
Gene Dosage of F5 c.3481C>T Stop-Codon (p.R1161Ter) Switches the Clinical Phenotype from Severe Thrombosis to Recurrent Haemorrhage: Novel Hypotheses for Readthrough Strategy

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Blood Sample Collection and DNA Extraction
2.2. PCR Amplification and Sanger Sequencing
2.3. Global and Specific Anticoagulant Response to APC
3. Results
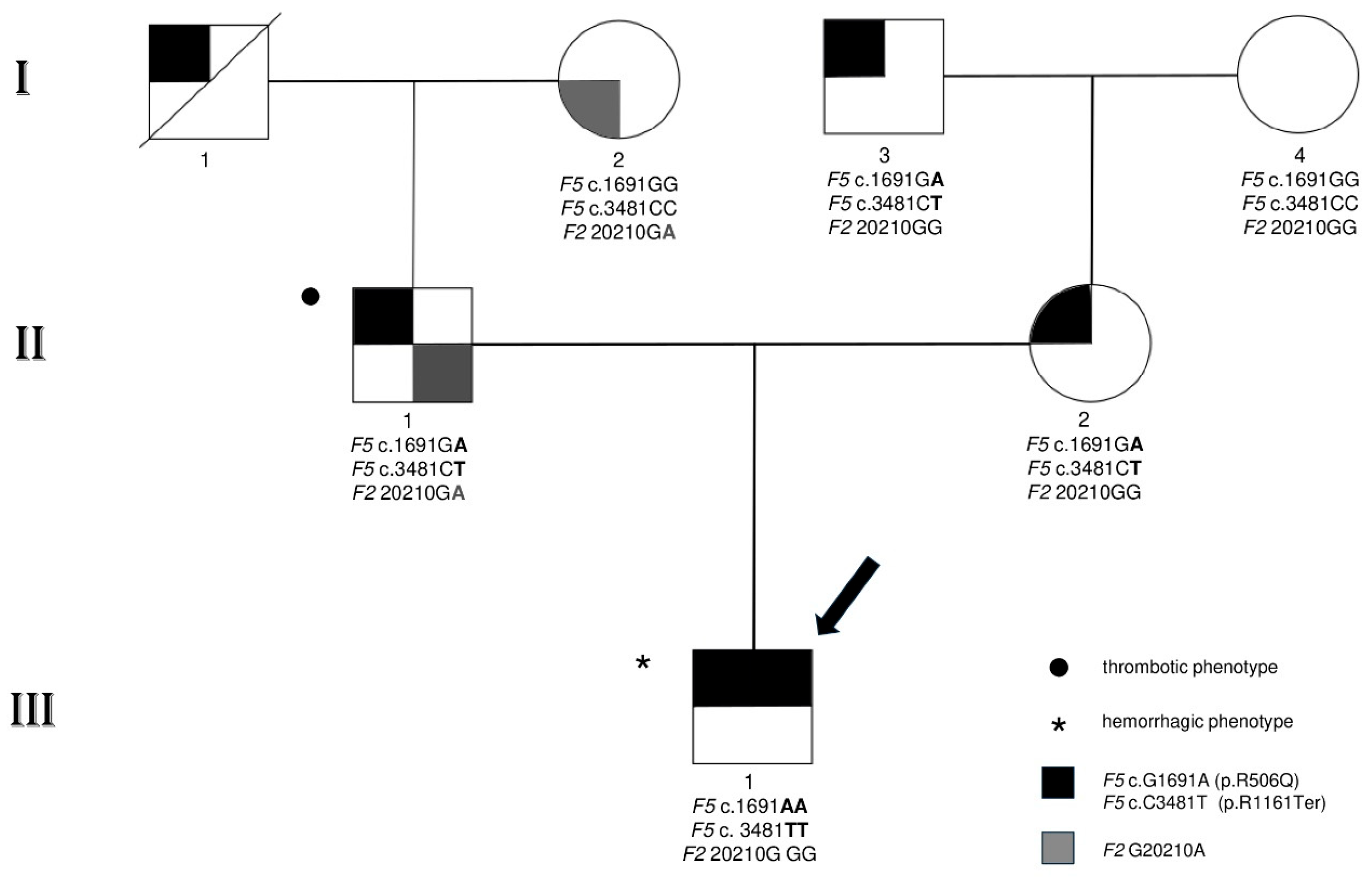
3.1. Family Characteristics and Clinical and Laboratory Phenotypic Findings
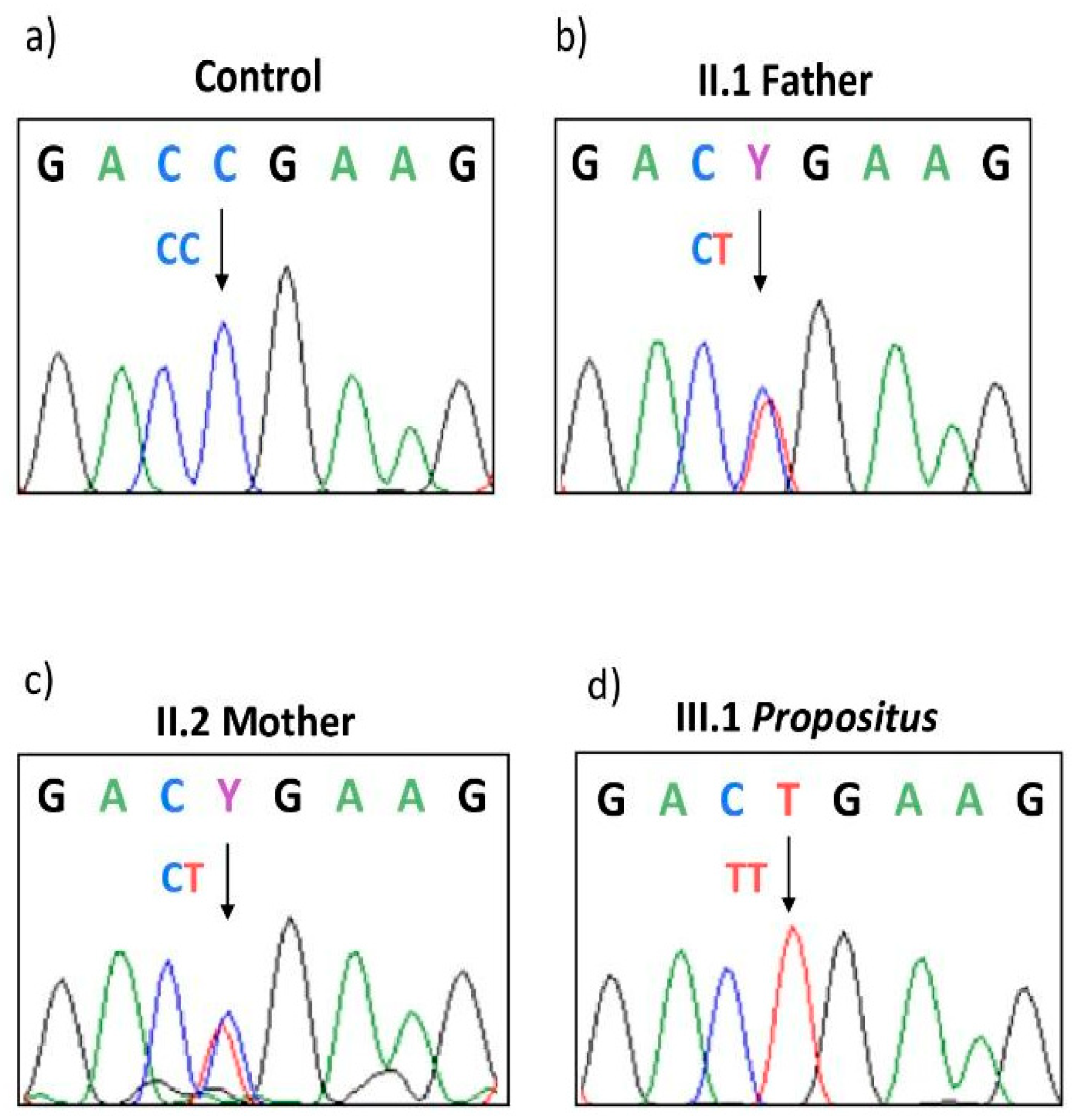
3.2. Genetic Investigations
3.3. In Vitro Assessment of the Anticoagulant Power of FV by APC-sr
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Asselta, R.; Tenchini, M.L.; Duga, S. Inherited defects of coagulation factor V: The hemorrhagic side. J. Thromb. Haemost. 2006, 4, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Vos, H.L. Inherited defects of coagulation Factor V: The thrombotic side. J. Thromb. Haemost. 2006, 4, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Foster, W.B.; Nesheim, M.E.; Mann, K.G. The factor Xa-catalyzed activation of factor V. J. Biol. Chem. 1983, 258, 13970–13977. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Dahlback, B.; Stenflo, J. Thrombin-catalyzed activation of human coagulation factor V. J. Biol. Chem. 1982, 257, 6556–6564. [Google Scholar] [CrossRef] [PubMed]
- Kalafatis, M.; Rand, M.D.; Mann, K.G. The mechanism of inactivation of human factor V and human factor Va by activated protein C. J. Biol. Chem. 1994, 269, 31869–31880. [Google Scholar] [CrossRef] [PubMed]
- Lak, M.; Sharifian, R.; Peyvandi, F.; Mannucci, P.M. Symptoms of inherited factor V deficiency in 35 Iranian patients. Br. J. Haematol. 1998, 103, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Mannucci, P.M. Rare coagulation disorders. Thromb. Haemost. 1999, 82, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, I.; Hong, D.W.; Mandle, L.; Jones, C.D.; DiMichele, D.; Monahan, P.E.; Zehnder, J.L. High frequency of premature termination mutations in the factor V gene: Three factor V deficiency case reports and a mutation review. Thromb. Haemost. 2005, 93, 610–611. [Google Scholar] [PubMed]
- Ajzner, E.E.; Balogh, I.; Szabo, T.; Marosi, A.; Haramura, G.; Muszbek, L. Severe coagulation factor V deficiency caused by 2 novel frameshift mutations: 2952delT in exon 13 and 5493insG in exon 16 of factor 5 gene. Blood 2002, 99, 702–705. [Google Scholar] [CrossRef]
