
Gollop–Wolfgang Complex Is Associated with a Monoallelic Variation in WNT11
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and DNA Isolation
2.2. Whole-Genome Sequencing
2.3. Alignment and Variant Calling
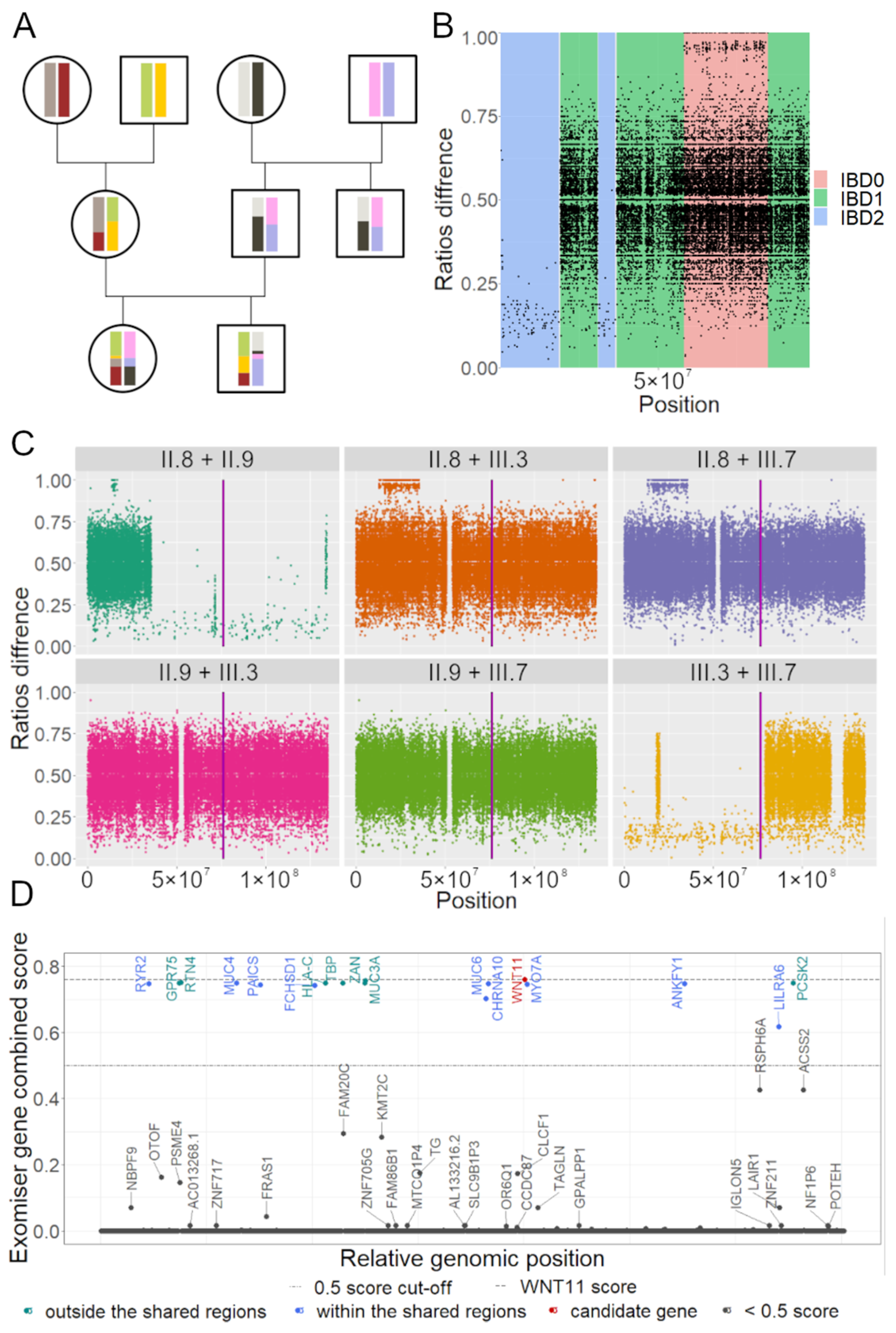
2.4. Identical by Descent Analysis
2.5. Variant Annotation and Prioritization
2.6. Variant Confirmation Using Sanger Sequencing
2.7. Structural Variants
2.8. Modelling Wnt11 Mut-Frizzled 8 Interaction
2.9. Cell Transfection and Luciferase Assay
3. Results
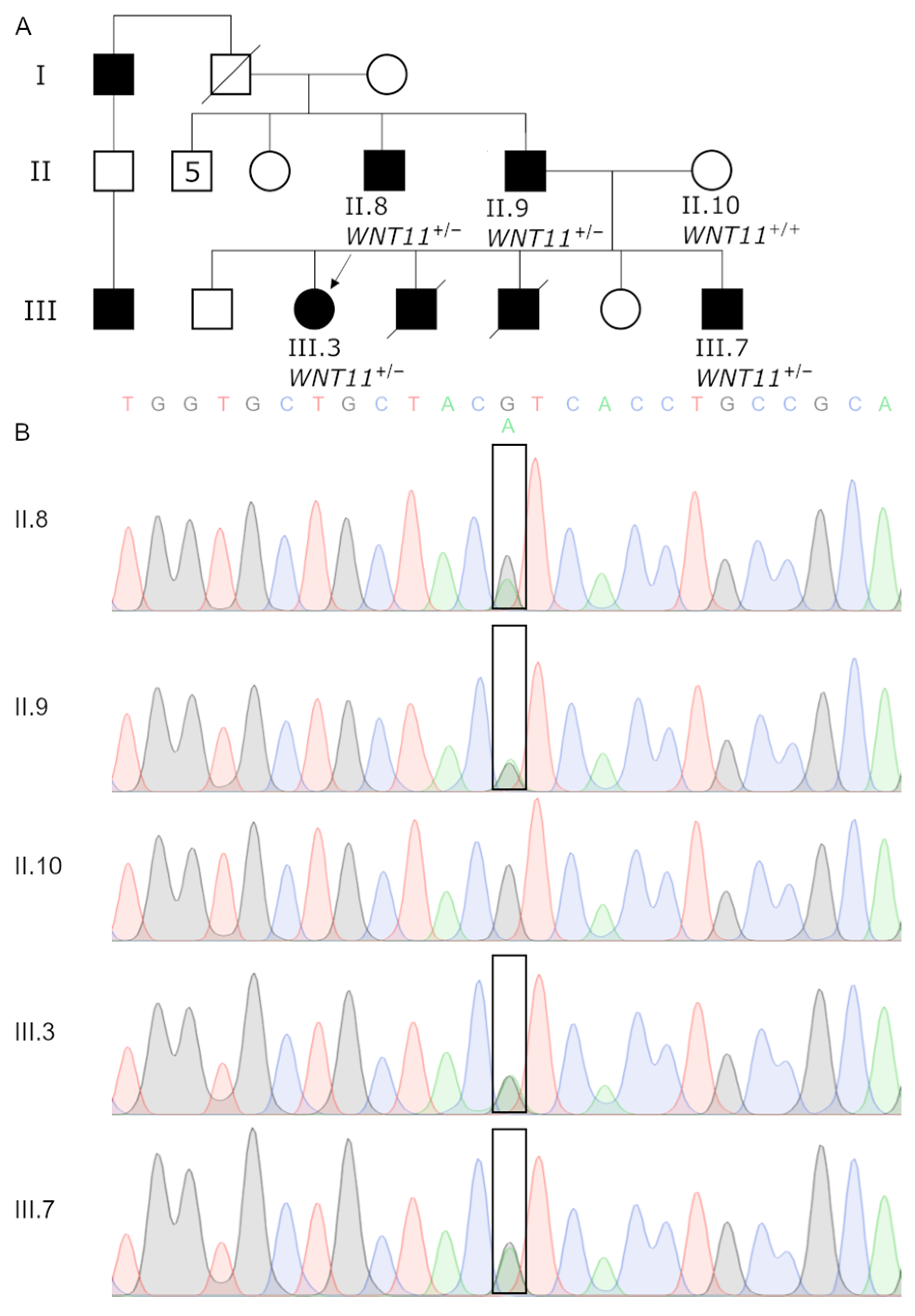
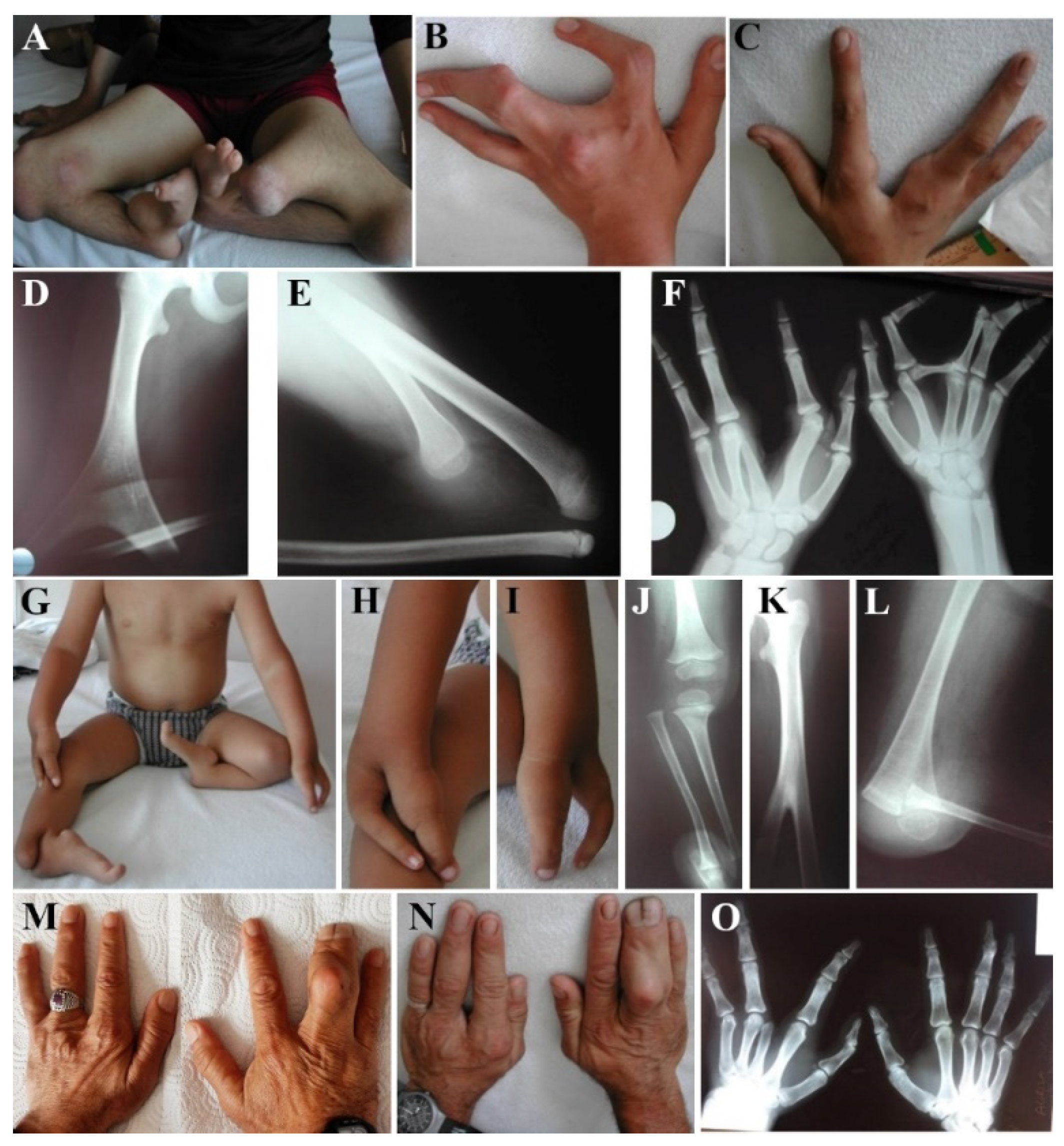
3.1. Clinical Examination of GWC Patients
3.2. IBD Analysis Provided Regions Shared among Affected
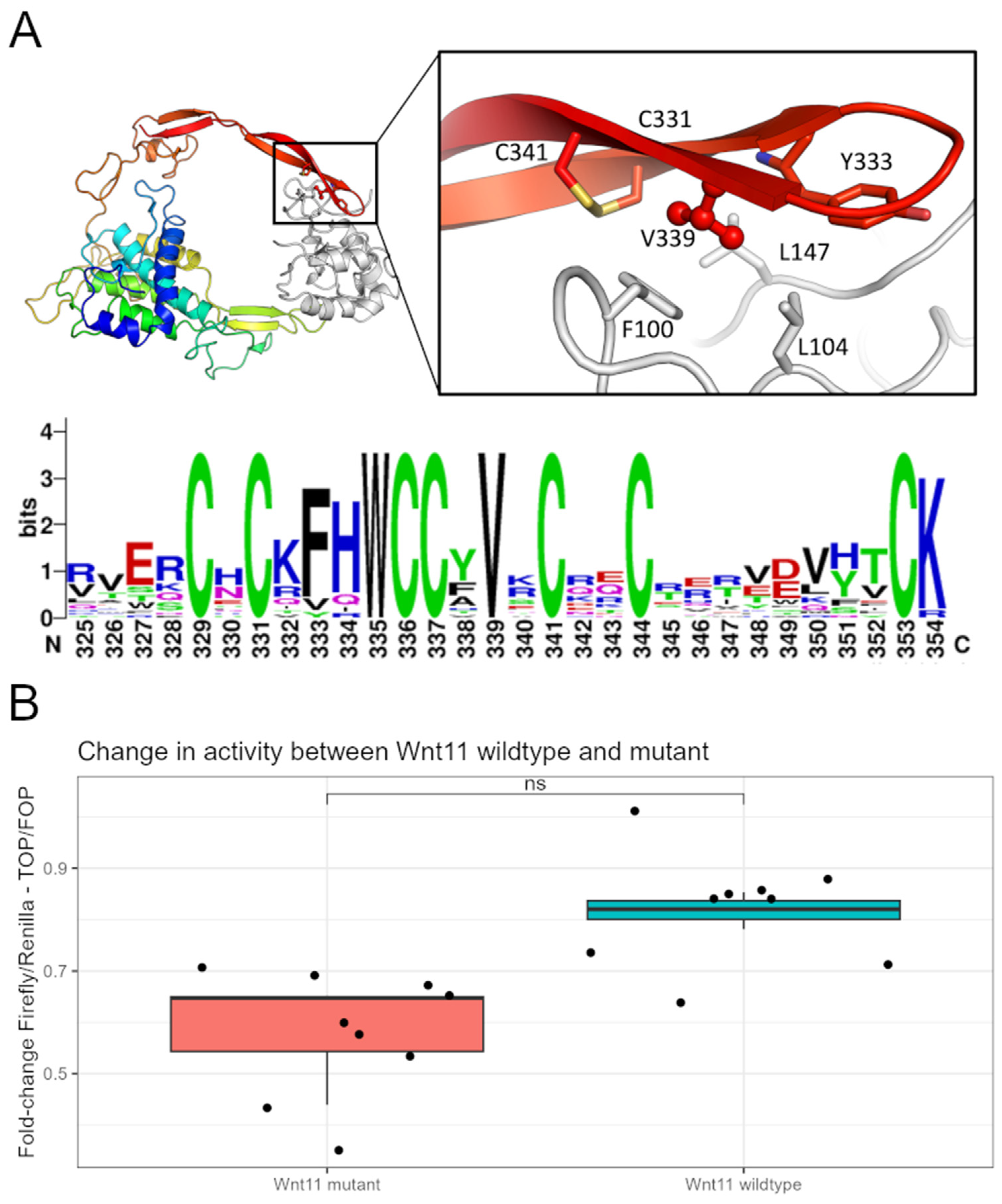
3.3. Wnt11 Variant Influences Interaction with Frizzled 8 Protein
3.4. Wnt11 Variant Weakens a Cellular Response to the Ligand
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sowińska-Seidler, A.; Socha, M.; Jamsheer, A. Split-Hand/Foot Malformation—Molecular Cause and Implications in Genetic Counseling. J. Appl. Genet. 2014, 55, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Holder-Espinasse, M.; Jamsheer, A.; Escande, F.; Andrieux, J.; Petit, F.; Sowinska-Seidler, A.; Socha, M.; Jakubiuk-Tomaszuk, A.; Gerard, M.; Mathieu-Dramard, M.; et al. Duplication of 10q24 Locus: Broadening the Clinical and Radiological Spectrum. Eur. J. Hum. Genet. 2019, 27, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Majewski, F.; Küster, W.; ter Haar, B.; Goecke, T. Aplasia of Tibia with Split-Hand/Split-Foot Deformity. Report of Six Families with 35 Cases and Consideration about Variability and Penetrance. Hum. Genet. 1985, 70, 136–147. [Google Scholar] [CrossRef] [PubMed]
