202.3 | |
| Xvv NCPPB1326 | Zimbabwe 1962 | 63× | SRR494491.5 | |
| Xvv NCPPB1381 | Zimbabwe 1962 | 66× | SRR494499.3 | |
2.2. Distinguishing Xcm from Xvv
| RefSeq Locus tag | Predicted Gene Product |
|---|---|
| XcampmN_010100002667 | hypothetical protein |
| XcampmN_010100009057 | general secretion pathway protein D |
| XcampmN_010100016989 | transposase |
| XcampmN_010100016984 | phage-related integrase |
| XcampmN_010100014552 | hypothetical protein |
| XcampmN_010100013878 | DNA-cytosine methyltransferase |
| XcampmN_010100013483 | hypothetical protein |
| XcampmN_010100011643 | conjugal transfer relaxosome component TraJ |
| XcampmN_010100011578 | hypothetical protein |
| XcampmN_010100011573 | Fis family transcriptional regulator |
| XcampmN_010100011558 | hypothetical protein |
| XcampmN_010100011553 | hypothetical protein |
| XcampmN_010100010854 | hypothetical protein |
| XcampmN_010100010849 | XRE family transcriptional regulator |
| XcampmN_010100006985 | hypothetical protein |
| XcampmN_010100004971 | exported protein |
| XcampmN_010100004961 | virulence regulator |
| XcampmN_010100004956 | hypothetical protein |
| XcampmN_010100004736 | hypothetical protein |
| XcampmN_010100001342 | ISXo2 putative transposase |
| XcampmN_010100001332 | ABC-type antimicrobial peptide transport system ATPase component |
| XcampmN_010100001327 | RND family efflux transporter MFP subunit |
| XcampmN_010100013888 | ISxac1 transposase |
| XcampmN_010100011563 | putative DNA methylase |
| XcampmN_010100004966 | integrase |
| XcampmN_010100001337 | peptide ABC transporter permease |
| XcampmN_010100013883 | restriction endonuclease-like protein |
| XcampmN_010100000225 | putative secreted protein |
| XcampmN_010100000622 | fimbrillin |
