A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLST of Campylobacter

{kind=link}
{kind=link}
Abstract
:1. The Campylobacter Problem
2. Campylobacter Ecology and Population Structure
3. Genomic Analysis of Campylobacter Isolates
4. The Challenges of Analyzing Multiple Bacterial Genomes
5. Analyzing Genome Sequence Variation—The Reference Genome Approach
6. The ‘Reference Gene’ Approach to Genome Analysis
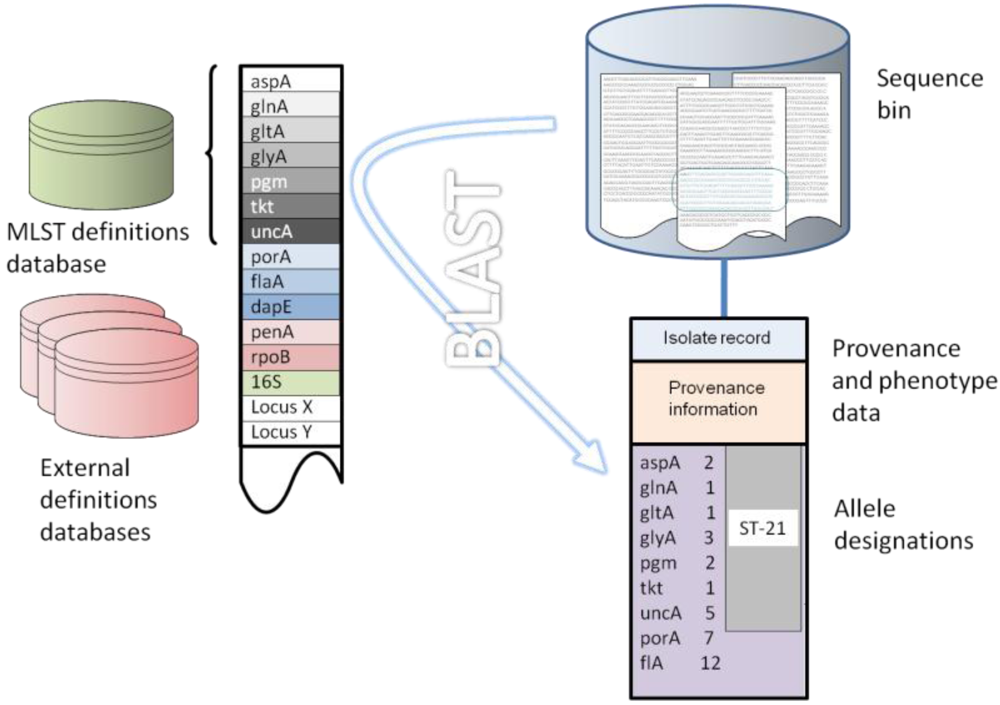
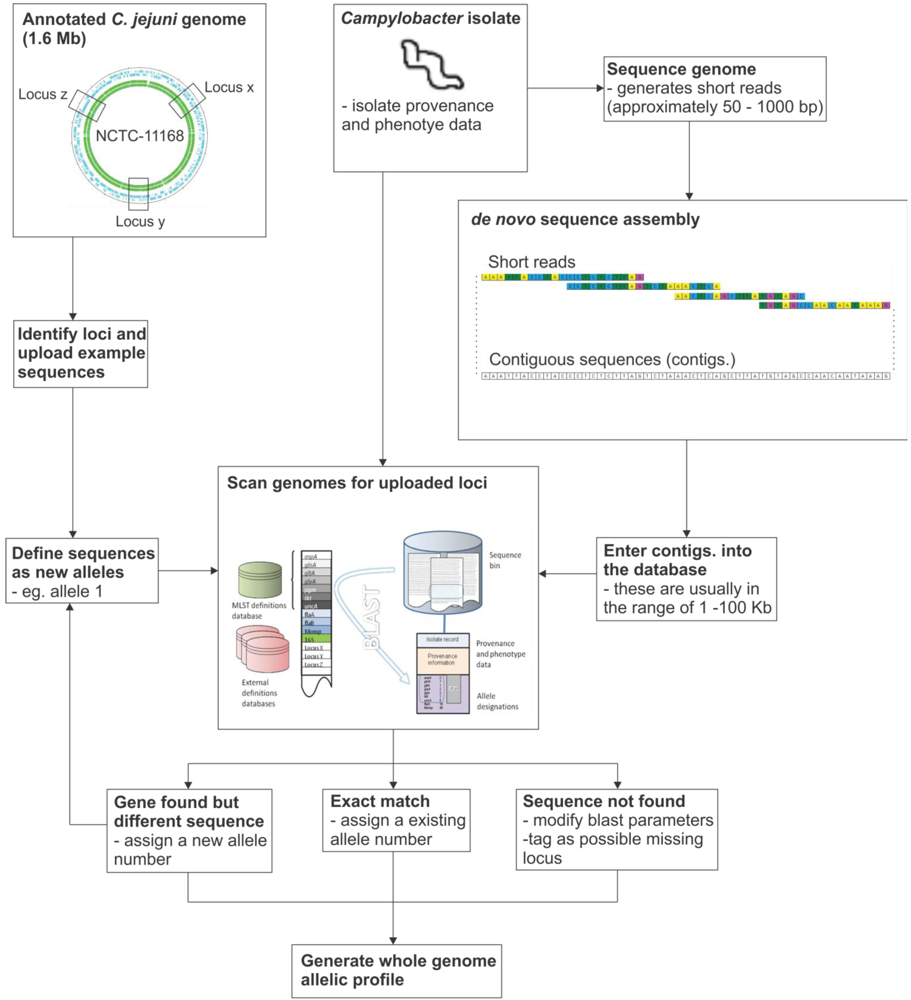
7. Implementation of the Reference Gene Approach
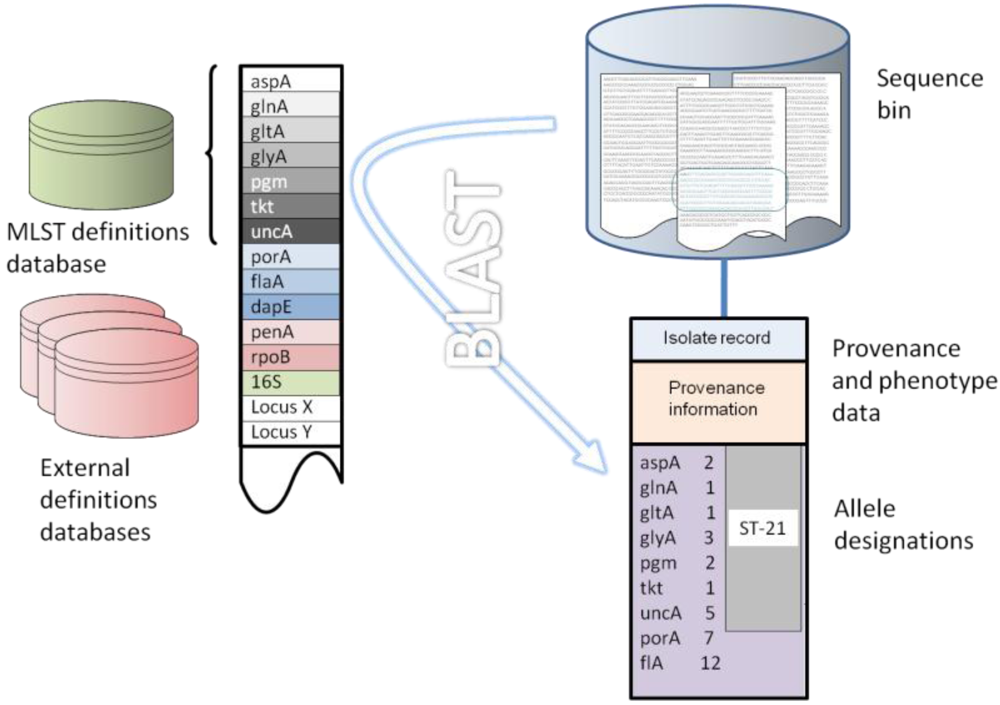
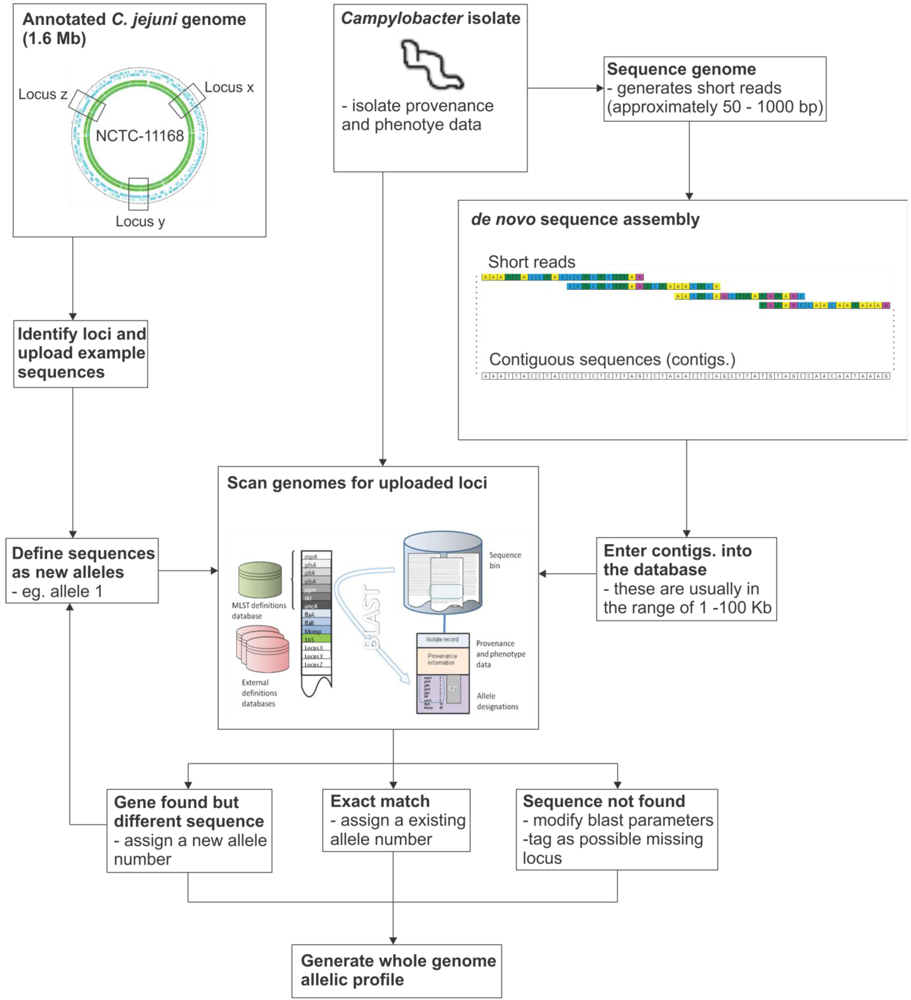
8. Concluding Remarks
Acknowledgements
References
- Newell, D.G.; Koopmans, M.; Verhoef, L.; Duizer, E.; Aidara-Kane, A.; Sprong, H.; Opsteegh, M.; Langelaar, M.; Threfall, J.; Scheutz, F.; et al. Food-borne diseases—The challenges of 20 years ago still persist while new ones continue to emerge. Int. J. Food Microbiol. 2010, 139, S3–S15. [Google Scholar] [CrossRef]
- Strachan, N.J.C.; Forbes, K.J. The growing UK epidemic of human campylobacteriosis. Lancet 2010, 376, 665–667. [Google Scholar]
- Nachamkin, I. Chronic effects of Campylobacter infection. Microbes Infect. 2002, 4, 399–403. [Google Scholar]
- Skirrow, M.B. Campylobacter enteritis: A “new” disease. BMJ 1977, 2, 9–11. [Google Scholar]
- Young, K.T.; Davis, L.M.; Dirita, V.J. Campylobacter jejuni: Molecular biology and pathogenesis. Nat. Rev. Microbiol. 2007, 5, 665–679. [Google Scholar]
- Medini, D.; Serruto, D.; Parkhill, J.; Relman, D.A.; Donati, C.; Moxon, R.; Falkow, S.; Rappuoli, R. Microbiology in the post-genomic era. Nat. Rev. Microbiol. 2008, 6, 419–430. [Google Scholar]
- Dingle, K.E.; Colles, F.M.; Wareing, D.R.A.; Ure, R.; Fox, A.J.; Bolton, F.J.; Bootsma, H.J.; Willems, R.J.L.; Urwin, R.; Maiden, M.C.J. Multilocus sequence typing system for Campylobacter jejuni. J. Clin. Microbiol. 2001, 39, 14–23. [Google Scholar]
- Dingle, K.E.; McCarthy, N.D.; Cody, A.J.; Peto, T.E.; Maiden, M.C. Extended sequence typing of Campylobacter spp., United Kingdom. Emerg. Infect. Dis. 2008, 14, 1620–1622. [Google Scholar] [CrossRef]