- Duga, S.; Montefusco, M.C.; Asselta, R.; Malcovati, M.; Peyvandi, F.; Santagostino, E.; Mannucci, P.M.; Tenchini, M.L. Arg2074Cys missense mutation in the C2 domain of factor V causing moderately severe factor V deficiency: Molecular characterization by expression of the recombinant protein. Blood 2003, 101, 173–177. [Google Scholar] [CrossRef]
- Montefusco, M.C.; Duga, S.; Asselta, R.; Santagostino, E.; Mancuso, G.; Malcovati, M.; Mannucci, P.M.; Tenchini, M.L. A novel two base pair deletion in the factor V gene associated with severe factor V deficiency. Br. J. Haematol. 2000, 111, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, M.C.; Duga, S.; Asselta, R.; Malcovati, M.; Peyvandi, F.; Santagostino, E.; Mannucci, P.M.; Tenchini, M.L. Clinical and molecular characterization of 6 patients affected by severe deficiency of coagulation factor V: Broadening of the mutational spectrum of factor V gene and in vitro analysis of the newly identified missense mutations. Blood 2003, 102, 3210–3216. [Google Scholar] [CrossRef]
- Castoldi, E.; Lunghi, B.; Mingozzi, F.; Muleo, G.; Redaelli, R.; Mariani, G.; Bernardi, F. A missense mutation (Y1702C) in the coagulation factor V gene is a frequent cause of factor V deficiency in the Italian population. Haematologica 2001, 86, 629–633. [Google Scholar] [PubMed]
- Asselta, R.; Montefusco, M.C.; Duga, S.; Malcovati, M.; Peyvandi, F.; Mannucci, P.M.; Tenchini, M.L. Severe factor V deficiency: Exon skipping in the factor V gene causing a partial deletion of the C1 domain. J. Thromb. Haemost. 2003, 1, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Bossone, A.; D’Angelo, F.; Santacroce, R.; De Lucia, D.; Margaglione, M. Factor V Arg2074Cys: A novel missense mutation in the C2 domain of factor V. Thromb. Haemost. 2002, 87, 923–924. [Google Scholar] [CrossRef]
- Vos, H.L. An online database of mutations and polymorphisms in and around the coagulation factor V gene. J. Thromb. Haemost. 2007, 5, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, E.; Simioni, P.; Kalafatis, M.; Lunghi, B.; Tormene, D.; Girelli, D.; Girolami, A.; Bernardi, F. Combinations of 4 mutations (FV R506Q, FV H1299R, FV Y1702C, PT 20210G/A) affecting the prothrombinase complex in a thrombophilic family. Blood 2000, 96, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- van Wijk, R.; Montefusco, M.C.; Duga, S.; Asselta, R.; van Solinge, W.; Malcovati, M.; Tenchini, M.L.; Mannucci, P.M. Coexistence of a novel homozygous nonsense mutation in exon 13 of the factor V gene with the homozygous Leiden mutation in two unrelated patients with severe factor V deficiency. Br. J. Haematol. 2001, 114, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.J.; Wang, Z.Y.; Su, Y.H.; Yang, H.Y.; Zhao, X.J.; Zhang, W.; Yu, Z.Q.; Bai, X.; Ruan, C.G. Gene analysis of five inherited factor V deficiency cases. Zhonghua Xue Ye Xue Za Zhi 2008, 29, 145–148. [Google Scholar]
