
PSTPIP1-Associated Myeloid-Related Proteinemia Inflammatory (PAMI) Syndrome: A Systematic Review

 , , ,
, , ,
Abstract
:1. Introduction
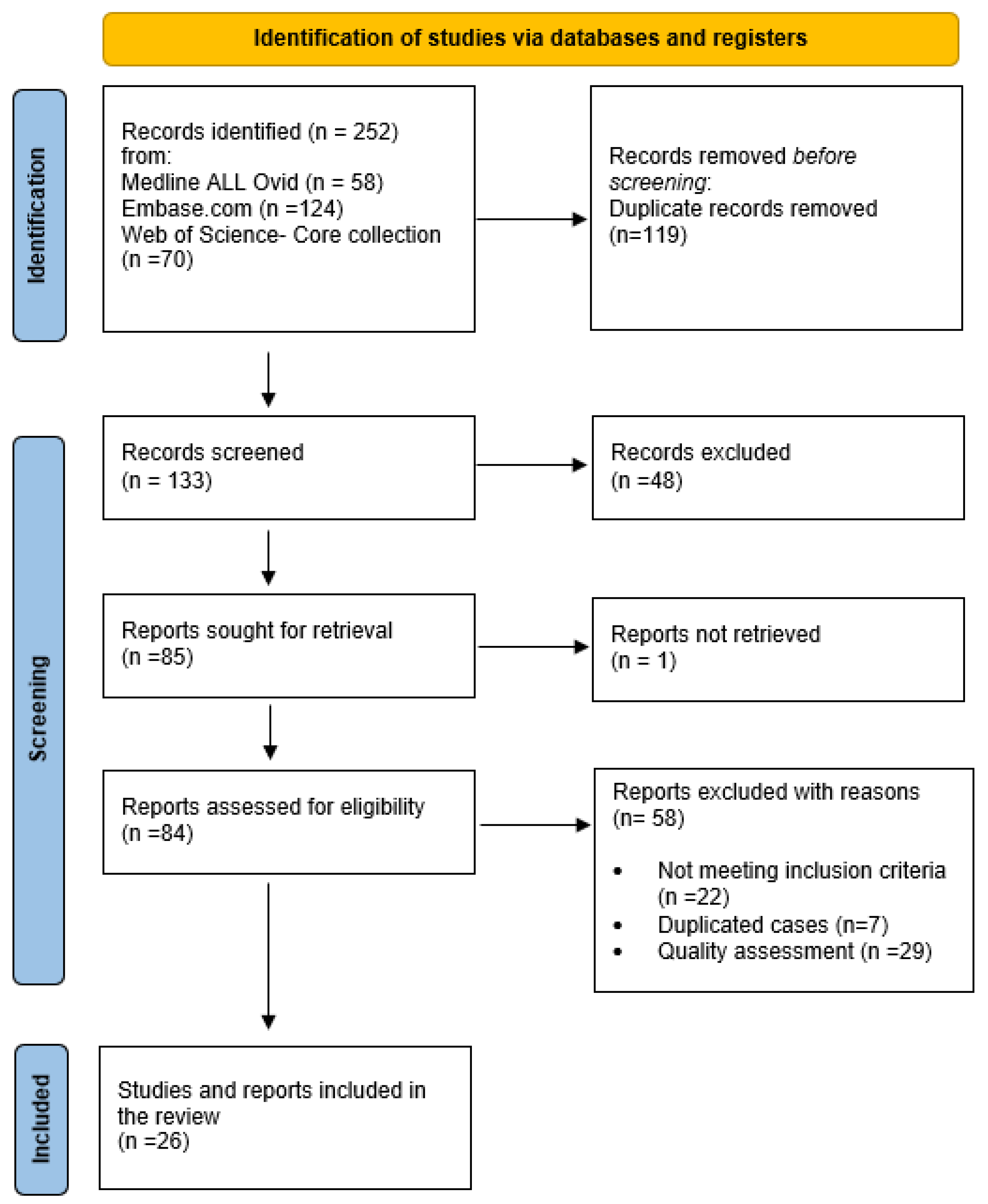
2. Methods
2.1. Declaration and Protocol
2.2. Inclusion and Exclusion Criteria
2.2.1. Inclusion Criteria
Types of Studies
Types of Participants
- Confirmed PAMI syndrome in the presence of one of the PAMI-specific PSTPIP1 gene deleterious variants (E250K or E257K).