- Maiden, M.C.; Dingle, K.E. Population Biology of Campylobacter jejuni and Related Organisms. In Campylobacter, 3rd ed; ASM Press: Washington, DC, USA, 2008; pp. 27–40. [Google Scholar]
- Dingle, K.E.; Colles, F.M.; Ure, R.; Wagenaar, J.; Duim, B.; Bolton, F.J.; Fox, A.J.; Wareing, D.R.A.; Maiden, M.C.J. Molecular characterisation of Campylobacter jejuni clones: A rational basis for epidemiological investigations. Emerg. Infect. Dis. 2002, 8, 949–955. [Google Scholar]
- Miller, W.G.; On, S.L.; Wang, G.; Fontanoz, S.; Lastovica, A.J.; Mandrell, R.E. Extended multilocus sequence typing system for Campylobacter coli, C. lari, C. upsaliensis, and C. helveticus. J. Clin. Microbiol. 2005, 43, 2315–2329. [Google Scholar]
- van Bergen, M.A.; Dingle, K.E.; Maiden, M.C.; Newell, D.G.; van der Graaf-Van Bloois, L.; van Putten, J.P.; Wagenaar, J.A. Clonal nature of Campylobacter fetus as defined by multilocus sequence typing. J. Clin. Microbiol. 2005, 43, 5888–5898. [Google Scholar]
- Wilson, D.J.; Gabriel, E.; Leatherbarrow, A.J.; Cheesbrough, J.; Gee, S.; Bolton, E.; Fox, A.; Hart, C.A.; Diggle, P.J.; Fearnhead, P. Rapid evolution and the importance of recombination to the gastroenteric pathogen Campylobacter jejuni. Mol. Biol. Evol. 2009, 26, 385–397. [Google Scholar]
- Sheppard, S.K.; Colles, F.; Richardson, J.; Cody, A.J.; Elson, R.; Lawson, A.; Brick, G.; Meldrum, R.; Little, C.L.; Owen, R.J.; et al. Host association of Campylobacter genotypes transcends geographic variation. Appl. Environ. Microbiol. 2010, 76, 5269–5277. [Google Scholar]
- Sheppard, S.K.; Dallas, J.F.; Wilson, D.J.; Strachan, N.J.C.; McCarthy, N.D.; Jolley, K.A.; Colles, F.M.; Rotariu, O.; Ogden, I.D.; Forbes, K.J.; et al. Evolution of an agriculture-associated disease causing Campylobacter coli clade: Evidence from national surveillance data in Scotland. PLoS One 2010, 5, e15708. [Google Scholar]
- Dingle, K.E.; Colles, F.M.; Falush, D.; Maiden, M.C. Sequence typing and comparison of population biology of Campylobacter coli and Campylobacter jejuni. J. Clin. Microbiol. 2005, 43, 340–347. [Google Scholar]