- Delev, D.; Pavlova, A.; Heinz, S.; Seifried, E.; Oldenburg, J. Factor 5 mutation profile in German patients with homozygous and heterozygous factor V deficiency. Haemophilia 2009, 15, 1143–1153. [Google Scholar] [CrossRef]
- Song, J.; Guella, I.; Kwon, K.Y.; Cho, H.; Park, R.; Asselta, R.; Choi, J.R. A novel in-frame deletion in the factor V C1 domain associated with severe coagulation factor V deficiency in a Korean family. Blood Coagul. Fibrinolysis 2009, 20, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Paraboschi, E.M.; Menegatti, M.; Rimoldi, V.; Borhany, M.; Abdelwahab, M.; Gemmati, D.; Peyvandi, F.; Duga, S.; Asselta, R. Profiling the mutational landscape of coagulation factor V deficiency. Haematologica 2020, 105, e180–e185. [Google Scholar] [CrossRef] [PubMed]
- Todaro, A.M.; Radu, C.M.; Ciccone, M.; Toffanin, S.; Serino, M.L.; Campello, E.; Bulato, C.; Lunghi, B.; Gemmati, D.; Cuneo, A.; et al. In vitro and ex vivo rescue of a nonsense mutation responsible for severe coagulation factor V deficiency. J. Thromb. Haemost. 2024, 22, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B.; Carlsson, M.; Svensson, P.J. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: Prediction of a cofactor to activated protein C. Proc. Natl. Acad. Sci. USA 1993, 90, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Bertina, R.M.; Koeleman, B.P.; Koster, T.; Rosendaal, F.R.; Dirven, R.J.; de Ronde, H.; van der Velden, P.A.; Reitsma, P.H. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994, 369, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, B.; Scanavini, D.; Girelli, D.; Legnani, C.; Bernardi, F. Does factor V Asp79His (409 G/C) polymorphism influence factor V and APC resistance levels? J. Thromb. Haemost. 2005, 3, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, B.; Iacoviello, L.; Gemmati, D.; Dilasio, M.G.; Castoldi, E.; Pinotti, M.; Castaman, G.; Redaelli, R.; Mariani, G.; Marchetti, G.; et al. Detection of new polymorphic markers in the factor V gene: Association with factor V levels in plasma. Thromb. Haemost. 1996, 75, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, B.; Scanavini, D.; Castoldi, E.; Gemmati, D.; Tognazzo, S.; Redaelli, R.; Ghirarduzzi, A.; Ieran, M.; Pinotti, M.; Bernardi, F. The factor V Glu1608Lys mutation is recurrent in familial thrombophilia. J. Thromb. Haemost. 