- Confirmed Hyperzinceamia and Hypercalprotectineamia syndrome in the presence of one of the PAMI-specific PSTPIP1 gene deleterious variants (E250K or E257K).
- Clinical features of PAPA syndrome in the presence of one of the PAMI-specific PSTPIP1 gene deleterious variants (E250K or E257K).
- Clinical features of PAID in the presence of one of the PAMI-specific PSTPIP1 gene deleterious variants (E250K or E257K).
2.2.2. Exclusion Criteria
- Patients with of one of the PAMI-specific PSTPIP1 gene deleterious variants (E250K or E257K), but without any clinical or biological manifestations.
- Patients reported as carrying the clinical diagnosis PAMI syndrome, but without evidence of PAMI-specific PSTPIP1 gene deleterious variants (E250K or E257K) (i.e., genetic analysis not performed, or alternative deleterious variants identified).
2.3. Search Strategy and Selection Process
2.4. Data Collection and Data Items
3. Results
3.1. Demographics
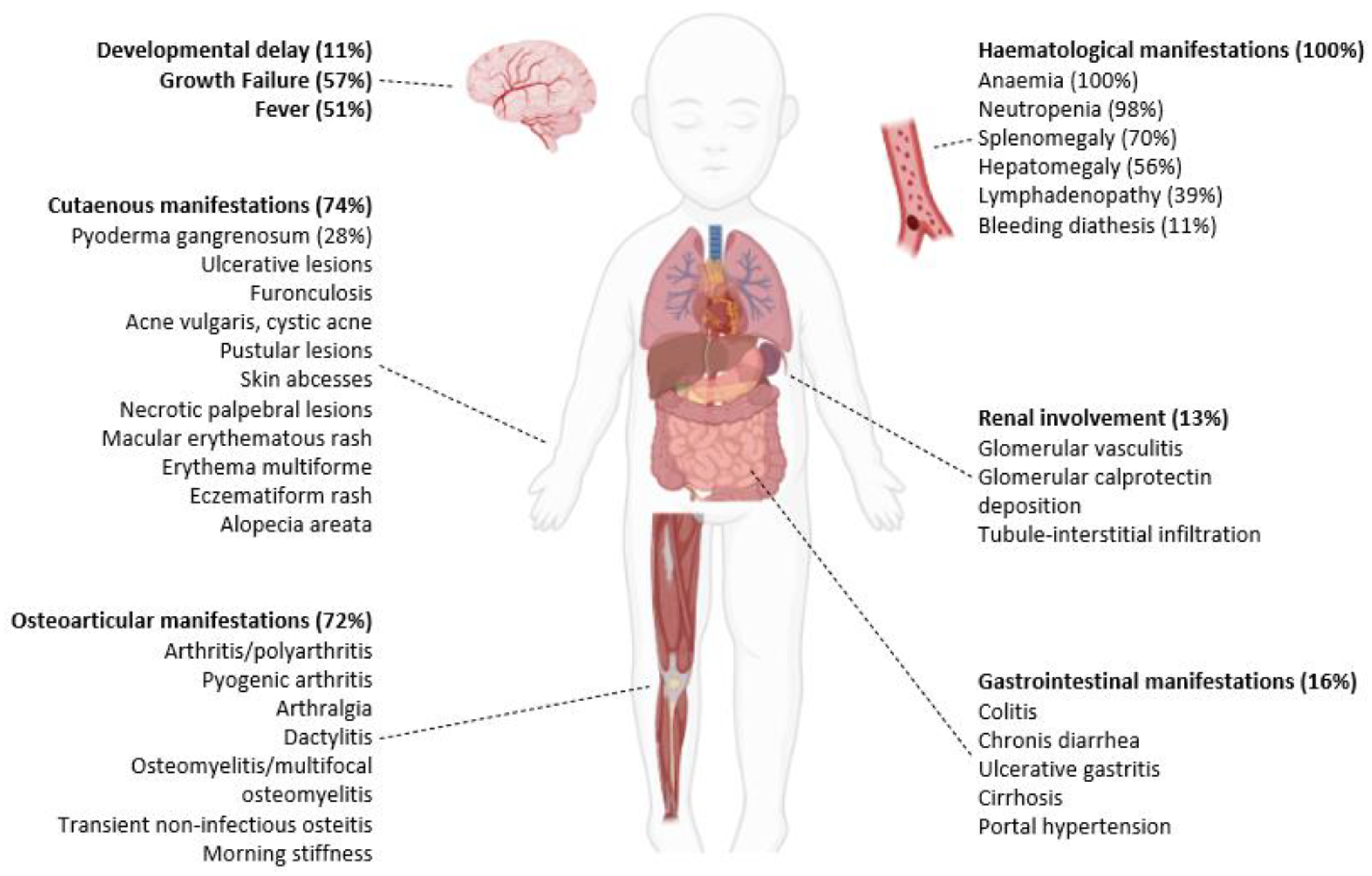
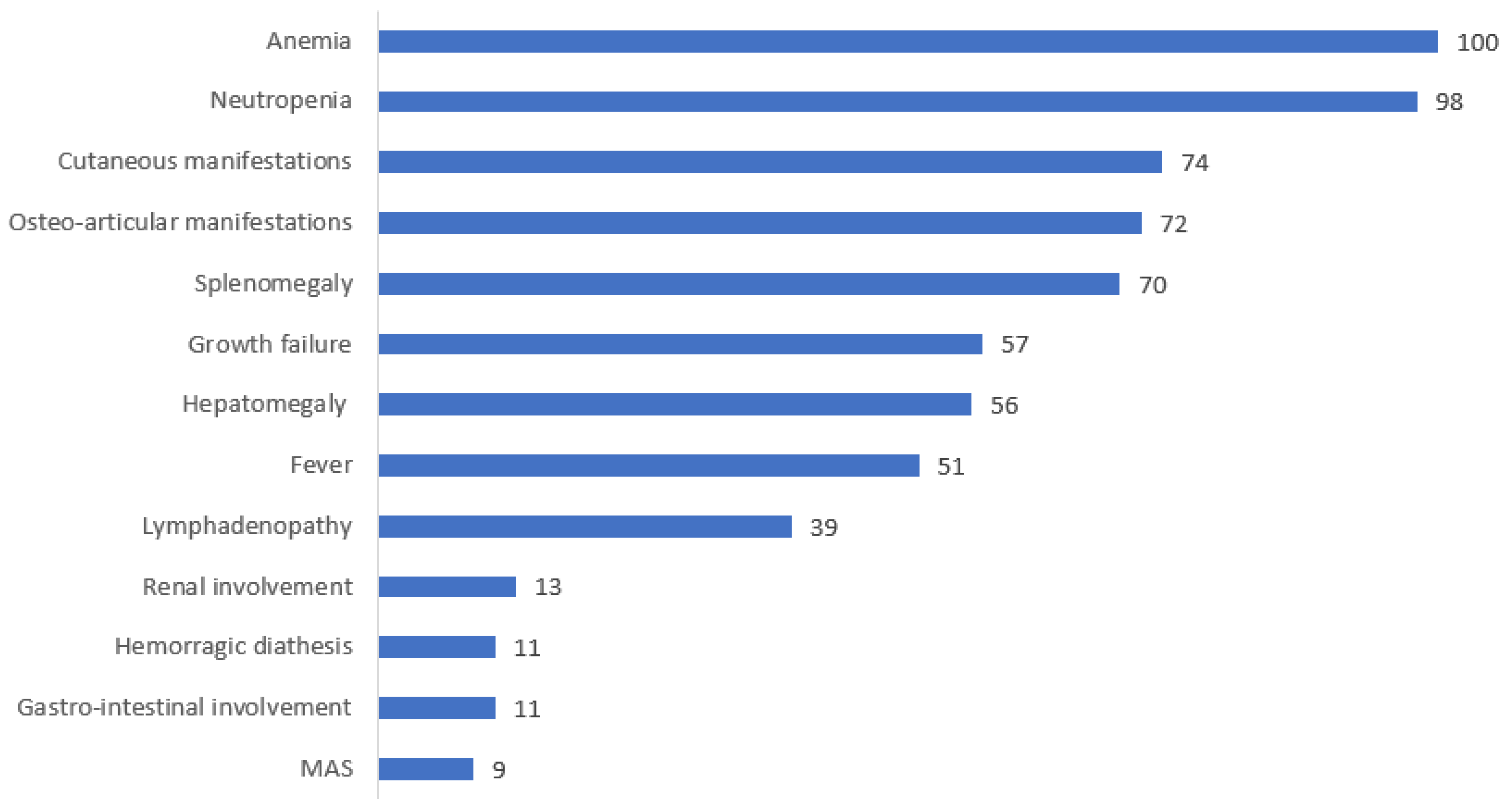