- Sheppard, S.K.; McCarthy, N.D.; Falush, D.; Maiden, M.C. Convergence of Campylobacter species: Implications for bacterial evolution. Science 2008, 320, 237–239. [Google Scholar]
- French, N.; Barrigas, M.; Brown, P.; Ribiero, P.; Williams, N.; Leatherbarrow, H.; Birtles, R.; Bolton, E.; Fearnhead, P.; Fox, A. Spatial epidemiology and natural population structure of Campylobacter jejuni colonizing a farmland ecosystem. Environ. Microbiol. 2005, 7, 1116–1126. [Google Scholar]
- Colles, F.M.; Jones, K.; Harding, R.M.; Maiden, M.C. Genetic diversity of Campylobacter jejuni isolates from farm animals and the farm environment. Appl. Environ. Microbiol. 2003, 69, 7409–7413. [Google Scholar]
- Strachan, N.J.; Gormley, F.J.; Rotariu, O.; Ogden, I.D.; Miller, G.; Dunn, G.M.; Sheppard, S.K.; Dallas, J.F.; Reid, T.M.; Howie, H.; et al. Attribution of Campylobacter infections in northeast Scotland to specific sources by use of multilocus sequence typing. J. Infect. Dis. 2009, 199, 1205–1208. [Google Scholar] [CrossRef]
- Sheppard, S.K.; Dallas, J.F.; Strachan, N.J.; MacRae, M.; McCarthy, N.D.; Wilson, D.J.; Gormley, F.J.; Falush, D.; Ogden, I.D.; Maiden, M.C.; et al. Campylobacter genotyping to determine the source of human infection. Clin. Infect. Dis. 2009, 48, 1072–1078. [Google Scholar] [CrossRef]
- Mullner, P.; Spencer, S.E.; Wilson, D.J.; Jones, G.; Noble, A.D.; Midwinter, A.C.; Collins-Emerson, J.M.; Carter, P.; Hathaway, S.; French, N.P. Assigning the source of human campylobacteriosis in New Zealand: A comparative genetic and epidemiological approach. Infect. Genet. Evol. 2009, 6, 1311–1319. [Google Scholar]
- Wilson, D.J.; Gabriel, E.; Leatherbarrow, A.J.H.; Cheesbrough, J.; Gee, S.; Bolton, E.; Fox, A.; Fearnhead, P.; Hart, A.; Diggle, P.J. Tracing the source of campylobacteriosis. PLoS Genet. 2008, 26, e1000203. [Google Scholar]
- McCarthy, N.D.; Colles, F.M.; Dingle, K.E.; Bagnall, M.C.; Manning, G.; Maiden, M.C.; Falush, D. Host-associated genetic import in Campylobacter jejuni. Emerg. Infect. Dis. 2007, 13, 267–272. [Google Scholar]
- Maiden, M.C. Multilocus sequence typing of bacteria. Annu. Rev. Microbiol. 2006, 60, 561–588. [Google Scholar]