2005, 3, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Hoekema, L.; Castoldi, E.; Tans, G.; Girelli, D.; Gemmati, D.; Bernardi, F.; Rosing, J. Functional properties of factor V and factor Va encoded by the R2-gene. Thromb. Haemost. 2001, 85, 75–81. [Google Scholar] [CrossRef]
- Norstrom, E.; Thorelli, E.; Dahlback, B. Functional characterization of recombinant FV Hong Kong and FV Cambridge. Blood 2002, 100, 524–530. [Google Scholar] [CrossRef]
- Zammiti, W.; Mtiraoui, N.; Mercier, E.; Abboud, N.; Saidi, S.; Mahjoub, T.; Almawi, W.Y.; Gris, J.C. Association of factor V gene polymorphisms (Leiden; Cambridge; Hong Kong and HR2 haplotype) with recurrent idiopathic pregnancy loss in Tunisia. A case-control study. Thromb. Haemost. 2006, 95, 612–617. [Google Scholar] [CrossRef]
- Simioni, P.; Scudeller, A.; Radossi, P.; Gavasso, S.; Girolami, B.; Tormene, D.; Girolami, A. “Pseudo homozygous” activated protein C resistance due to double heterozygous factor V defects (factor V Leiden mutation and type I quantitative factor V defect) associated with thrombosis: Report of two cases belonging to two unrelated kindreds. Thromb. Haemost. 1996, 75, 422–426. [Google Scholar] [CrossRef]
- Delahousse, B.; Iochmann, S.; Pouplard, C.; Fimbel, B.; Charbonnier, B.; Gruel, Y. Pseudo-homozygous activated protein C resistance due to coinheritance of heterozygous factor V Leiden mutation and type I factor V deficiency. Variable expression when analyzed by different activated protein C resistance functional assays. Blood Coagul. Fibrinolysis 1997, 8, 503–509. [Google Scholar] [CrossRef]
- Gemmati, D.; Longo, G.; Franchini, E.; Araujo Silva, J.; Gallo, I.; Lunghi, B.; Moratelli, S.; Maestri, I.; Serino, M.L.; Tisato, V. Cis-Segregation of c.1171C>T Stop Codon (p.R391*) in SERPINC1 Gene and c.1691G>A Transition (p.R506Q) in F5 Gene and Selected GWAS Multilocus Approach in Inherited Thrombophilia. Genes 2021, 12, 934. [Google Scholar] [CrossRef]
- Bernal, S.; Pelaez, I.; Alias, L.; Baena, M.; De Pablo-Moreno, J.A.; Serrano, L.J.; Camero, M.D.; Tizzano, E.F.; Berrueco, R.; Liras, A. High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies. Int. J. Mol. Sci. 2021, 22, 9705. [Google Scholar] [CrossRef]
