Gene Duplication and Ectopic Gene Conversion in Drosophila

{kind=link}
{kind=link}
Abstract
:1. Introduction
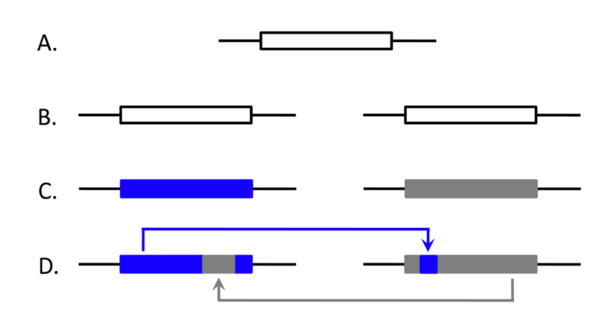
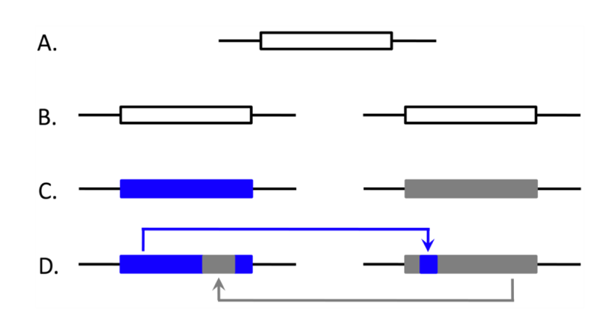
- What is the relative contribution of EGC to patterns of concerted evolution?
- How does genomic context (e.g., nucleotide base composition; linkage relationships between duplicates) affect EGC?
- How do selection and EGC interact to influence adaptation?
- Does gene conversion between duplicates bias estimates of the gene duplication rate and the tempo of paralog differentiation?
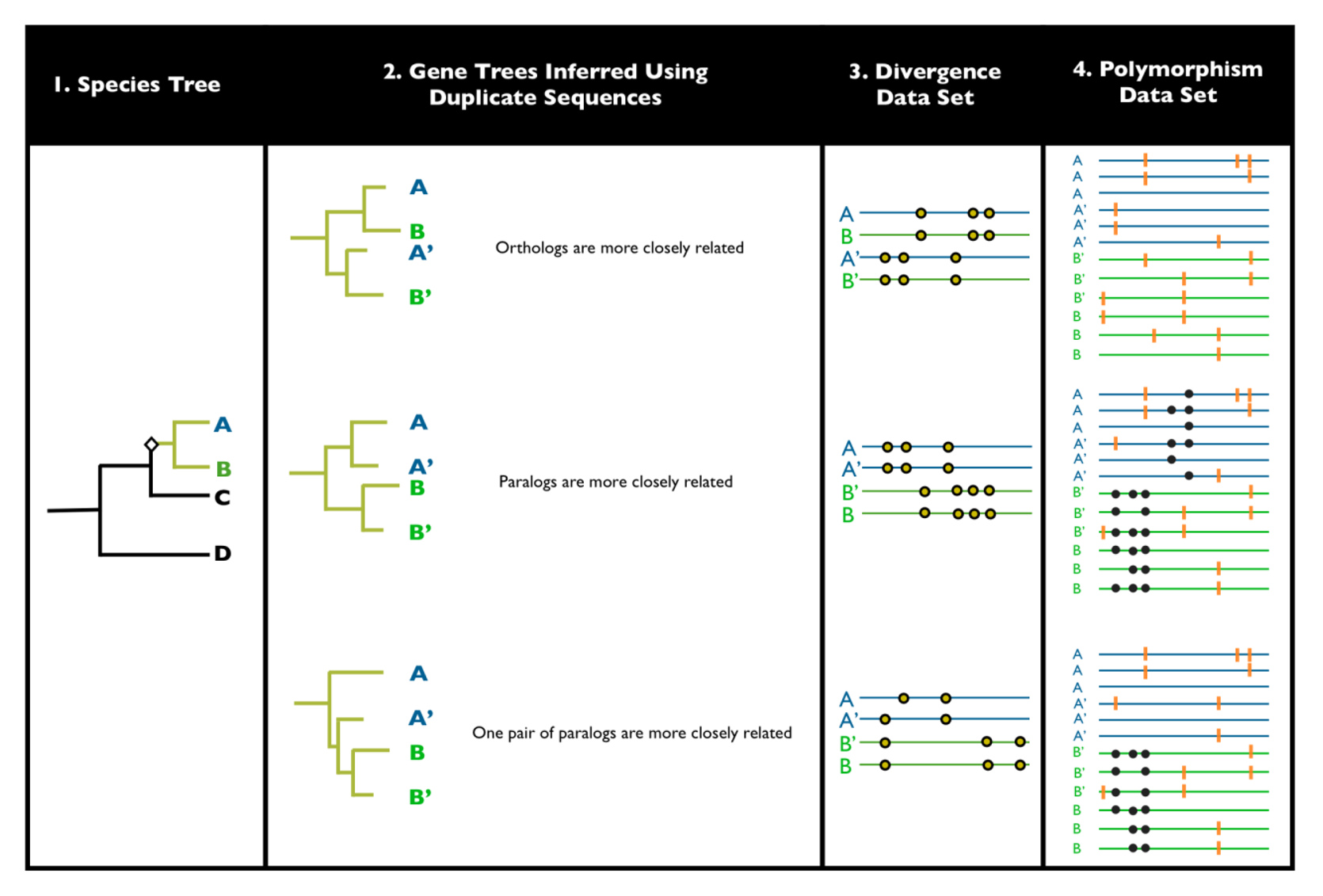
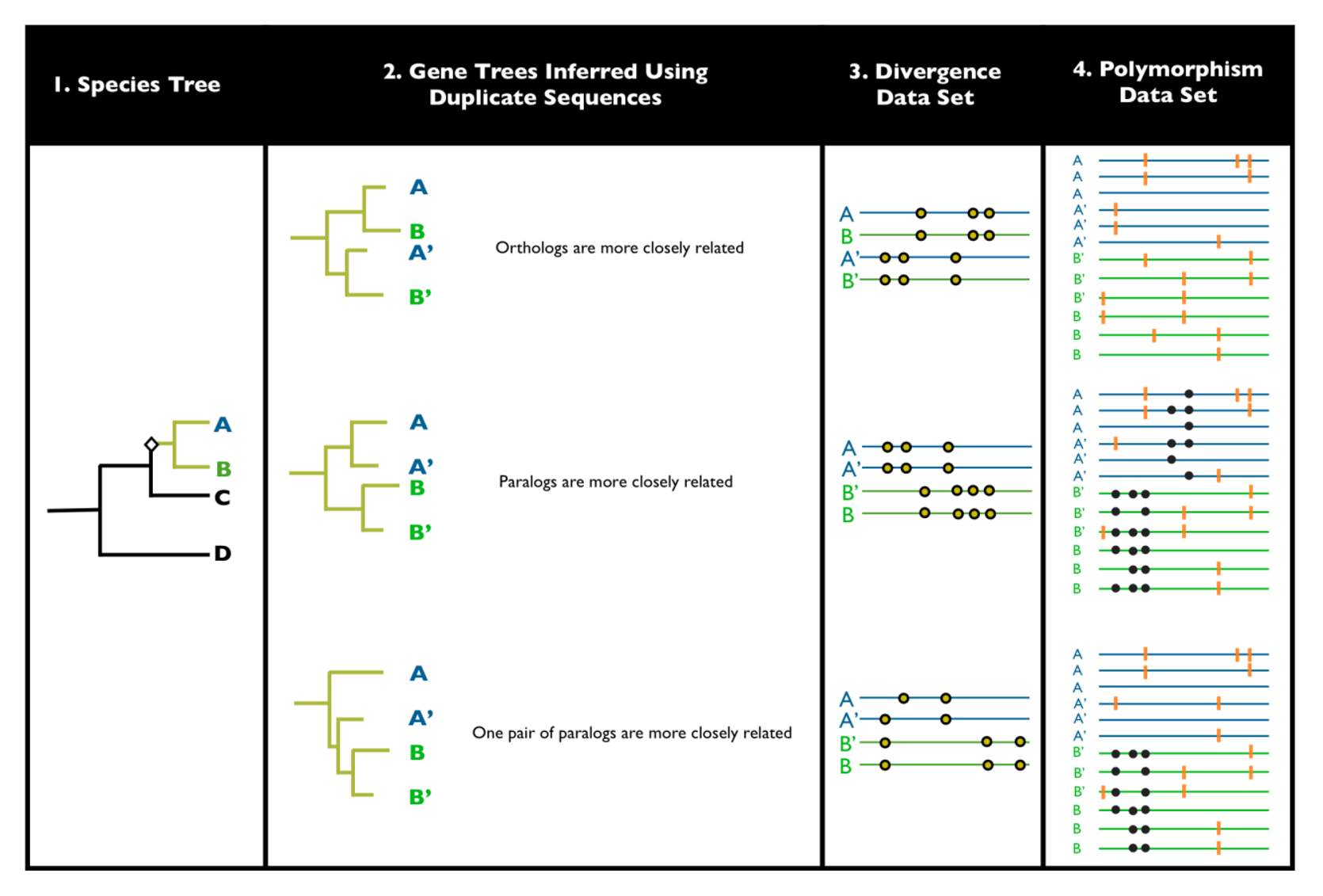
2. Detecting Ectopic Gene Conversion
3. Historical Background to Ectopic Gene Conversion
3.1. Repetitive DNA and the Origin of Genome Size Variation
3.2. Concerted Evolution, rDNA, and Drosophila
4. Distinguishing Ectopic Gene Conversion and Gene Turnover as Drivers of Concerted Evolution in Drosophila
4.1. Ectopic Conversion from a Case Studies Perspective
Heat Shock Proteins (HSP)
Amylase
4.2. Ectopic Gene Conversion from a Genome-Wide Perspective
5. The Genomic Context of Duplication and Ectopic Gene Conversion
6. Interaction between Selection, Ectopic Gene Conversion and Evolutionary Divergence between Paralogs
7. Temporal Dynamics of Duplication and Paralog Divergence
8. Conclusions
Acknowledgments
References
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