- van Bokhoven, H.; Hamel, B.C.J.; Bamshad, M.; Sangiorgi, E.; Gurrieri, F.; Duijf, P.H.G.; Vanmolkot, K.R.J.; van Beusekom, E.; van Beersum, S.E.C.; Celli, J.; et al. P63 Gene Mutations in EEC Syndrome, Limb-Mammary Syndrome, and Isolated Split Hand–Split Foot Malformation Suggest a Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2001, 69, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xin, Q.; Li, L.; Li, J.; Zhang, C.; Qiu, R.; Qian, C.; Zhao, H.; Liu, Y.; Shan, S.; et al. Exome Sequencing Reveals a Heterozygous DLX5 Mutation in a Chinese Family with Autosomal-Dominant Split-Hand/Foot Malformation. Eur. J. Hum. Genet. 2014, 22, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Hammid, A.; Umair, M.; Ahmad, W. A Novel Heterozygous Intragenic Sequence Variant in DLX6 Probably Underlies First Case of Autosomal Dominant Split-Hand/Foot Malformation Type 1. Mol. Syndr. 2017, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Faden, M.A.; Alashram, W.; Alkuraya, F.S. Identification of a Novel DLX5 Mutation in a Family with Autosomal Recessive Split Hand and Foot Malformation. J. Med. Genet. 2012, 49, 16–20. [Google Scholar] [CrossRef]
- Ugur, S.A.; Tolun, A. Homozygous WNT10b Mutation and Complex Inheritance in Split-Hand/Foot Malformation. Hum. Mol. Genet. 2008, 17, 2644–2653. [Google Scholar] [CrossRef]
- Moses, M.A.; George, A.L.; Sakakibara, N.; Mahmood, K.; Ponnamperuma, R.M.; King, K.E.; Weinberg, W.C. Molecular Mechanisms of P63-Mediated Squamous Cancer Pathogenesis. Int. J. Mol. Sci. 2019, 20, 3590. [Google Scholar] [CrossRef]
- Robledo, R.F.; Rajan, L.; Li, X.; Lufkin, T. The Dlx5 and Dlx6 Homeobox Genes Are Essential for Craniofacial, Axial, and Appendicular Skeletal Development. Genes. Dev. 2002, 16, 1089–1101. [Google Scholar] [CrossRef]