- Marchetti, G.; Gemmati, D.; Patracchini, P.; Pinotti, M.; Bernardi, F. PCR detection of a repeat polymorphism within the F7 gene. Nucleic Acids Res. 1991, 19, 4570. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Scapoli, G.L. A modified functional global test to measure protein C, protein S activities and the activated protein C-resistance phenotype. Thromb. Res. 1998, 92, 141–148. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Verzola, I.; Mari, R.; Moratelli, S.; Ballerini, G. Resistance to activated protein C and low levels of protein S activity in nine thrombophilic families: A correct diagnosis. Blood Coagul. Fibrinolysis 1997, 8, 118–123. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Tognazzo, S.; Ongaro, A.; Moratelli, S.; Gilli, G.; Forini, E.; De Mattei, M.; Scapoli, G.L. The reduced sensitivity of the ProC Global test in protein S deficient subjects reflects a reduction in the associated thrombotic risk. Blood Coagul. Fibrinolysis 2001, 12, 691–697. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.; Mari, R.; Verzola, I.; Moratelli, S.; Ballerini, G. Different Anticoagulant Response to Activated Protein C (APC test) and to Agkistrodon Contortix Venom (ACV test) in a Family with FV-R506Q Substitution. Clin. Appl. Thromb. Hemost. 1997, 3, 168–173. [Google Scholar] [CrossRef]
- Scanavini, D.; Girelli, D.; Lunghi, B.; Martinelli, N.; Legnani, C.; Pinotti, M.; Palareti, G.; Bernardi, F. Modulation of factor V levels in plasma by polymorphisms in the C2 domain. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 200–206. [Google Scholar] [CrossRef]
- Ay, C.; Frenzel, L.; Pinachyan, K.; Le Quellec, S. Gene therapy for haemophilia A and B, from basic principles to clinical implementation: An illustrated review. Haemophilia 2024, 30, 5–15. [Google Scholar] [CrossRef]
- Lo Faro, V.; Johansson, T.; Hoglund, J.; Hadizadeh, F.; Johansson, A. Polygenic risk scores and risk stratification in deep vein thrombosis. Thromb. Res. 2023, 228, 151–162. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Moratelli, S.; Tognazzo, S.; Ongaro, A.; Scapoli, G.L. Coexistence of factor V G1691A and factor II G20210A gene mutations in a thrombotic family is associated with recurrence and early onset of venous thrombosis. Haemostasis 2001, 31, 99–105. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Moratelli, S.; Mari, R.; Ballerini, G.; Scapoli, G.L. Coexistence of antithrombin deficiency, factor V Leiden and hyperhomocysteinemia in a thrombotic family. Blood Coagul. Fibrinolysis 1998, 9, 173–176. [Google Scholar] [CrossRef]
- Dorgalaleh, A.; Bahraini, M.; Shams, M.; Parhizkari, F.; Dabbagh, A.; Naderi, T.; Fallah, A.; Fazeli, A.; Ahmadi, S.E.; Samii, A.; et al. Molecular basis of rare congenital bleeding disorders. Blood Rev. 2023, 59, 101029. [Google Scholar] [CrossRef]