- Xiao, H.; Jiang, N.; Schaffner, E.; Stockinger, E.J.; van der Knaap, E.A. Retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science 2008, 14, 1527–1530. [Google Scholar]
- Pronk, J.C.; Frants, R.R.; Jansen, W.; Eriksson, A.W.; Tonino, G.J.M. Evidence for duplication of the human salivary amylase gene. Hum. Genet. 1982, 60, 32–35. [Google Scholar]
- Zhang, J.Z.; Zhang, Y.; Rosenberg, H.F. Adaptive evolution of a dupicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat. Genet. 2002, 30, 411–415. [Google Scholar]
- Perry, G.H.; Dominy, N.J.; Claw, K.G.; Lee, A.S.; Fiegler, H.; Redon, R.; Werner, J.; Villanea, F.A.; Mountain, J.L.; Misra, R.; Carter, N.P.; Lee, C.; Stone, A.C. Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 2007, 39, 1256–1260. [Google Scholar]
- Sackton, T.B.; Lazzaro, B.P.; Schlenke, T.A.; Evans, J.D.; Hultmark, D.; Clark, A.G. Dynamic evolution of the innate immune system in Drosophila. Nat. Genet. 2007, 39, 1461–1468. [Google Scholar]
- Lynch, M.; Force, A.G. The origin of interspecific genomic incompatibility via gene duplication. Am. Nat. 2007, 156, 590–605. [Google Scholar]
- Ting, C.-T.; Tsaur, S.C.; Sun, S.; Browne, W.E.; Chen, Y.-C.; Patel, N.H.; Wu, C.-I. Gene duplication and speciation in Drosophila: Evidence from the Odysseus locus. Proc. Natl. Acad. Sci. USA 2004, 101, 12232–12235. [Google Scholar]
- Moyle, L.C.; Muir, C.D.; Han, M.V.; Hahn, M.W. The contribution of gene movement to the “two rules of speciation”. Evolution 2010, 64, 1541–1557. [Google Scholar]