- Perkins, R.S.; Singh, R.; Abell, A.N.; Krum, S.A.; Miranda-Carboni, G.A. The Role of WNT10B in Physiology and Disease: A 10-Year Update. Front. Cell Dev. Biol. 2023, 11, 1120365. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Jacobson-Dickman, E.; Xu, C.; Manouvrier, S.; Dwyer, A.A.; Sykiotis, G.P.; Beenken, A.; Liu, Y.; Tommiska, J.; Hu, Y.; et al. Congenital Hypogonadotropic Hypogonadism with Split Hand/Foot Malformation: A Clinical Entity with a High Frequency of FGFR1 Mutations. Genet. Med. 2015, 17, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Jin, M.; Chen, L. Role of FGF/FGFR Signaling in Skeletal Development and Homeostasis: Learning from Mouse Models. Bone Res. 2014, 2, 14003. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Nath, S.K.; Gaines, M.; Al-Ali, M.T.; Al-Khaja, N.; Hutchings, D.; Golla, J.; Deutsch, S.; Bottani, A.; Antonarakis, S.E.; et al. Genomewide Linkage Scan for Split–Hand/Foot Malformation with Long-Bone Deficiency in a Large Arab Family Identifies Two Novel Susceptibility Loci on Chromosomes 1q42.2-Q43 and 6q14.1. Am. J. Hum. Genet. 2007, 80, 105–111. [Google Scholar] [CrossRef]
- Armour, C.M.; Bulman, D.E.; Jarinova, O.; Rogers, R.C.; Clarkson, K.B.; DuPont, B.R.; Dwivedi, A.; Bartel, F.O.; McDonell, L.; Schwartz, C.E.; et al. 17p13.3 Microduplications Are Associated with Split-Hand/Foot Malformation and Long-Bone Deficiency (SHFLD). Eur. J. Hum. Genet. 2011, 19, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Klopocki, E.; Lohan, S.; Doelken, S.C.; Stricker, S.; Ockeloen, C.W.; Soares Thiele de Aguiar, R.; Lezirovitz, K.; Mingroni Netto, R.C.; Jamsheer, A.; Shah, H.; et al. Duplications of BHLHA9 Are Associated with Ectrodactyly and Tibia Hemimelia Inherited in Non-Mendelian Fashion. J. Med. Genet. 2012, 49, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Petit, F.; Jourdain, A.-S.; Andrieux, J.; Baujat, G.; Baumann, C.; Beneteau, C.; David, A.; Faivre, L.; Gaillard, D.; Gilbert-Dussardier, B.; et al. Split Hand/Foot Malformation with Long-Bone Deficiency and BHLHA9 Duplication: Report of 13 New Families: SHFLD and BHLHA9 Duplication. Clin. Genet. 2014, 85, 464–469. [Google Scholar] [CrossRef]