- Branchini, A.; Morfini, M.; Lunghi, B.; Belvini, D.; Radossi, P.; Bury, L.; Serino, M.L.; Giordano, P.; Cultrera, D.; Molinari, A.C.; et al. F9 missense mutations impairing factor IX activation are associated with pleiotropic plasma phenotypes. J. Thromb. Haemost. 2022, 20, 69–81. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Ongaro, A.; Tognazzo, S.; Moratelli, S.; Resca, R.; Moretti, M.; Scapoli, G.L. A common mutation in the gene for coagulation factor XIII-A (VAL34Leu): A risk factor for primary intracerebral hemorrhage is protective against atherothrombotic diseases. Am. J. Hematol. 2001, 67, 183–188. [Google Scholar] [CrossRef]
- Nicolaes, G.A.; Dahlback, B. Factor V and thrombotic disease: Description of a janus-faced protein. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Weyand, A.C.; Grzegorski, S.J.; Rost, M.S.; Lavik, K.I.; Ferguson, A.C.; Menegatti, M.; Richter, C.E.; Asselta, R.; Duga, S.; Peyvandi, F.; et al. Analysis of factor V in zebrafish demonstrates minimal levels needed for early hemostasis. Blood Adv. 2019, 3, 1670–1680. [Google Scholar] [CrossRef]
- Unar, A.; Bertolino, L.; Patauner, F.; Gallo, R.; Durante-Mangoni, E. Pathophysiology of Disseminated Intravascular Coagulation in Sepsis: A Clinically Focused Overview. Cells 2023, 12, 2120. [Google Scholar] [CrossRef]
- Unar, A.; Bertolino, L.; Patauner, F.; Gallo, R.; Durante-Mangoni, E. Decoding Sepsis-Induced Disseminated Intravascular Coagulation: A Comprehensive Review of Existing and Emerging Therapies. J. Clin. Med. 2023, 12, 6128. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, A.K.; Todaro, A.M.; Castoldi, E. Factor V variants in bleeding and thrombosis. Res. Pract. Thromb. Haemost. 2024, 8, 102330. [Google Scholar] [CrossRef]
- Bouchard, B.A.; Chapin, J.; Brummel-Ziedins, K.E.; Durda, P.; Key, N.S.; Tracy, P.B. Platelets and platelet-derived factor Va confer hemostatic competence in complete factor V deficiency. Blood 2015, 125, 3647–3650. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Greenberg, C.S. Importance of platelet activation in the regulation of whole blood coagulation in the presence of a factor V inhibitor. Haemophilia 2019, 25, e307–e310. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B. Novel insights into the regulation of coagulation by factor V isoforms, tissue factor pathway inhibitoralpha, and protein S. J. Thromb. Haemost. 