- Zhang, J.; Dean, A.M.; Brunet, F.; Long, M. Evolving protein functional diversity in new genes of Drosophila. Proc. Natl. Acad. Sci. USA 2004, 101, 16246–16250. [Google Scholar]
- Burki, F.; Kaessmann, K. Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter flux. Nat. Genet. 2004, 36, 1061–1063. [Google Scholar]
- Zhang, J.Z. Evolution by gene duplication. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar]
- Lupski, J.R. Genomic Disorders: Recombination-based disease resulting from Genome architecture. Am. J. Hum. Genet. 2003, 72, 246–252. [Google Scholar]
- Chen, J.M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion; mechanisms, evolution, and human disease. Nat. Rev. Genet. 2007, 8, 762–775. [Google Scholar]
- Conrad, B.; Antonarakis, S.E. Gene duplication: A drive for phenotypic diversity and cause of human disease. Annu. Rev. Genom. Hum. Genet. 2007, 8, 17–35. [Google Scholar]
- Lange, J.; Skaletsky, H.; van Daalen, S.K.M.; Embry, S.L.; Korver, C.M.; Brown, L.G.; Oate, R.D.; Silder, S.; Repping, S.; David, C.; et al. Isodicentric Y chromosomes and sex disorders as byproducts of homologous recombination that maintains palindromes. Cell 2009, 138, 855–869. [Google Scholar]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar]
- Teshima, K.M.; Innan, H. The effect of gene conversion on the divergence between duplicated genes. Genetics 2004, 166, 1553–1560. [Google Scholar]
- Innan, H. Population genetic models of duplicated genes. Genetica 2009, 137, 19–37. [Google Scholar]