3.2. Clinical Presentation and Disease Course
3.3. Laboratory, Histology and Genetic Findings
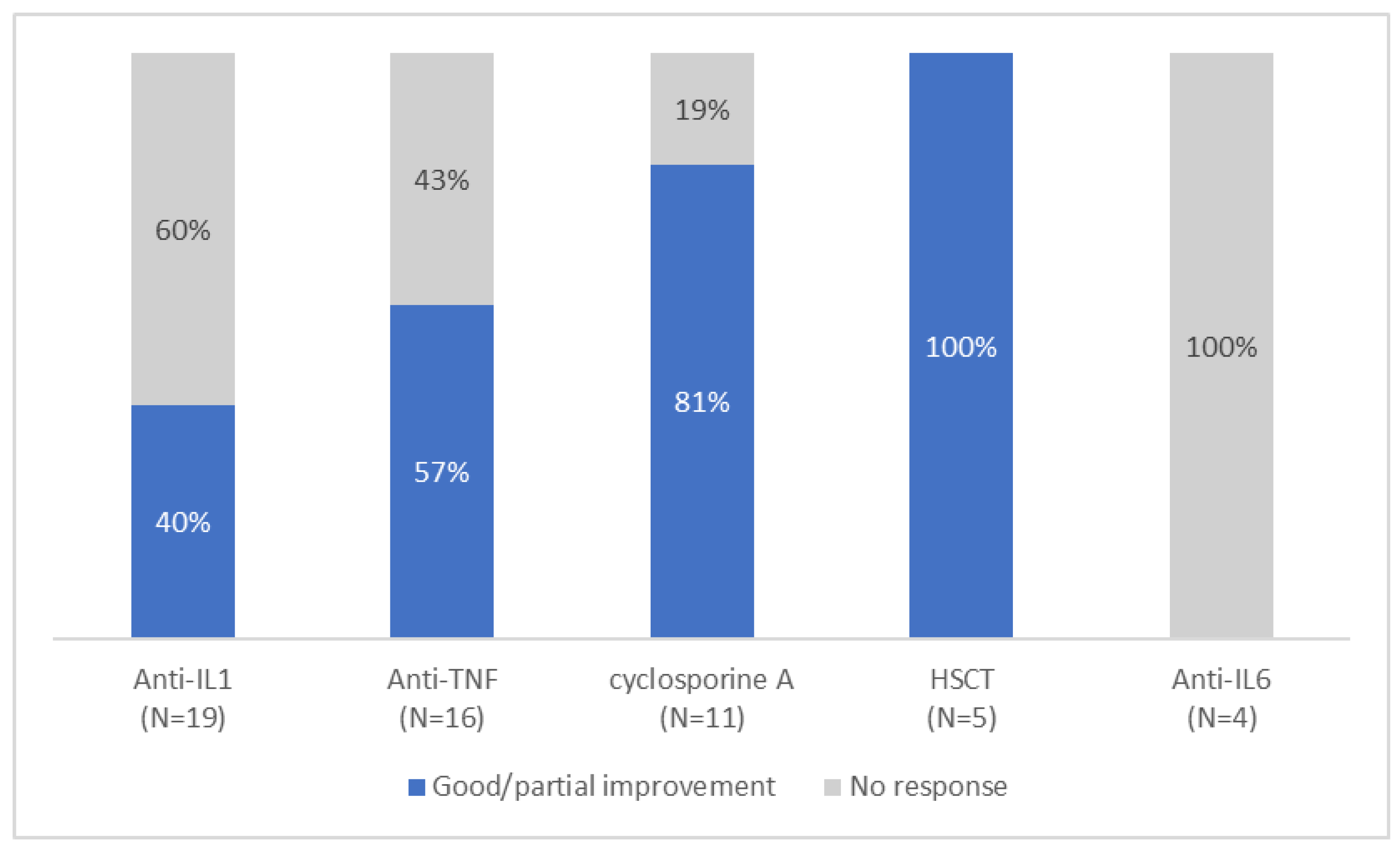
3.4. Treatment and Outcome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hashmi, S.; Diaz, R.; Despotovic, J.; Muscal, E.; Bergstrom, K.; Bertuch, A. Neutropenia as a presenting feature of hyperzincemia due to mutation in proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1). Pediatr. Blood Cancer 2017, 64, S28. [Google Scholar]
- Belelli, E.; Passarelli, C.; Pardeo, M.; Holzinger, D.; De Benedetti, F.; Insalaco, A. Haematological involvement associated with a mild autoinflammatory phenotype, in two patients carrying the E250K mutation of PSTPIP1. Clin. Exp. Rheumatol. 2017, 35, S113–S115. [Google Scholar]
- Holzinger, D.; Fassl, S.K.; de Jager, W.; Lohse, P.; Röhrig, U.F.; Gattorno, M.; Omenetti, A.; Chiesa, S.; Schena, F.; Austermann, J.; et al. Single amino acid charge switch defines clinically distinct proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1)-associated inflammatory diseases. J. Allergy Clin. Immunol. 