2017, 15, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Camire, R.M. Rethinking events in the haemostatic process: Role of factor V and TFPI. Haemophilia 2016, 22 (Suppl. S5), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Tisato, V. Chapter 24—Genomic and epigenomic signature at the branch-point among genome, phenome, and sexome in health and disease: A multiomics approach. In Principles of Gender-Specific Medicine, 4th ed.; Legato, M.J., Ed.; Academic Press: Cambridge, MA, USA, 2023; pp. 393–408. [Google Scholar]
- Diamond, S.L. Systems biology to predict blood function. J. Thromb. Haemost. 2009, 7 (Suppl. S1), 177–180. [Google Scholar] [CrossRef] [PubMed]
- Hockin, M.F.; Jones, K.C.; Everse, S.J.; Mann, K.G. A model for the stoichiometric regulation of blood coagulation. J. Biol. Chem. 2002, 277, 18322–18333. [Google Scholar] [CrossRef]
- Ayers, D.; Day, P.J. Systems Medicine: The Application of Systems Biology Approaches for Modern Medical Research and Drug Development. Mol. Biol. Int. 2015, 2015, 698169. [Google Scholar] [CrossRef]
- Morais, P.; Adachi, H.; Yu, Y.T. Suppression of Nonsense Mutations by New Emerging Technologies. Int. J. Mol. Sci. 2020, 21, 4394. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Mutyam, V.; Thakerar, A.; Mobley, J.; Bridges, R.J.; Rowe, S.M.; Keeling, K.M.; Bedwell, D.M. Identification of the amino acids inserted during suppression of CFTR nonsense mutations and determination of their functional consequences. Hum. Mol. Genet. 2017, 26, 3116–3129. [Google Scholar] [CrossRef] [PubMed]
- Wangen, J.R.; Green, R. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. Elife 2020, 9, e52611. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, S.; Testa, M.F.; Pinotti, M.; Branchini, A. Molecular Insights into Determinants of Translational Readthrough and Implications for Nonsense Suppression Approaches. Int. J. Mol. Sci. 2020, 21, 9449. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Morrill, C.; Friesen, W.J.; Babu, S.; Baiazitov, R.Y.; Du, W.; Karloff, D.B.; Lee, C.S.; Moon, Y.C.; Ren, H.; Sierra, J.; et al. Guanidino quinazolines and pyrimidines promote readthrough of premature termination codons in cells with native nonsense mutations. Bioorg. Med. Chem. Lett. 2022, 76, 128989. [Google Scholar] [CrossRef] [PubMed]
- Ensinck, M.M.; Carlon, M.S. One Size Does Not Fit All: The Past, Present and Future of Cystic Fibrosis Causal Therapies. Cells 2022, 11, 1868. [Google Scholar] [CrossRef] [PubMed]