- Sawyer, S. Statistical tests for detecting gene conversion. J. Mol. Evol. 1989, 6, 526–538. [Google Scholar]
- Leigh, B.A.J.; Ish-Horowicz, D. Evolution of the 87A and 87C heat-shock loci in Drosophila. Nature 1981, 290, 677–682. [Google Scholar]
- Osada, N.; Innan, H. Duplication and gene conversion in the Drosophila melanogaster genome. PLoS Genet. 2008, 4, e1000305. [Google Scholar]
- Innan, H. A two-locus gene conversion model with selection and its application to the human RHCE and RHD genes. Proc. Natl. Acad. Sci. USA 2003, 100, 8793–8798. [Google Scholar]
- Sturtevant, A.H. The effects of unequal crossing over at the Bar locus in Drosophila. Genetics 1925, 10, 117–147. [Google Scholar]
- Bridges, C.B. The Bar “gene” a duplication. Science 1936, 83, 210–211. [Google Scholar]
- Brown, D.D.; Wensink, P.C.; Jordan, E. A comparison of the Ribosomal DNA's of Xenopus mulleri: The Evoluiton of Tandem Genes. J. Mol. Biol. 1972, 63, 57–73. [Google Scholar]
- Mirsky, A.E.; Ris, H. The deoxyribonucleic acid content of animal cells and its evolutionary significance. J. Gen. Phys. 1951, 34, 451. [Google Scholar]
- Britten, R.J.; Kohne, D.E. Repeated Sequences in DNA. Science 1968, 161, 529–540. [Google Scholar]
- Muller, H.J.; Prokofyeva-Belgovskaya, A.A.; Kossikov, K.V. Unequal crossing-over in the Bar mutant as a result of duplication of a minute chromosome section. CR Acad. Sci. U.S.S.R. 1936, 1, 87–88. [Google Scholar]
- Keyl H.-G. A demonstrable local and geometric increase in the chromosomal DNA of Chironomus. Experientia 1965, 21, 191–193.