| XcampmN_010100015677 | methyltransferase |
| XcampmN_010100016677 | Putative acetylhydrolase |
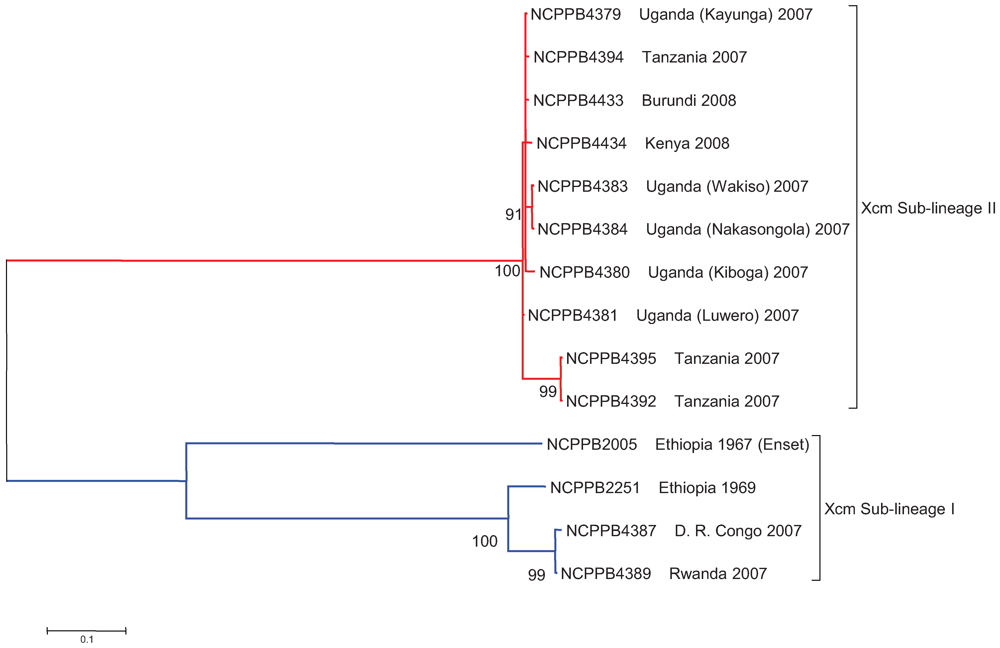
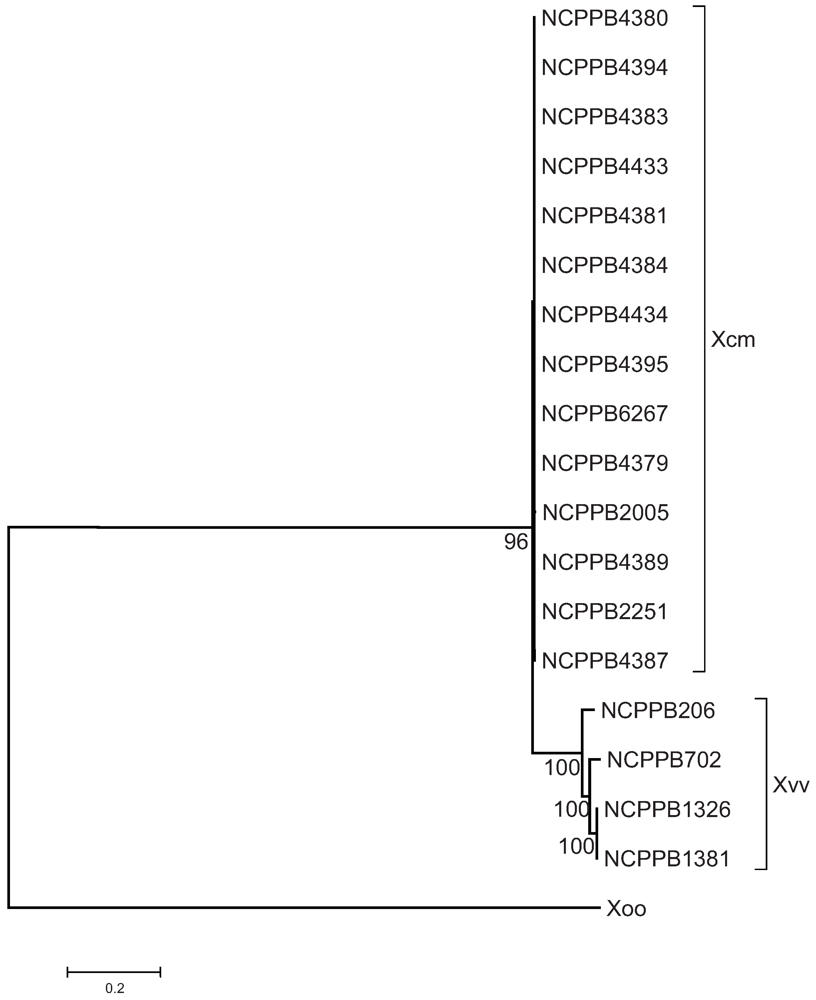
2.3. The Sequenced Xcm Isolates Comprise a Single Monophyletic Clade
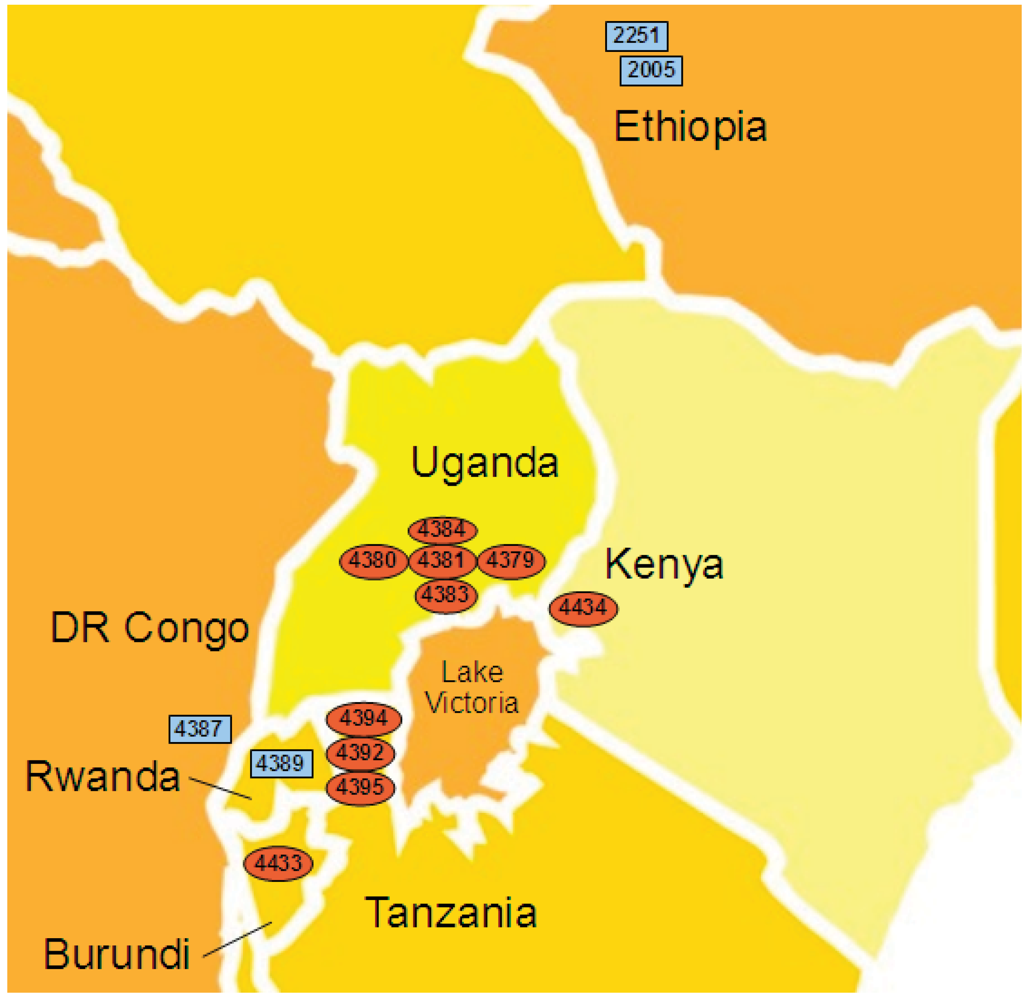
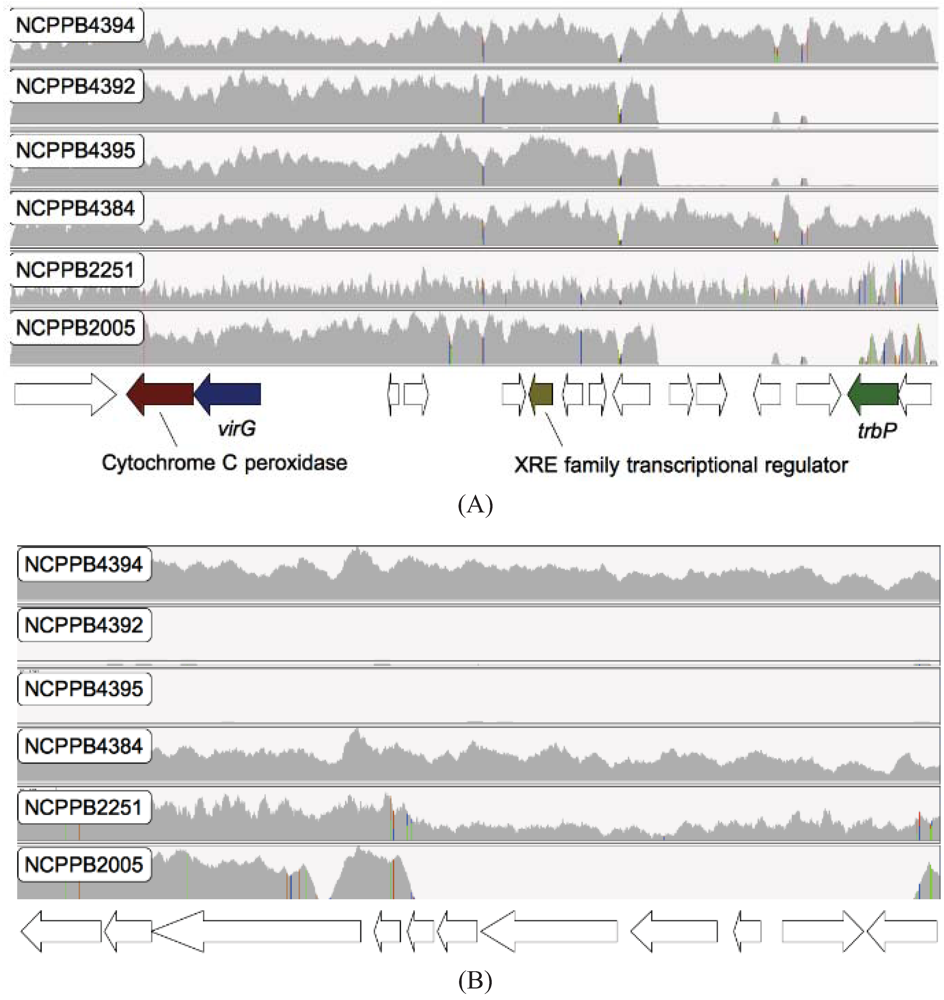
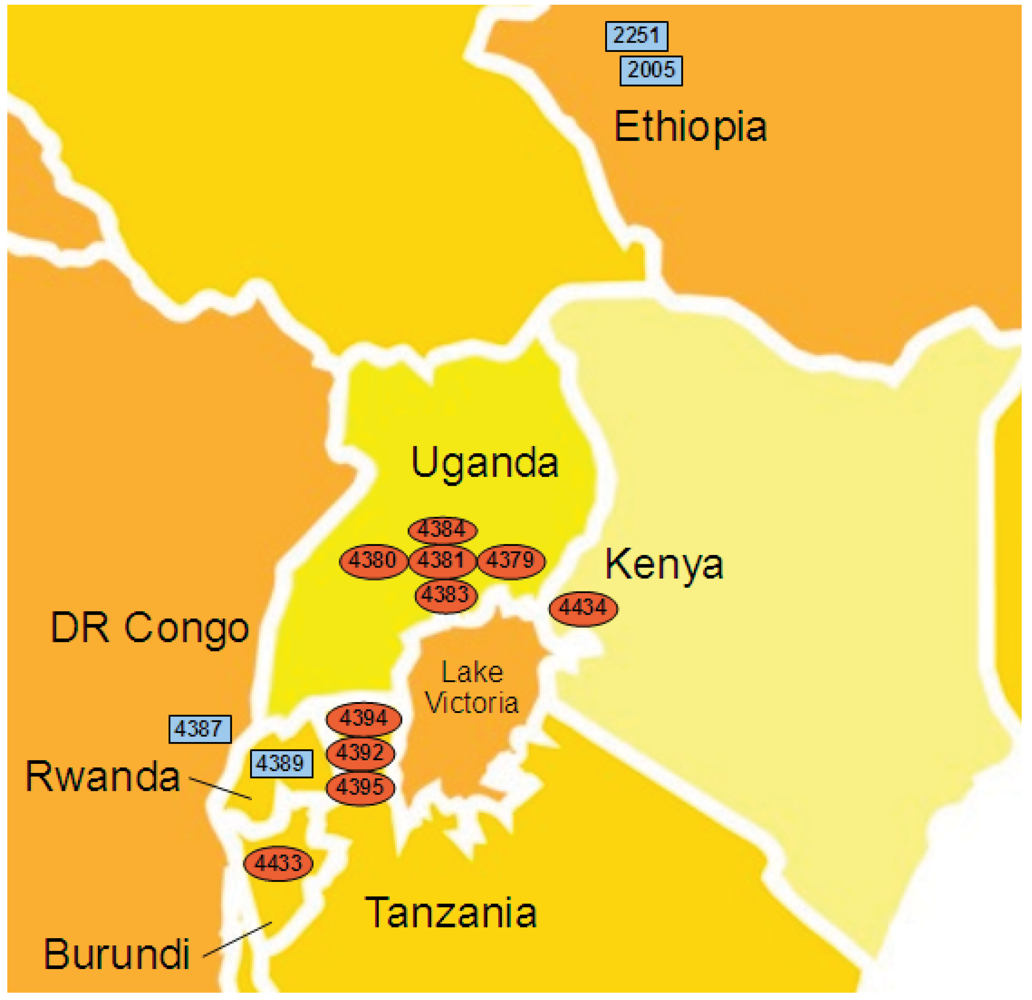
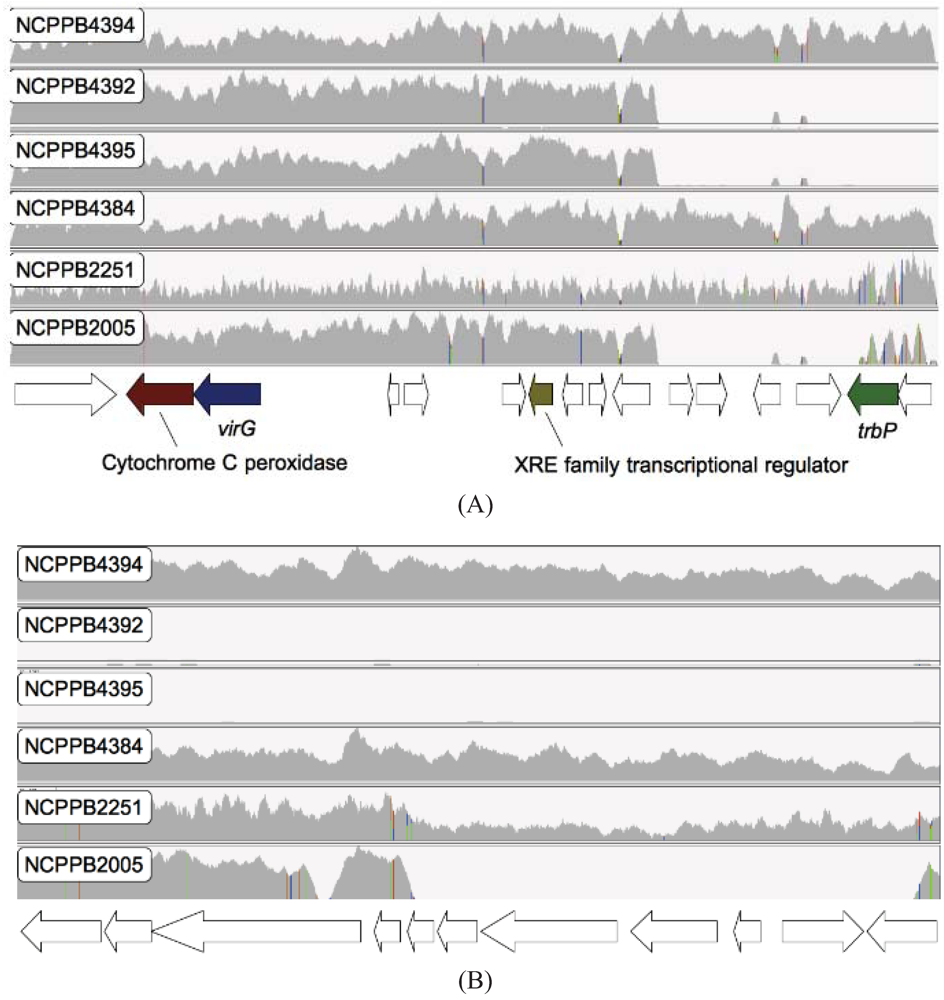
2.4. Xcm Isolates from Uganda, Kenya, Tanzania and Burundi are Genetically Distinct from Isolates from Ethiopia, DR Congo and Rwanda

| RefSeq Accession | Position | I | II | Locus Tag and Predicted Gene Product |
|---|---|---|---|---|
| NZ_ACHT01000013 | 861 | g | c | XcampmN_010100000120 putative ISXo8 transposase |
| NZ_ACHT01000014 | 6000 | a | g | XcampmN_010100000165 putative monovalent cation/H+ antiporter subunit A |
| NZ_ACHT01000034 | 8898 | t | c | XcampmN_010100000807 putative integrase protein |
| NZ_ACHT01000045 | 1261 | a | g | XcampmN_010100001162 bifunctional aspartate kinase/diaminopimelate decarboxylase protein |
| NZ_ACHT01000045 | 45,548 | a | c | XcampmN_010100001377 chemotaxis protein |
| NZ_ACHT01000059 | 1907 | c | t | XcampmN_010100001687 putative sugar transporter component |
| NZ_ACHT01000101 | 995 | g | t | XcampmN_010100003517 soluble lytic murein transglycosylase |
| NZ_ACHT01000104 | 13,081 | t | g | XcampmN_010100003612 GTP-dependent nucleic acid-binding protein EngD |
| NZ_ACHT01000113 | 10,410 | t | g | XcampmN_010100004062 acetyltransferase (GNAT) family protein |
| NZ_ACHT01000236 | 10,652 | t | c | XcampmN_010100007340 metallopeptidase |
| NZ_ACHT01000242 | 10,465 | a | c | XcampmN_010100007585 dihydrolipoamide acetyltransferase |
| NZ_ACHT01000294 | 2184 | g | t | XcampmN_010100009424 xanthan biosynthesis glucuronosyltransferase GumK |