- Falush, D. Toward the use of genomics to study microevolutionary change in bacteria. PLoS Genet. 2009, 5, e1000627. [Google Scholar]
- Didelot, X.; Maiden, M.C.J. Impact of recombination on bacterial evolution. Trends Microbiol. 2010, 18, 315–322. [Google Scholar]
- Maiden, M.C.J.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar]
- Fearnhead, P.; Smith, N.G.; Barrigas, M.; Fox, A.; French, N. Analysis of recombination in Campylobacter jejuni from MLST population data. J. Mol. Evol. 2005, 61, 333–340. [Google Scholar]
- Schouls, L.M.; Reulen, S.; Duim, B.; Wagenaar, J.A.; Willems, R.J.; Dingle, K.E.; Colles, F.M.; van Embden, J.D. Comparative genotyping of Campylobacter jejuni by amplified fragment length polymorphism, multilocus sequence typing, and short repeat sequencing: Strain diversity, host range, and recombination. J. Clin. Microbiol. 2003, 41, 15–26. [Google Scholar]
- Didelot, X.; Falush, D. Inference of bacterial microevolution using multilocus sequence data. Genetics 2007, 175, 1251–1266. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Corander, J.; Marttinen, P. Bayesian identification of admixture events using multilocus molecular markers. Mol. Ecol. 2006, 15, 2833–2843. [Google Scholar]
- de Haan, C.P.; Kivisto, R.I.; Hakkinen, M.; Corander, J.; Hanninen, M.L. Multilocus sequence types of Finnish bovine Campylobacter jejuni isolates and their attribution to human infections. BMC Microbiol. 2010, 10, 200. [Google Scholar]
- Sheppard, S.K.; Colles, F.M.; McCarthy, N.D.; Strachan, N.J.; Ogden, I.D.; Forbes, K.J.; Dallas, J.F.; Maiden, M.C. Niche segregation and genetic structure of Campylobacter jejuni populations from wild and agricultural host species. Mol. Ecol. 2011, 20, 3484–3490. [Google Scholar]
- Champion, O.L.; Gaunt, M.W.; Gundogdu, O.; Elmi, A.; Witney, A.A.; Hinds, J.; Dorrell, N.; Wren, B.W. Comparative phylogenomics of the food-borne pathogen Campylobacter jejuni reveals genetic markers predictive of infection source. Proc. Natl. Acad. Sci. USA 2005, 102, 16043–16048. [Google Scholar]