- Callan, H.G. The organization of genetic units in chromosomes. J. Cell Sci. 1967, 2, 1–7. [Google Scholar]
- Whitehouse, H.L.K. A cycloid model for the chromosome. J. Cell Sci. 1967, 2, 9–22. [Google Scholar]
- Straus, N.A. Comparative DNA renaturation kinetics in amphibians. Proc. Nat. Acad. Sci. USA 1971, 68, 799–802. [Google Scholar]
- Callan, H.G.; Lloyd, L. Lampbrush chromosomes of crested newts Triturus cristatus (Laurenti). Phil. Trans. R. Soc. B 1960, 243, 135–219. [Google Scholar]
- Thomas, C.A., Jr. The Theory of the Master Gene. In The Neurosciences: Evolution of Eukaryotic Genes; Schmitt, F.O., Ed.; Rockefeller University Press: New York, NY, USA, 1970; p. 973. [Google Scholar]
- Edelman, G.M.; Gally, J.A. Somatic recombination of duplicated genes—A hypothesis on origin of antibody diversity. Proc. Natl. Acad. Sci. USA 1967, 57, 353–358. [Google Scholar]
- Edelman, G.M.; Gally, J.A. Arrangement and evolution of eukaryotic genes. In The Neurosciences: Evolution of Eukaryotic Genes; Schmitt, F.O., Ed.; Rockefeller University Press: New York, NY, USA, 1970; p. 962. [Google Scholar]
- Long, E.O.; Dawid, I.B. Repeated genes in eukaryotes. Ann. Rev. Biochem. 1980, 49, 727–764. [Google Scholar]
- Eickbush, T.H.; Eickbush, D.G. Finely orchestrated movements: Evolution of the ribosomal RNA genes. Genetics 2007, 175, 447–485. [Google Scholar]
- Brown, D.D.; Sugimoto, K. 5 S DNAs of Xenopus laevis and Xenopus mulleri: Evolution of a gene family. J. Mol. Biol. 1973, 78, 397–415. [Google Scholar]
- Smith, G.P. Unequal crossover and the evolution of multigene Families. Cold Spring Harbor Symp. Quant. Biol. 1974, 38, 507–513. [Google Scholar]
- Dover, G.A. Molecular drive: a cohesive mode of species evolution. Nature 1982, 199, 111–117. [Google Scholar]
- Ritossa, F.M.; Atwood, K.C.; Lindsley, D.L.; Spiegelman, S. On the chromosomal distribution of DNA complementary to ribosomal and soluble RNA. Nat. Cancer Inst. Monogr. 1966, 23, 449. [Google Scholar]
- Ritossa, F.M. Unstable redundancy of genes for ribosomal RNA. Proc. Nat. Acad. Sci. USA 1968, 60, 509–516. [Google Scholar]
- Tartoff, K.D. Unequal crossing over then and now. Genetics 1988, 120, 1–6. [Google Scholar]
- Hawley, R.S.; Marcus, C.H. Recombinational controls of rDNA redundancy in Drosophila. Annu. Rev. Mol. Cell Biol. 1989, 4, 641–649. [Google Scholar]
- Tartoff, K.D. Unequal mitotic sister chromatid exchange and disproportionate replication as mechanisms regulating ribosomal RNA gene redundancy. Proc. Nat. Acad. Sci. USA 1974, 71, 1272–1276. [Google Scholar]
- Tartoff, K.D. Evolution of transcribed and spacer sequences in the ribosomal RNA genes of drosophila. Cell 1979, 3, 607–614. [Google Scholar]
- Renkawitz-Pohl, R.; Glätzer, K.H.; Kunz, W. Characterization of cloned ribosomal DNA from wheat and barley. Nucleic Acid. Res. 1980, 8, 4593–4611. [Google Scholar]
- Schlötterer, C.; Tautz, D. Chromosomal homogeneity of Drosophila ribosomal DNA arrays suggests intrachromosomal exchanges drive concerted evolution. Curr. Biol. 1994, 4, 777–783. [Google Scholar]
- Lyckegaard, E.M.S.; Clark, A.G. Evolution of ribosomal RNA gene copy number on the sex chromosomes of Drosophila melanogaster. Mol. Biol. Evol. 1991, 8, 458–474. [Google Scholar]
- Zhang, X.; Eickbush, T.H. Characterization of active R2 retrotransposition in the rDNA locus of Drosophila simulans. Genetics 2005, 170, 195–205. [Google Scholar]
- Stage, D.E.; Eickbush, T.H. Sequence variation within the rRNA gene loci of 12 Drosophila species. Genome Res. 2007, 17, 1888–1897. [Google Scholar]