| NZ_ACHT01000345 | 1576 | t | c | XcampmN_010100010814 cytochrome C peroxidase |
| NZ_ACHT01000402 | 4858 | t | c | XcampmN_010100012145 heavy metal transporter |
| NZ_ACHT01000404 | 632 | g | a | XcampmN_010100012200 tryptophan halogenase |
| NZ_ACHT01000500 | 23,584 | a | g | XcampmN_010100016057 putative polysaccharide deacetylase |
| NZ_ACHT01000520 | 5360 | a | g | XcampmN_010100016692 5-methyltetrahydrofolate-homocysteine methyl transferase |
| NZ_ACHT01000549 | 7371 | a | c | XcampmN_010100018271 two-component system sensor protein |
| NZ_ACHT01000560 | 4001 | t | c | XcampmN_010100018673 exodeoxyribonuclease III |
| NZ_ACHT01000590 | 927 | c | t | XcampmN_010100019303 RNA polymerase sigma factor |
| NZ_ACHT01000626 | 10,220 | t | c | XcampmN_010100019733 putative glutathionylspermidine synthase |
| NZ_ACHT01000634 | 2345 | t | c | XcampmN_010100019848 beta-mannosidase precursor |
| NZ_ACHT01000644 | 2590 | g | a | XcampmN_010100020168 two-component system sensor protein |
| NZ_ACHT01000694 | 10,665 | a | t | XcampmN_010100022153 peptide-acetyl-coenzyme A transporter family protein |
| NZ_ACHT01000720 | 19,485 | t | c | XcampmN_010100023003 drug:proton antiporter (19121–20371) |
2.5. Comparison of Xcm Isolated from Enset Versus Xcm Isolated from Banana
| Refseq Accession | Position | NCPPB 2005 (enset) | NCPPB 2251 (Banana) | NCPPB 4389 (Banana) | Locus Tag and Predicted Gene Product | |
|---|---|---|---|---|---|---|
| NZ_ACHT01000041 | 15,615 | c | t | t | XcampmN_010100000977 hemolysin III | |
| NZ_ACHT01000072 | 4507 | a | c | c | XcampmN_010100002109 VirB3 protein | |
| NZ_ACHT01000140 | 1116 | c | t | t | XcampmN_010100004536 LacI family transcription regulator | |