- Dorrell, N.; Mangan, J.A.; Laing, K.G.; Hinds, J.; Linton, D.; Al-Ghusein, H.; Barrell, B.G.; Parkhill, J.; Stoker, N.G.; Karlyshev, A.V.; et al. Whole genome comparison of Campylobacter jejuni human isolates using a low-cost microarray reveals extensive genetic diversity. Genome Res. 2001, 11, 1706–1715. [Google Scholar] [CrossRef]
- Leonard, E.E., 2nd; Takata, T.; Blaser, M.J.; Falkow, S.; Tompkins, L.S.; Gaynor, E.C. Use of an open-reading frame-specific Campylobacter jejuni DNA microarray as a new genotyping tool for studying epidemiologically related isolates. J. Infect. Dis. 2003, 187, 691–694. [Google Scholar] [CrossRef]
- Taboada, E.N.; Mackinnon, J.M.; Luebbert, C.C.; Gannon, V.P.; Nash, J.H.; Rahn, K. Comparative genomic assessment of Multi-Locus Sequence Typing: Rapid accumulation of genomic heterogeneity among clonal isolates of Campylobacter jejuni. BMC Evol. Biol. 2008, 8, 229. [Google Scholar]
- Pearson, B.M.; Pin, C.; Wright, J.; I’Anson, K.; Humphrey, T.; Wells, J.M. Comparative genome analysis of Campylobacter jejuni using whole genome DNA microarrays. FEBS Lett. 2003, 554, 224–230. [Google Scholar]
- Taboada, E.N.; Acedillo, R.R.; Carrillo, C.D.; Findlay, W.A.; Medeiros, D.T.; Mkytczuk, O.L.; Roberts, M.J.; Valencia, C.A.; Farber, J.M.; Nash, J.H.E. Large-scale comparative genomics meta-analysis of Campylobacter jejuni isolates reveals low level of genome plasticity. J. Clin. Microbiol. 2004, 42, 4566–4576. [Google Scholar]
- Parker, C.T.; Quinones, B.; Miller, W.G.; Horn, S.T.; Mandrell, R.E. Comparative genomic analysis of Campylobacter jejuni strains reveals diversity due to genomic elements similar to those present in C. jejuni strain RM1221. J. Clin. Microbiol. 2006, 44, 4125–4135. [Google Scholar] [CrossRef]
- Quinones, B.; Guilhabert, M.R.; Miller, W.G.; Mandrell, R.E.; Lastovica, A.J.; Parker, C.T. Comparative genomic analysis of clinical strains of Campylobacter jejuni from South Africa. PLoS One 2008, 3, e2015. [Google Scholar]
- Lefebure, T.; Bitar, P.D.P.; Suzuki, H.; Stanhope, M.J. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol. Evol. 2010, 2, 646–655. [Google Scholar]