2015, 136, 1337–1345. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Int. J. Surg. 2021, 88, 105906. [Google Scholar] [CrossRef] [PubMed]
- Isidor, B.; Poignant, S.; Corradini, N.; Fouassier, M.; Quartier, P.; Roth, J.; Picherot, G. Hyperzincemia and hypercalprotectinemia: Unsuccessful treatment with tacrolimus. Acta Paediatr. 2009, 98, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Goto, K.; Ito, K.; Ban, K.; Okada, S.; Moriyama, A.; Togari, H. Effects of cyclosporine A in hyperzincaemia and hypercalprotectinaemia. Acta Paediatr. 2006, 95, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Fessatou, S.; Fagerhol, M.K.; Roth, J.; Stamoulakatou, A.; Kitra, V.; Hadarean, M.; Paleologos, G.; Chandrinou, H.; Sampson, B.; Papassotiriou, I. Severe anemia and neutropenia associated with hyperzincemia and hypercalprotectinemia. J. Pediatr. Hematol./Oncol. 2005, 27, 477–480. [Google Scholar] [CrossRef]
- Sampson, B.; Fagerhol, M.K.; Sunderkötter, C.; Golden, B.E.; Richmond, P.; Klein, N.; Kovar, I.Z.; Beattie, J.H.; Wolska-Kusnierz, B.; Saito, Y.; et al. Hyperzincaemia and hypercalprotectinaemia: A new disorder of zinc metabolism. Lancet 2002, 360, 1742–1745. [Google Scholar] [CrossRef]
- Demidowich, A.P.; Freeman, A.F.; Kuhns, D.B.; Aksentijevich, I.; Gallin, J.I.; Turner, M.L. Brief report: Genotype, phenotype, and clinical course in five patients with PAPA syndrome (pyogenic sterile arthritis, pyoderma gangrenosum, and acne). Arthritis Rheum. 2012, 64, 2022–2027. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan—A web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- The Joanna Briggs Institute Critical Appraisal Tools for Use in JBI Systematic Reviews Checklist for Case Reports. 2017. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwi-9LO9kOCAAxVBcGwGHWJbAacQFnoECA4QAQ&url=https%3A%2F%2Fjbi.global%2Fsites%2Fdefault%2Ffiles%2F2019-05%2FJBI_Critical_Appraisal-Checklist_for_Case_Reports2017_0.pdf&usg=AOvVaw2Lu-IQRqxstRAOanORXSCG&opi=89978449 (accessed on 14 August 2023).