| NZ_ACHT01000199 | 8012 | g | t | g | XcampmN_010100006143 type III secreted effector HopW1 | |
| NZ_ACHT01000215 | 3229 | c | t | c | XcampmN_010100006660 HrpF protein | |
| NZ_ACHT01000236 | 9512 | c | t | t | XcampmN_010100007340 metallopeptidase | |
| NZ_ACHT01000294 | 31,553 | g | a | a | XcampmN_010100009559 MFS transporter | |
| NZ_ACHT01000303 | 7530 | a | c | c | XcampmN_010100009850 histidine kinase/response regulator hybrid protein | |
| NZ_ACHT01000332 | 2191 | a | g | g | XcampmN_010100010574 putative filamentous hemagglutinin-like protein | |
| NZ_ACHT01000360 | 1961 | a | g | g | XcampmN_010100011266 two-component system sensor protein | |
| NZ_ACHT01000374 | 12,027 | t | c | c | XcampmN_010100011573 Fis family transcriptional regulator | |
| NZ_ACHT01000388 | 5277 | t | g | g | XcampmN_010100011860 AraC family transcriptional regulator | |
| NZ_ACHT01000396 | 3578 | c | g | g | XcampmN_010100011920 catalase | |
| NZ_ACHT01000439 | 5166 | c | g | g | XcampmN_010100013743 ECF subfamily RNA polymerase sigma factor | |
| NZ_ACHT01000532 | 743 | c | t | t | XcampmN_010100017284 beta-glucosidase | |
| NZ_ACHT01000560 | 2783 | c | t | t | XcampmN_010100018663 molybdopterin biosynthesis | |
| NZ_ACHT01000668 | 1036 | c | a | a | XcampmN_010100021383 ABC transporter permease | |
| NZ_ACHT01000690 | 6284 | t | g | g | XcampmN_010100022008 isocitrate dehydrogenase | |
2.6. Loss of Phage-Associated Genes in Some Xcm Isolates
2.7. Experimental Validation of Genetic Polymorphisms

| Primer Sequences | Target Sequence RefSeq Accession Number and Coordinates | Restriction Enzyme |
|---|---|---|
| GAGCTCCTGCGCCGATGCGTGAGCGT AAAGGCGGCTATTCTA | NZ_ACHT01000081: 5900–6398 | AluI |
| CGGCGTGGTTTTGCCTTTGCCGTACGG CCTGGCGGTGAT | NZ_ACHT01000112: 10863–11347 | FokI |