- Nei, M.; Gu, X.; Sitnikova, T. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Nat. Acad. Sci. USA 1997, 94, 7799–7806. [Google Scholar]
- Nei, M.; Rooney, A.P. Concerted and birth-and-death evolution of multigene families. Annu. Rev. Genet. 2005, 39, 121–152. [Google Scholar]
- Bettencourt, B.R.; Feder, M.E. Rapid concerted evolution via gene conversion at the Drosophila hsp70 genes. J. Mol. Evol. 2002, 54, 569–586. [Google Scholar]
- Hogan, C.C.; Bettencourt, B.R. Duplicate gene evolution toward multiple fates at the Drosophila melanogaster HIP/HIP-Replacement locus. J. Mol. Evol. 2009, 68, 337–350. [Google Scholar]
- Payant, V.; Abukashawa, S.; Sasseville, M.; Benkel, B.F.; Hickey, D.A.; David, J. Evolutionary conservation of the chromosomal configuration and regulation of amylase genes among eight species of the Drosophila melanogaster species subgroup. Mol. Biol. Evol. 1988, 5, 560–567. [Google Scholar]
- Bally-Cuif, L.; Payant, V.; Abukashawa, S.; Benkel, B.F.; Hickey, D.A. Molecular cloning and partial sequence characterization of the duplicated amylase genes from Drosophila erecta. Genet. Sci. Evol. 1990, 22, 57–64. [Google Scholar]
- Hickey, D.A.; Bally-Cuif, L.; Abukashawa, S.; Payant, V.; Benkel, B.F. Concerted evolution of duplicated protein-coding genes in Drosophila. Proc. Natl. Acad. Sci. USA 1991, 88, 1611–1615. [Google Scholar]
- Shibata, H.; Yamazaki, T. Molecular evolution of the duplicated Amy locus in the Drosophila melanogaster species subgroup: Concerted evolution only in the coding region and an excess of nonsynonymous substitutions in speciation. Genetics 1995, 141, 223–236. [Google Scholar]
- Okuyama, E.; Shibata, H.; Tachida, H.; Yamazaki, T. Molecular evolution of the 5′-flanking regions of the duplicated Amy genes in Drosophila melanogaster species subgroup. Mol. Biol. Evol. 1996, 13, 574–583. [Google Scholar]
- Zhang, Z.; Inomata, N.; Yamazaki, T.; Kishino, H. Evolutionary history and mode of the amylase multigene family in Drosophila. J. Mol. Evol. 2003, 57, 702–709. [Google Scholar]
- Brown, C.J.; Aquadro, C.F.; Anderson, W.W. DNA sequence evolution of the amylase multigene family in Drosophila pseudoobscura. Genetics 1990, 126, 131–138. [Google Scholar]
- Popadić, A.; Anderson, W.W. Evidence for gene conversion in the amylase multigene family of Drosophila pseudoobscura. Mol. Biol. Evol. 1995, 12, 564–572. [Google Scholar]
- Inomata, N.; Yamazaki, T. Evolution of nucleotide substitutions and gene regulation in the amylase multigenes in Drosophila kikkawai and its sibling species. Mol. Biol. Evol. 2000, 17, 601–615. [Google Scholar]
- Araki, H.; Inomata, N.; Yamazaki, T. Molecular evolution of duplicated amylase gene regions in Drosophila melanogaster: Evidence of positive selection in the coding regions and selective constraints in the cis-regulatory regions. Genetics 2001, 157, 667–677. [Google Scholar]
- Inomata, N.; Yamazaki, T. Nucleotide variation of the duplicated Amylase genes in Drosophila kikkawai. Mol. Biol. Evol. 2002, 19, 678–688. [Google Scholar]
- Charles, J.P.; Chihara, C.; Nejad, S.; Riddiford, L.M. A cluster of cuticle protein genes of Drosophila melanogaster at 65A: Sequence, structure and evolution. Genetics 1997, 147, 1213–1224. [Google Scholar]
- Žurovcová, M.; Ayala, F.J. Polymorphism patterns in two tightly linked developmental genes, Idgf1 and Idgf3, of Drosophila melanogaster. Genetics 2002, 162, 177–188. [Google Scholar]
- King, L.M. The role of gene conversion in determining sequence variation and divergence in the Est-5 gene family in Drosophila pseudoobscura. Genetics 1998, 148, 305–315. [Google Scholar]