- Ondari, J.; Kinyanjui, J.; Miano, P.; Sang, E.; Oburu, E.; Maru, M. Femoral Bifurcation and Bilateral Tibial Hemimelia: Case Report. Pan Afr. Med. J. 2018, 30, 99. [Google Scholar] [CrossRef]
- Vanderberg, R.H.; Block, T.; Gates, T.; Gomez, J. Gollop-Wolfgang Complex: Clinical and Imaging Implications. Indian. J. Radiol. Imaging 2021, 31, 721–724. [Google Scholar] [CrossRef]
- Gollop, T.R.; Lucchesi, E.; Martins, R.M.; Nione, A.S. Familial Occurrence of Bifid Femur and Monodactylous Ectrodactyly. Am. J. Med. Genet. 1980, 7, 319–322. [Google Scholar] [CrossRef]
- Wolfgang, G.L. Complex Congenital Anomalies of the Lower Extremities: Femoral Bifurcation, Tibial Hemimelia, and Diastasis of the Ankle. Case Report and Review of the Literature. J. Bone Jt. Surg. Am. 1984, 66, 453–458. [Google Scholar] [CrossRef]
- Richieri-Costa, A.; Brunoni, D.; Filho, J.L.; Kasinski, S.; Opitz, J.M.; Reynolds, J.F. Tibial Aplasia-ectrodactyly as Variant Expression of the Gollop-Wolfgang Complex: Report of a Brazilian Family. Am. J. Med. Genet. 1987, 28, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Kohn, G.; el Shawwa, R.; Grunebaum, M. Aplasia of the Tibia with Bifurcation of the Femur and Ectrodactyly: Evidence for an Autosomal Recessive Type. Am. J. Med. Genet. 1989, 33, 172–175. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A Framework for Variation Discovery and Genotyping Using Next-Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv 2017, preprint. [Google Scholar] [CrossRef]