| TCACCTGTTCGATGCGGCCGCTACTGG CTGTCGCGGC | NZ_ACHT01000124: 5385–5873 | AluI |
| ATGTTTGCCGATACCTGGATGCGCATG CTTGCCGGTTTCGACGA | NZ_ACHT01000304: 10080–10567 | NdeI |
3. Experimental Section
4. Conclusions
Acknowledgments
References
- Biruma, M.; Pillay, M.; Tripathi, L.; Blomme, G.; Abele, S.; Mwangi, M.; Bandyopadhyay, R.; Muchunguzi, P.; Kassim, S.; Nyine, M. Banana Xanthomonas wilt: A review of the disease, management strategies and future research directions. Afr. J. Biotechnol. 2007, 6, 953–962. [Google Scholar]
- Yirgou, D.; Bradbury, J.F. Bacterial wilt of Enset (Ensete ventricosum) incited by Xanthomonas musacearum sp. n . Phytopathology 1968, 58, 111–112. [Google Scholar]
- Yirgou, D.; Bradbury, J.F. A note on wilt of banana caused by the enset wilt organism Xanthomonas musacearum. East Afr. Agric. Forestry J. 1974, 40, 111–114. [Google Scholar]
- Tushemereirwe, W.; Kangire, A.; Ssekiwoko, F.; Offord, L.C.; Crozier, J.; Boa, E.; Rutherford, M.; Smith, J.J. First report of Xanthomonas campestris pv. musacearum on banana in Uganda. Plant Pathol. 2004, 53, 802. [Google Scholar] [CrossRef]
- Ndungo, V.; Eden-Green, S.; Blomme, G.; Crozier, J.; Smith, J.J. Presence of banana Xanthomonas wilt (Xanthomonas campestris pv. musacearum) in the Democratic Republic of Congo (DRC). Plant Pathol. 2006, 55, 294. [Google Scholar]
- Reeder, R.H.; Muhinyuza, J.B.; Opolot, O.; Aritua, V.; Crozier, J.; Smith, J. Presence of banana bacterial wilt (Xanthomonas campestris pv. musacearum) in Rwanda. Plant Pathol. 2007, 56, 1038. [Google Scholar]
- 7. Carter, B.A.; Reeder, R.; Mgenzi, S.R.; Kinyua, M.; Mbaka, J.N.; Doyle, K.; Nakato, V.; Mwangi, M.; Beed, F.; Aritua, V.; et al. Identification of Xanthomonas vasicola (formerly X. campestris pv. musacearum), causative organism of banana Xanthomonas wilt, in Tanzania, Kenya and Burundi. 2010. [Google Scholar]