- The Joanna Briggs Institute Critical Appraisal Tools for Use in JBI Systematic Reviews Checklist for Case Series. 2017. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwi-9LO9kOCAAxVBcGwGHWJbAacQFnoECA8QAQ&url=https%3A%2F%2Fjbi.global%2Fsites%2Fdefault%2Ffiles%2F2019-05%2FJBI_Critical_Appraisal-Checklist_for_Case_Series2017_0.pdf&usg=AOvVaw3vdcMs9W_668l0pxCY_P0K&opi=89978449 (accessed on 14 August 2023).
- Mejbri, M.; Theodoropoulou, K.; Hofer, M. PSTPIP1-associated myeloid-related proteinemia inflammatory syndrome/PAMI syndrome: Case report and review of the literature. Ann. Rheum. Dis. 2019, 78, 977–978. [Google Scholar]
- Zheng, W.; Fan, X.; Yang, Z.; Shangguan, Y.; Jin, T.; Liu, Y.; Huang, J.; Ye, X.; Zhou, Q.; Li, X. Strong inflammatory signatures in the neutrophils of PAMI syndrome. Front. Immunol. 2022, 13, 926087. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, B.G.; Titheradge, H.; Al-Abadi, E. PSTPIP1-associated myeloid-related proteinaemia inflammatory (PAMI) syndrome; a case presenting as a perinatal event with early central nervous system involvement? Pediatr. Rheumatol. Online J. 2022, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.; Bigley, V.; Irvine, A.; Leahy, R.; Conlon, N. PAMI Syndrome: Two Cases of an Autoinflammatory Disease with an ALPS-Like Phenotype. J. Clin. Immunol. 2022, 42, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Su, G.; Liu, Y.; Lai, J. Clinical and genetic characteristics of PSTPIP1-associated myeloid-related proteinemia inflammatory syndrome. Pediatr. Rheumatol. Online J. 2021, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.M.; Huang, H.; Ding, F.; Yang, Z.; Wang, J.; Jin, Y.L. PAMI syndrome: A rare cause that can be easily misdiagnosed. Am. J. Med. Genet. Part A 2021, 185, 3074–3082. [Google Scholar] [CrossRef]
- Mendonça, L.O.; Terreri, M.T.; Osaku, F.M.; Barros, S.F.; Köhler, K.F.; Prado, A.I.; Barros, M.T.; Kalil, J.; Castro, F.F.M. Immunological repertoire linked to PSTPIP1-associated myeloid-related inflammatory (PAMI) syndrome. Pediatr. Rheumatol. Online J. 2021, 19, 126. [Google Scholar] [CrossRef] [PubMed]
- Laberko, A.; Burlakov, V.; Maier, S.; Abinun, M.; Skinner, R.; Kozlova, A.; Suri, D.; Lehmberg, K.; Müller, I.; Balashov, D.; et al. HSCT is effective in patients with PSTPIP1-associated myeloid-related proteinemia inflammatory (PAMI) syndrome. J. Allergy Clin. Immunol. 2021, 148, 250–255.e1. [Google Scholar] [CrossRef]
- Huang, X.; Xu, M.; Dai, S.; Wang, M.; Zheng, H.; Zeng, K.; Li, L. Rare cases of PAMI syndrome in both father and son with the same missense mutation in PSTPIP1 gene and literature review. J. Dermatol. 2021, 48, 519–528. [Google Scholar] [CrossRef]
- Del Borrello, G.; Guardo, D.; Micalizzi, C.; Ceccherini, I.; Miano, M.; Gattorno, M.; Dufour, C. Hemolysis and Neurologic Impairment in PAMI Syndrome: Novel Characteristics of an Elusive Disease. Pediatrics 2021, 147, e20200784. [Google Scholar] [CrossRef]
- Borgia, P.; Papa, R.; D’Alessandro, M.; Caorsi, R.; Piaggio, G.; Angeletti, A.; Ceccherini, I.; Ghiggeri, G.M.; Gattorno, M. Kidney Involvement in PSTPIP1 Associated Inflammatory Diseases (PAID): A Case Report and Review of the Literature. Front. Med. 2021, 8, 759092. [Google Scholar] [CrossRef] [PubMed]