- Pagnamenta, A.T.; Howard, M.F.; Wisniewski, E.; Popitsch, N.; Knight, S.J.L.; Keays, D.A.; Quaghebeur, G.; Cox, H.; Cox, P.; Balla, T.; et al. Germline Recessive Mutations in PI4KA Are Associated with Perisylvian Polymicrogyria, Cerebellar Hypoplasia and Arthrogryposis. Hum. Mol. Genet. 2015, 24, 3732–3741. [Google Scholar] [CrossRef]
- Smedley, D.; Jacobsen, J.O.B.; Jäger, M.; Köhler, S.; Holtgrewe, M.; Schubach, M.; Siragusa, E.; Zemojtel, T.; Buske, O.J.; Washington, N.L.; et al. Next-Generation Diagnostics and Disease-Gene Discovery with the Exomiser. Nat. Protoc. 2015, 10, 2004–2015. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid Detection of Structural Variants and Indels for Germline and Cancer Sequencing Applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A Probabilistic Framework for Structural Variant Discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An Approach to Discover, Genotype, and Characterize Typical and Atypical CNVs from Family and Population Genome Sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-Wide Integration of Enhancers and Target Genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A Structural Variation Reference for Medical and Population Genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.A.; Van Laer, L.; Alaerts, M.; Ardeshirdavani, A.; Moreau, Y.; Laukens, K.; Loeys, B.; Vandeweyer, G. pBRIT: Gene Prioritization by Correlating Functional and Phenotypic Annotations through Integrative Data Fusion. Bioinformatics 2018, 34, 2254–2262. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.C.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A Curated Collection of Structural Variation in the Human Genome. Nucl. Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2023.
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham III, T.E.; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber 2023; University of California: San Francisco, CA, USA, 2023. [Google Scholar]