- Coller, J.; Ignatova, Z. tRNA therapeutics for genetic diseases. Nat. Rev. Drug Discov. 2024, 23, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Aghajan, M.; Quesenberry, T.; Low, A.; Murray, S.F.; Monia, B.P.; Guo, S. Targeting Translation Termination Machinery with Antisense Oligonucleotides for Diseases Caused by Nonsense Mutations. Nucleic Acid Ther. 2019, 29, 175–186. [Google Scholar] [CrossRef]
- Lejeune, F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017, 50, 175–185. [Google Scholar] [CrossRef]
- Chen, R.; Snyder, M. Systems biology: Personalized medicine for the future? Curr. Opin. Pharmacol. 2012, 12, 623–628. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| F5 Gene | Exon | FV Protein | SNP | Primer Sequence | Size (bp) |
|---|---|---|---|---|---|
| c.1691G>A | 10 | p.R506Q | rs6025 | Fw: CATACTACAGTGACGTGGAC | 206 |
| Rv: TGTTCTCTTGAAGGAAATGC | |||||
| c.3481C>T | 13 | p.R1161Ter | rs118203909 | Fw: GACACTGGTCAGGCAAGCTG | 307 |
| Rv: TGAGGTCTGGAGAGAGGTTTGT |
| Family Members | F5 c.3481 C>T | F5 c.1691G>A | F2 G20210A | FV:C (%) | APC-sr |
|---|---|---|---|---|---|
| I1 | CT | GA | GG | n.e. | n.e. |
| I2 | CC | GG | GA | 115 | 2.9 |
| I3 | CT | GA | GG | 48 | 2.1 |
| I4 | CC | GG | GG | 110 | 3.1 |
| II1 | CT | GA | GA | 38 | 1.55 |
| II2 | CT | GA | GG | 50 | 2.0 |
| III1 | TT | AA | GG | <1 | 1.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gemmati, D.; D’Aversa, E.; Antonica, B.; Grisafi, M.; Salvatori, F.; Pizzicotti, S.; Pellegatti, P.; Ciccone, M.; Moratelli, S.; Serino, M.L.; et al. Gene Dosage of F5 c.3481C>T Stop-Codon (p.R1161Ter) Switches the Clinical Phenotype from Severe Thrombosis to Recurrent Haemorrhage: Novel Hypotheses for Readthrough Strategy. Genes 2024, 15, 432. https://doi.org/10.3390/genes15040432
Gemmati D, D’Aversa E, Antonica B, Grisafi M, Salvatori F, Pizzicotti S, Pellegatti P, Ciccone M, Moratelli S, Serino ML, et al. Gene Dosage of F5 c.3481C>T Stop-Codon (p.R1161Ter) Switches the Clinical Phenotype from Severe Thrombosis to Recurrent Haemorrhage: Novel Hypotheses for Readthrough Strategy. Genes. 2024; 15(4):432. https://doi.org/10.3390/genes15040432
Chicago/Turabian StyleGemmati, Donato, Elisabetta D’Aversa, Bianca Antonica, Miriana Grisafi, Francesca Salvatori, Stefano Pizzicotti, Patrizia Pellegatti, Maria Ciccone, Stefano Moratelli, Maria Luisa Serino, and et al. 2024. "Gene Dosage of F5 c.3481C>T Stop-Codon (p.R1161Ter) Switches the Clinical Phenotype from Severe Thrombosis to Recurrent Haemorrhage: Novel Hypotheses for Readthrough Strategy" Genes 15, no. 4: 432. https://doi.org/10.3390/genes15040432