- Resende, L.O.; Jorge, M.F.S.; Schmitt, J.V. Extensive pyoderma gangrenosum-like lesions revealing a case of hyperzincemia and hypercalprotectinemia: When to suspect it? Anais Brasileiros de Dermatologia 2019, 94, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, S.K.; Bergstrom, K.; Bertuch, A.A.; Despotovic, J.M.; Muscal, E.; Xia, F.; Bi, W.; Marcogliese, A.; Diaz, R. PSTPIP1-associated myeloid-related proteinemia inflammatory syndrome: A rare cause of childhood neutropenia associated with systemic inflammation and hyperzincemia. Pediatr. Blood Cancer 2019, 66, e27439. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Furlong, T.; Gracie, G.; Huang, M.L.; Yang, T.; Wu, K.H.; Danta, M.; Wong, M.; Williams, A.; March, L.; et al. Autoinflammation Masquerading as Autoimmunity in an Adult with Heterozygous p.E250K PSTPIP1 Mutation. J. Clin. Immunol. 2019, 39, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Klötgen, H.W.; Beltraminelli, H.; Yawalkar, N.; Van Gijn, M.E.; Holzinger, D.; Borradori, L. The expanding spectrum of clinical phenotypes associated with PSTPIP1 mutations: From PAPA to PAMI syndrome and beyond. Br. J. Dermatol. 2018, 178, 982–983. [Google Scholar] [CrossRef]
- Lindwall, E.; Singla, S.; Davis, W.E.; Quinet, R.J. Novel PSTPIP1 gene mutation in a patient with pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome. Semin. Arthritis Rheum. 2015, 45, 91–93. [Google Scholar] [CrossRef]
- Holzinger, D.; Roth, J. Alarming consequences—Autoinflammatory disease spectrum due to mutations in proline-serine-threonine phosphatase-interacting protein 1. Curr. Opin. Rheumatol. 2016, 28, 550–559. [Google Scholar] [CrossRef]
- Sologuren, I.; Ruiz-Ortiz, E.; Barrios, Y.; Herrera-Ramos, E.; Martinez-Saavedra, M.; Gonzalez-Quevedo, N.; Lopez-Almaraz, R.; Colino, E.; Arostegui, J.; Rodriguez Gallego, C. Clinical, genetic and immunological characterization of a patient with the new pstpip1-associated autoinflammatory disease hyperzincemia with hypercalprotectinemia. J. Clin. Immunol. 2014, 34, S198. [Google Scholar]
- Su, G.; Lai, J.; Zhu, J.; Zhang, D.; Hou, J.; Xu, Y.; Zhou, Z. Analysis of five cases of monogenic lupus related to primary immunodeficiency diseases. Inflamm. Res. 2021, 70, 1211–1216. [Google Scholar] [CrossRef]
- Wang, W.; Yu, Z.; Gou, L.; Zhong, L.; Li, J.; Ma, M.; Wang, C.; Zhou, Y.; Ru, Y.; Sun, Z.; et al. Single-Center Overview of Pediatric Monogenic Autoinflammatory Diseases in the Past Decade: A Summary and Beyond. Front. Immunol. 2020, 11, 565099. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Author | Age of Onset | Age in Years | Sex | Family History | Fever | Growth Failure | Lymphadenopathy Hepatosplenomegaly | Dermatologic Signs | Osteoarticular Signs | Hematologic Signs | Other Symptoms |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Zheng et al., 2022 [14] | 6 months | 6 | F | Chinese, healthy parents | Yes | NA | Hepatosplenomegaly | No | Recurrent pyogenic arthritis | Anemia Neutropenia MAS | No |
| 2 | Zheng et al., 2022 [14] | 3 years | 7 | F | NA | NA | NA | Splenomegaly | Skin ulceration | No | Anemia Pancytopenia | NA |
| 3 | Whiteside et al., 2022 [15] | Neonatal period | 3 | F | African descent and healthy parents | No | No | No | No | No | Neutropenia | Seizures, speech, and cognitive delay secondary to bilateral infarction in the neonatal period with history of fetal distress, high inflammatory markers |