- Hirai, H.; Matoba, K.; Mihara, E.; Arimori, T.; Takagi, J. Crystal Structure of a Mammalian Wnt–Frizzled Complex. Nat. Struct. Mol. Biol. 2019, 26, 372–379. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator: Figure 1. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Veeman, M.T.; Slusarski, D.C.; Kaykas, A.; Louie, S.H.; Moon, R.T. Zebrafish Prickle, a Modulator of Noncanonical Wnt/Fz Signaling, Regulates Gastrulation Movements. Curr. Biol. 2003, 13, 680–685. [Google Scholar] [CrossRef]
- Andre, P.; Song, H.; Kim, W.; Kispert, A.; Yang, Y. Wnt5a and Wnt11 Regulate Mammalian Anterior-Posterior Axis Elongation. Development 2015, 142, 1516–1527. [Google Scholar] [CrossRef]
- Biswas, T.; Jaswal, A.P.; Yadav, U.S.; Bandyopadhyay, A. Simultaneous Differentiation of Articular and Transient Cartilage: WNT-BMP Interplay and Its Therapeutic Implication. Int. J. Dev. Biol. 2020, 64, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Lim, X. The Wnt Homepage. Available online: http://web.stanford.edu/group/nusselab/cgi-bin/wnt (accessed on 16 December 2023).
- Huang, Y.; Peng, Y.; Li, H.; Li, C.; Wu, Y.; Wang, X.; Chang, J.; Miao, C. Wilforine Inhibits Rheumatoid Arthritis Pathology through the Wnt11/β-Catenin Signaling Pathway Axis. Arthritis Res. Ther. 2023, 25, 243. [Google Scholar] [CrossRef] [PubMed]
- Bisevac, J.; Katta, K.; Petrovski, G.; Moe, M.C.; Noer, A. Wnt/β-Catenin Signaling Activation Induces Differentiation in Human Limbal Epithelial Stem Cells Cultured Ex Vivo. Biomedicines 2023, 11, 1829. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.-L. Planar Cell Polarity Regulators in Asymmetric Organogenesis during Development and Disease. J. Genet. Genom. 