- Fouts, D.E.; Mongodin, E.F.; Mandrell, R.E.; Miller, W.G.; Rasko, D.A.; Ravel, J.; Brinkac, L.M.; DeBoy, R.T.; Parker, C.T.; Daugherty, S.C.; et al. Major structural differences and novel potential virulence mechanisms from the genomes of multiple campylobacter species. PLoS Biol. 2005, 3, e15. [Google Scholar]
- Nauta, M.J.; Jacobs-Reitsma, W.F.; Havelaar, A.H. A risk assessment model for Campylobacter in broiler meat. Risk Anal. 2007, 27, 845–861. [Google Scholar]
- Peabody, R.; Ryan, M.J.; Wall, P.G. Outbreaks of Campylobacter infection: Rare events for a common pathogen. Commun. Dis. Rep. 1997, 7, R33–R37. [Google Scholar]
- Frost, J.A.; Gillespie, I.A.; O’Brien, S.J. Public health implications of Campylobacter outbreaks in England and Wales, 1995-9: Epidemiological and microbiological investigations. Epidemiol. Infect. 2002, 128, 111–118. [Google Scholar]
- Neimann, J.; Engberg, J.; Molbak, K.; Wegener, H.C. A case-control study of risk factors for sporadic Campylobacter infections in Denmark. Epidemiol. Infect. 2003, 130, 353–366. [Google Scholar]
- Pearson, T.; Okinaka, R.T.; Foster, J.T.; Keim, P. Phylogenetic understanding of clonal populations in an era of whole genome sequencing. Infect. Genet. Evol. 2009, 9, 1010–1019. [Google Scholar]
- Caro-Quintero, A.; Rodriguez-Castano, G.P.; Konstantinidis, K.T. Genomic insights into the convergence and pathogenicity factors of Campylobacter jejuni and Campylobacter coli species. J. Bacteriol. 2009, 191, 5824–5831. [Google Scholar]
- Biggs, P.J.; Fearnhead, P.; Hotter, G.; Mohan, V.; Collins-Emerson, J.; Kwan, E.; Besser, T.E.; Cookson, A.; Carter, P.E.; French, N.P. Whole-genome comparison of two Campylobacter jejuni isolates of the same sequence type reveals multiple loci of different ancestral lineage. PLoS One 2011, 6, e27121. [Google Scholar]
- Harris, S.R.; Feil, E.J.; Holden, M.T.; Quail, M.A.; Nickerson, E.K.; Chantratita, N.; Gardete, S.; Tavares, A.; Day, N.; Lindsay, J.A.; et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science 2010, 327, 469–474. [Google Scholar]