- Kubiriba, J.; Karamura, E.B.; Jogo, W.; Tushemereirwe, W.K.; Tinzaara, W. Community mobilization: A key to effective control of banana Xanthomonas wilt. J. Dev. Agric. Econ. 2012, 4, 125–131. [Google Scholar]
- Namukwaya, B.; Tripathi, L.; Tripathi, J.N.; Arinaitwe, G.; Mukasa, S.B.; Tushemereirwe, W.K. Transgenic banana expressing Pflp gene confers enhanced resistance to Xanthomonas wilt disease. Transgenic. Res. 2011. [Google Scholar] [CrossRef]
- Tripathi, L.; Mwaka, H.; Tripathi, J.N.; Tushemereirwe, W.K. Expression of sweet pepper Hrap gene in banana enhances resistance to Xanthomonas campestris pv. musacearum. Mol. Plant Pathol. 2010, 11, 721–731. [Google Scholar]
- Aritua, V.; Parkinson, N.; Thwaites, R.; Heeney, J.V.; Jones, D.R.; Tushemereirwe, W.; Crozier, J.; Reeder, R.; Stead, D.E.; Smith, J. Characterization of the Xanthomonas sp. causing wilt of enset and banana and its proposed reclassification as a strain of X. vasicola. Plant Pathol. 2008, 57, 170–177. [Google Scholar]
- Studholme, D.J.; Kemen, E.; MacLean, D.; Schornack, S.; Aritua, V.; Thwaites, R.; Grant, M.; Smith, J.; Jones, J.D. Genome-wide sequencing data reveals virulence factors implicated in banana Xanthomonas wilt. FEMS Microbiol. Lett. 2010, 310, 182–192. [Google Scholar] [CrossRef]
- Aritua, V.; Parkinson, N.; Thwaites, R.; Jones, D.R.; Tushemereirwe, W.; Smith, J.J. Molecular epidemiology of Xanthomonas campestris pv. musacearum, the causal agent of xanthomonas wilt of banana and enset. Acta Hort. (ISHS) 2009, 828, 219–226. [Google Scholar]