- Balakirev, E.S.; Ayala, F.J. Molecular population genetics of the beta-esterase gene cluster of Drosophila melanogaster. J. Genet. 2003, 82, 115–131. [Google Scholar]
- Matsuo, Y.; Yamazaki, T. Nucleotide variation and divergence in the histone multigene family in Drosophila melanogaster. Genetics 1989, 122, 87–97. [Google Scholar]
- Lazarro, B.P.; Clark, A.G. Evidence for recurrent paralogous gene conversion and exceptional allelic divergence in the Attacin genes of Drosophila melanogaster. Genetics 2001, 159, 659–671. [Google Scholar]
- Kelleher, E.S.; Markow, T.A. Duplication, selection and gene conversion in a Drosophila mojavensis female reproductive protein family. Genetics 2009, 181, 1451–1465. [Google Scholar]
- Thornton, K.; Long, M. Excess of amino acid substitutions relative to polymorphism between X-linked duplications in Drosophila. Mol. Biol. Evol. 2005, 22, 273–284. [Google Scholar]
- Clement, Y.; Tavares, R.; Marais, G.A.B. Does lack of recombination enhance asymmetric evolution among duplicate genes? Insights from the Drosophila melanogaster genome. Gene 2006, 385, 89–95. [Google Scholar]
- Meisel, M.P. Evolutionary dynamics of recently duplicated genes: Selective constraints on diverging paralogs in the Drosophila pseudoobscura genome. J. Mol. Evol. 2009, 69, 81–93. [Google Scholar]
- Arguello, J.R.; Chen, Y.; Yang, S.; Wang, W.; Long, M. Origination of an X-linked testes chimeric gene by illegitimate recombination in Drosophila. PLoS Genet. 2006, 2, e77. [Google Scholar]
- Skaletsky, H.; Kuroda-Kawaguchi, T.; Minx, P.J.; Cordum, H.S.; Hillier, L.D.; Brown1, L.G.; Repping, S.; Pyntikova, T.; Ali, J.; Bieri, T. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 2003, 423, 825–837. [Google Scholar]
- Rozen, S.; Skaletsky, H.; Marszalek, J.D.; Minx, P.J.; Cordum, H.S.; Waterston, R.H.; Wilson, R.K.; Page, D.C. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 2003, 423, 873–876. [Google Scholar]
- Hahn, H.W.; Han, M.; Han, S.G. Gene family evolution across 12 Drosophila genomes. PLoS Genet. 2007, 3, e197. [Google Scholar]
- Casola, G.; Ganote, C.L.; Hahn, M.W. Nonallelic gene conversion in the genus Drosophila. Genetics 2010, 185, 95–103. [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007, 450, 203–218. [Google Scholar] [Green Version]
- Emerson, J.J.; Cardoso-Moreira, M.; Borevitz, J.O.; Long, M. Natural selection shapes genome wide patterns of copy number polymorphism in D. melanogaster. Science 2008, 320, 1629–1631. [Google Scholar]
- Mansai, S.P.; Innan, H. The power of the methods for detecting interlocus gene conversion. Genetics 2010, 184, 517–527. [Google Scholar]
- Dopman, E.B.; Hartl, D.L. A portrait of copy-number polymorphism in Drosophila melanogaster. Proc. Nat. Acad. Sci. USA 2007, 104, 19920–19925. [Google Scholar]
- Cridland, J.M.; Thornton, K.R. Validation of rearrangement breakpoints identified by paired-end sequencing in natural populations of Drosophila melanogaster. Genome Biol. Evol. 2010, 2, 83–101. [Google Scholar]
- Heger, A.; Ponting, C.P. Evolutionary rate analyses of orthologs and paralogs from 12 Drosophila genomes. Genome Res. 2007, 17, 1837–1849. [Google Scholar]
- Zhou, Q.; Zhang, G.; Zhang, Y.; Xu, S.; Zhao, R.; Zhan, Z.; Li, X.; Ding, Y.; Yang, S.; Wang, W. On the origin of new genes in Drosophila. Genome Res. 2008, 18, 1446–1455. [Google Scholar]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar]