- Gutacker, M.M.; Mathema, B.; Soini, H.; Shashkina, E.; Kreiswirth, B.N.; Graviss, E.A.; Musser, J.M. Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J. Infect. Dis. 2006, 193, 121–128. [Google Scholar]
- Morelli, G.; Song, Y.; Mazzoni, C.J.; Eppinger, M.; Roumagnac, P.; Wagner, D.M.; Feldkamp, M.; Kusecek, B.; Vogler, A.J.; Li, Y.; et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 2010, 42, 1140–1143. [Google Scholar] [CrossRef]
- Bentley, S.D.; Croucher, N.J.; Harris, S.R.; Fraser, C.; Quail, M.A.; Burton, J.; van der Linden, M.; Mcgee, L.; von Gottberg, A.; Song, J.H.; et al. Rapid pneumococcal evolution in response to clinical interventions. Science 2011, 331, 430–434. [Google Scholar]
- Zhang, W.; Qi, W.; Albert, T.J.; Motiwala, A.S.; Alland, D.; Hyytia-Trees, E.K.; Ribot, E.M.; Fields, P.I.; Whittam, T.S.; Swaminathan, B. Probing genomic diversity and evolution of Escherichia coli O157 by single nucleotide polymorphisms. Genome Res. 2006, 16, 757–767. [Google Scholar]
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Traces of human migrations in Helicobacter pylori populations. Science 2003, 299, 1582–1585. [Google Scholar]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar]
- Cody, A.J.; Maiden, M.J.; Dingle, K.E. Genetic diversity and stability of the porA allele as a genetic marker in human Campylobacter infection. Microbiology 2009, 155, 4145–4154. [Google Scholar]
- Taha, M.K.; Vazquez, J.A.; Hong, E.; Bennett, D.E.; Bertrand, S.; Bukovski, S.; Cafferkey, M.T.; Carion, F.; Christensen, J.J.; Diggle, M.; et al. Target gene sequencing to characterize the penicillin G susceptibility of Neisseria meningitidis. Antimicrob. Agents Chemother. 2007, 51, 2784–2792. [Google Scholar]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.A.; Barrell, B.G.; Parkhill, J. ACT: The artemis comparison tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar]
- Darling, A.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS One 2010, 5, e11147. [Google Scholar]