- Ryan, R.P.; Vorhölter, F.J.; Potnis, N.; Jones, J.B.; van Sluys, M.A.; Bogdanove, A.J.; Dow, J.M. Pathogenomics of Xanthomonas: Understanding bacterium-plant interactions. Nat. Rev. Microbiol. 2011, 9, 344–355. [Google Scholar] [CrossRef]
- Aritua, V.; Nanyonjo, A.; Kumakech, F.; Tushemereirwe, W. Rep-PCR reveals a high genetic homogeneity among Ugandan isolates of Xanthomonas campestris pv musacearum. Afr. J. Biotechnol. 2007, 6, 179–183. [Google Scholar]
- Achtman, M. Insights from genomic comparisons of genetically monomorphic bacterial pathogens. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2012, 367, 860–867. [Google Scholar] [CrossRef]
- Cai, R.; Lewis, J.; Yanm, S.; Lium, H.; Clarkem, C.R.; Campanile, F.; Almeida, N.F.; Studholme, D.J.; Lindeberg, M.; Schneider, D.; Zaccardelli, M.; et al. The plant pathogen Pseudomonas syringae pv. tomato is genetically monomorphic and under strong selection to evade tomato immunity. PLoS Pathog. 2011, 7, e1002130. [Google Scholar]
- Kodama, Y.; Shumway, M.; Leinonen, R. International nucleotide sequence database collaboration. The sequence read archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef]
- Studholme, D.J.; Wasukira, A.; Paszkiewicz, K.; Aritua, V.; Thwaites, R.; Smith, J.; Grant, M. Draft Genome sequences of Xanthomonas sacchari and two banana-associated Xanthomonads reveal insights into the Xanthomonas Group 1 Clade. Genes 2011, 2, 1050–1065. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- 21. Benson, D.A.; Karsch-Mizrachi, I.; Clark, K.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 40, D48–D53. [Google Scholar]
- Lewis Ivey, M.L.; Tusiime, G.; Miller, S.A. A PCR assay for the detection of Xanthomonas campestris pv. musacearum in bananas. Plant Dis. 2010, 94, 109–114. [Google Scholar] [CrossRef]