2023, 50, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Bley, I.A.; Zwick, A.; Hans, M.C.; Thieser, K.; Wagner, V.; Ludwig, N.; Khalmurzaev, O.; Matveev, V.B.; Loertzer, P.; Pryalukhin, A.; et al. DKK1 Inhibits Canonical Wnt Signaling in Human Papillomavirus-Positive Penile Cancer Cells. Transl. Oncol. 2022, 15, 101267. [Google Scholar] [CrossRef] [PubMed]
- Caetano da Silva, C.; Ostertag, A.; Raman, R.; Muller, M.; Cohen-Solal, M.; Collet, C. Wnt11f2 Zebrafish, an Animal Model for Development and New Insights in Bone Formation. Zebrafish 2023, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Witte, F.; Dokas, J.; Neuendorf, F.; Mundlos, S.; Stricker, S. Comprehensive Expression Analysis of All Wnt Genes and Their Major Secreted Antagonists during Mouse Limb Development and Cartilage Differentiation. Gene Expr. Patterns 2009, 9, 215–223. [Google Scholar] [CrossRef]
- Summerhurst, K.; Stark, M.; Sharpe, J.; Davidson, D.; Murphy, P. 3D Representation of Wnt and Frizzled Gene Expression Patterns in the Mouse Embryo at Embryonic Day 11.5 (Ts19). Gene Expr. Patterns 2008, 8, 331–348. [Google Scholar] [CrossRef]
- Kawakami, Y.; Wada, N.; Nishimatsu, S.; Ishikawa, T.; Noji, S.; Nohno, T. Involvement of Wnt-5a in Chondrogenic Pattern Formation in the Chick Limb Bud. Dev. Growth Differ. 1999, 41, 29–40. [Google Scholar] [CrossRef]
- Sisson, B.E.; Dale, R.M.; Mui, S.R.; Topczewska, J.M.; Topczewski, J. A Role of Glypican4 and Wnt5b in Chondrocyte Stacking Underlying Craniofacial Cartilage Morphogenesis. Mech. Dev. 2015, 138, 279–290. [Google Scholar] [CrossRef]