- Jolley, K.A.; Maiden, M.C. BIGSDB: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 2010, 11, 595. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Chaudhuri, R.R.; Loman, N.J.; Snyder, L.A.; Bailey, C.M.; Stekel, D.J.; Pallen, M.J. xBASE2: A comprehensive resource for comparative bacterial genomics. Nucleic Acids Res. 2008, 36, D543–D546. [Google Scholar]
- Stalker, J.; Gibbins, B.; Meidl, P.; Smith, J.; Spooner, W.; Hotz, H.R.; Cox, A.V. The Ensembl Web site: Mechanics of a genome browser. Genome Res. 2004, 14, 951–955. [Google Scholar]
- Davidsen, T.; Beck, E.; Ganapathy, A.; Montgomery, R.; Zafar, N.; Yang, Q.; Madupu, R.; Goetz, P.; Galinsky, K.; White, O. The comprehensive microbial resource. Nucleic Acids Res 2009, D340–D345. [Google Scholar]
- Liolios, K.; Chen, I.; Mavromatis, K.; Tavernarakis, N.; Hugenholtz, P.; Markowitz, V.; Kyrpides, N. The Genomes On Line Database (GOLD) in 2009: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2010, 38, D346–D354. [Google Scholar]
- Parkhill, J.; Wren, B.W.; Mungall, K.; Ketley, J.M.; Churcher, C.; Basham, D.; Chillingworth, T.; Davies, R.M.; Feltwell, T.; Holroyd, S.; et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 2000, 403, 665–668. [Google Scholar]
- Stahl, M.; Friis, L.M.; Nothaft, H.; Liu, X.; Li, J.J.; Szymanski, C.M.; Stintzi, A. L-Fucose utilization provides Campylobacter jejuni with a competitive advantage. Proc. Natl. Acad. Sci. USA 2011, 108, 7194–7199. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sheppard, S.K.; Jolley, K.A.; Maiden, M.C.J. A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLST of Campylobacter. Genes 2012, 3, 261-277. https://doi.org/10.3390/genes3020261
Sheppard SK, Jolley KA, Maiden MCJ. A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLST of Campylobacter. Genes. 2012; 3(2):261-277. https://doi.org/10.3390/genes3020261
Chicago/Turabian StyleSheppard, Samuel K., Keith A. Jolley, and Martin C. J. Maiden. 2012. "A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLST of Campylobacter" Genes 3, no. 2: 261-277. https://doi.org/10.3390/genes3020261
APA StyleSheppard, S. K., Jolley, K. A., & Maiden, M. C. J. (2012). A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLST of Campylobacter. Genes, 3(2), 261-277. https://doi.org/10.3390/genes3020261