- Adikini, S.; Tripathi, L.; Beeda, F.; Tusiime, G.; Magembe, E.M.; Kim, D.J. Development of a specific molecular tool for detecting Xanthomonas campestris pv. musacearum. Plant Pathol. 2011, 60, 443–452. [Google Scholar] [CrossRef]
- Adriko, J.; Aritua, V.; Mortensen, C.N.; Tushemereirwe, W.K.; Kubiriba, J.; Lund, O.S. Multiplex PCR for specific and robust detection of Xanthomonas campestris pv. musacearum in pure culture and infected plant material. Plant Pathol. 2012, 61, 489–497. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Kuo, T.T.; Tan, M.S.; Su, M.T.; Yang, M.K. Complete nucleotide sequence of filamentous phage Cf1c from Xanthomonas campestris pv. citri. Nucleic Acids Res. 1991, 19, 2498. [Google Scholar]
- Robinson, J.Y; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wasukira, A.; Tayebwa, J.; Thwaites, R.; Paszkiewicz, K.; Aritua, V.; Kubiriba, J.; Smith, J.; Grant, M.; Studholme, D.J. Genome-Wide Sequencing Reveals Two Major Sub-Lineages in the Genetically Monomorphic Pathogen Xanthomonas Campestris Pathovar Musacearum. Genes 2012, 3, 361-377. https://doi.org/10.3390/genes3030361
Wasukira A, Tayebwa J, Thwaites R, Paszkiewicz K, Aritua V, Kubiriba J, Smith J, Grant M, Studholme DJ. Genome-Wide Sequencing Reveals Two Major Sub-Lineages in the Genetically Monomorphic Pathogen Xanthomonas Campestris Pathovar Musacearum. Genes. 2012; 3(3):361-377. https://doi.org/10.3390/genes3030361
Chicago/Turabian StyleWasukira, Arthur, Johnbosco Tayebwa, Richard Thwaites, Konrad Paszkiewicz, Valente Aritua, Jerome Kubiriba, Julian Smith, Murray Grant, and David J. Studholme. 2012. "Genome-Wide Sequencing Reveals Two Major Sub-Lineages in the Genetically Monomorphic Pathogen Xanthomonas Campestris Pathovar Musacearum" Genes 3, no. 3: 361-377. https://doi.org/10.3390/genes3030361