- Caetano Da Silva, C.; Edouard, T.; Fradin, M.; Aubert-Mucca, M.; Ricquebourg, M.; Raman, R.; Salles, J.P.; Charon, V.; Guggenbuhl, P.; Muller, M.; et al. WNT11, a New Gene Associated with Early Onset Osteoporosis, Is Required for Osteoblastogenesis. Hum. Mol. Genet. 2022, 31, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Geetha-Loganathan, P.; Nimmagadda, S.; Scaal, M. Wnt Signaling in Limb Organogenesis. Organogenesis 2008, 4, 109–115. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Numbers | III.3 | III.7 | II.9 | II.8 |
|---|---|---|---|---|
| Split of Hand | Bilateral | Bilateral | Unilateral (Left) | Unilateral (Left) |
| Number of fingers | ||||
| Right | Four | Two | Five | Five |
| Left | Four | Two | Four | Four |
| Syndactyly of finger | Right (between second and third finger) | - | Right (between second and third finger) | Right (between second and third finger) |
| Femur bifurcation | Bilateral | Unilateral (left) | - | - |
| Absent tibia | Bilateral | Unilateral (left) | - | - |
| Number of toes | ||||
| Right | Two | One | Five Five | Five Five |
| Left | One | One | ||
| Syndactyly of foot | Right (between first and second finger) | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odrzywolski, A.; Tüysüz, B.; Debeer, P.; Souche, E.; Voet, A.; Dimitrov, B.; Krzesińska, P.; Vermeesch, J.R.; Tylzanowski, P. Gollop–Wolfgang Complex Is Associated with a Monoallelic Variation in WNT11. Genes 2024, 15, 129. https://doi.org/10.3390/genes15010129
Odrzywolski A, Tüysüz B, Debeer P, Souche E, Voet A, Dimitrov B, Krzesińska P, Vermeesch JR, Tylzanowski P. Gollop–Wolfgang Complex Is Associated with a Monoallelic Variation in WNT11. Genes. 2024; 15(1):129. https://doi.org/10.3390/genes15010129
Chicago/Turabian StyleOdrzywolski, Adrian, Beyhan Tüysüz, Philippe Debeer, Erika Souche, Arnout Voet, Boyan Dimitrov, Paulina Krzesińska, Joris Robert Vermeesch, and Przemko Tylzanowski. 2024. "Gollop–Wolfgang Complex Is Associated with a Monoallelic Variation in WNT11" Genes 15, no. 1: 129. https://doi.org/10.3390/genes15010129