- Engels, W.R.; Preston, C.R.; Johnson-Schlitz, D.M. Long-range cis preference in DNA homology search over the length of a Drosophila chromosome. Science 1994, 263, 1623–1625. [Google Scholar]
- Galtier, N.; Piganeau, G.; Mouchiroud, D.; Duret, L. G-C content evolution in mammalian genomes: The biased gene conversion hypothesis. Genetics 2001, 159, 907–911. [Google Scholar]
- Ohta, T. The mutational load of a multigene family with uniform members. Genet. Res. 1989, 53, 141–145. [Google Scholar]
- Sugino, R.P.; Innan, H. Selection for more of the same product as a force to enhance concerted evolution of duplicated genes. Trends Genet. 2006, 22, 642–644. [Google Scholar]
- Connallon, T.; Clark, A.G. Gene duplication, gene conversion and the evolution of the Y chromosome. Genetics 2010, 186, 277–286. [Google Scholar]
- Mano, S.; Innan, H. The evolutionary rate of duplicated genes under concerted evolution. Genetics 2008, 180, 493–505. [Google Scholar]
- Ohno, S. Evolution by Gene Duplication; Springer-Verlag: Berlin, Germany, 1970. [Google Scholar]
- Walsh, J.B. How often do duplicated genes evolve new functions? Genetics 1995, 139, 421–428. [Google Scholar]
- Walsh, B. Population genetic models of the fates of duplicate genes. Genetica 2003, 118, 279–294. [Google Scholar]
- Walsh, J.B. Sequence-dependent gene conversion: can duplicated genes diverge fast enough to escape conversion? Genetics 1987, 117, 543–557. [Google Scholar]
- Teshima, K.M.; Innan, H. Neofunctionalization of duplicated genes under the pressure of gene conversion. Genetics 2008, 178, 1385–1398. [Google Scholar]
- Hansen, T.F.; Carter, A.J.R.; Chiu, C.H. Gene conversion may aid adaptive peak shifts. J. Theor. Biol. 2000, 207, 495–511. [Google Scholar]
- Thornton, K.; Long, M. Rapid divergence of gene duplicates on the Drosophila melanogaster X chromosome. Mol. Biol. Evol. 2002, 19, 918–925. [Google Scholar]
- Meisel, R.P.; Hilldorfer, B.B.; Koch, J.L.; Lockton, S.; Schaeffer, S.W. Adaptive evolution of genes duplicated from the Drosophila pseudoobscura neo-X chromosome. Mol. Biol. Evol. 2010, 27, 1963–1978. [Google Scholar]
- Smith, N.G.C.; Eyre-Walker, A. Adaptive protein evolution in Drosophila. Nature 2002, 415, 1022–1024. [Google Scholar]
- Bierne, N.; Eyre-Walker, A. The genomic rate of adaptive amino acid substitution in Drosophila. Mol. Biol. Evol. 2004, 21, 1350–1360. [Google Scholar]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar]
- Jordan, I.K.; Wolf, Y.I.; Koonin, E.V. Duplicated genes evolve slower than singletons despite the initial rate increase. BMC Evol. Biol. 2004, 4, 22. [Google Scholar]
- Begun, D.J.; Holloway, A.K.; Stevens, K.; Hillier, L.W.; Poh, Y.P.; Hahn, M.W.; Nista, P.M.; Jones, C.D.; Kern, A.D.; Dewey, C.N.; Pachter, L.; Myers, E.; Langley, C.H. Population genomics: Whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007, 5, 2534–2559. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arguello, J.R.; Connallon, T. Gene Duplication and Ectopic Gene Conversion in Drosophila. Genes 2011, 2, 131-151. https://doi.org/10.3390/genes2010131
Arguello JR, Connallon T. Gene Duplication and Ectopic Gene Conversion in Drosophila. Genes. 2011; 2(1):131-151. https://doi.org/10.3390/genes2010131
Chicago/Turabian StyleArguello, J. Roman, and Tim Connallon. 2011. "Gene Duplication and Ectopic Gene Conversion in Drosophila" Genes 2, no. 1: 131-151. https://doi.org/10.3390/genes2010131
APA StyleArguello, J. R., & Connallon, T. (2011). Gene Duplication and Ectopic Gene Conversion in Drosophila. Genes, 2(1), 131-151. https://doi.org/10.3390/genes2010131