| 4 | Cox et al., 2022 [16] | 2 years | 46 | M | Irish, nonconsanguineous healthy parent | No | Yes | Lymphadenopathy | Nodulo-cystic acne | Intermittent synovitis and arthritis | Thrombocytopenia Neutropenia | Flare of colitis, recurrent lower respiratory tract infections |
| 5 | Cox et al., 2022 [16] | 6 months | 7 | M | Irish, non-consanguineous parents, mother with arthritis, sister with alopecia areata | No | NA | Splenomegaly with lymphadenopathy | Alopecia areata, acneiform rash of face and dorsum of hands | Poly arthropathy | Severe anemia Neutropenia Thrombocytopenia | Recurrent respiratory tract infection, diarrhea |
| 6 | Zhang et al., 2021 [17] | 6 years | 8 | F | NA | Yes | No | Splenomegaly with lymphadenopathy | Systemic rash, pyoderma gangrenosum | No | Pancytopenia | NO HTAP, angina with swollen and painful jaw with dysphagia (Ludwig’s angina) |
| 7 | Zhang et al., 2021 [17] | 10 years | 13 | F | NA | No | Yes | Splenomegaly | Pyoderma gangrenosum | Pyogenic arthritis | Anemia Neutropenia | No |
| 8 | Zhang et al., 2021 [17] | 13 years | 19 | M | NA | Yes | No | Hepatosplenomegaly with lymphadenopathy | No | Pyogenic arthritis | Pancytopenia | Severe HTAP with right heart failure |
| 9 | Xu et al., 2021 [18] | 3 years | 8 | F | Healthy parents, non-consanguineous | No | No | Hepatosplenomegaly | No | Unsymmetrical arthritis, aseptic necrosis of femoral head | Anemia Neutropenia | Recurrent infection |
| 10 | Xu et al., 2021 [18] | 6 years | 2.5 | F | Healthy parents, non-consanguineous | Yes | No | Hepatosplenomegaly | Yes, eyelid lesions | No | Anemia Neutropenia | Severe pulmonary infection with features of MAS |
| 11 | Huang et al., 2021 [21] | 13 years | 44 | M | Healthy non-consanguineous parents | No | NA | NA | Acne, cystic acne, facial papulonodular and hand ulcers | Intermittent elbow pain | Leucopenia | No |
| 12 | Huang et al., 2021 [21] | 17 years | 21 | M | Father with PAMI syndrome | No | NA | NA | Cystic acne | Elbow pain | Leucopenia | No |
| 13 | Mendonca et al., 2021 [19] | First months | 4 | F | Healthy parents, non-consanguineous, German–Italian descent | Yes, recurrent | Yes | Splenomegaly | Macular skin rash | Arthritis | Anemia Neutropenia Thrombocytopenia | Diarrhea, severe CMV infection |
| 14 | Mendonca et al., 2021 [19] | NA | adulthood | F | NA | No | No | No | No | No | Anemia Neutropenia | During childhood recurrent fever and thrombocytopenia |
| 14 | Laberko et al., 2021 [20] | At birth | 11.9 | F | Russia | Yes | Yes | Hepatosplenomegaly with lymphadenopathy | Soft tissue abscesses, pyoderma, chronic gingivitis, aphtosus, stomatitis | No | Anemia Neutropenia Thrombocytopenia | Chronic gingivitis, stomatitis, X-chromosome derivate (46, X, i(Xp)) |
| 16 | Laberko et al., 2021 [20] | At birth | 1 | M | Russia | Yes | Yes | Hepatosplenomegaly with lymphadenopathy | Vasculitis, panniculitis | Arthritis | Anemia Neutropenia Thrombocytopenia | Non-active colitis, ulcerative gastritis, MAS, myocarditis |
| 17 | Laberko et al., 2021 [20] | 2.9 years | 7 | F | Russia | Yes | No | Hepatosplenomegaly with lymphadenopathy | Polymorphic rash, vasculitis | Arthralgia, aseptic osteomyelitis | Anemia Neutropenia Thrombocytopenia | MAS |
| 18 | Laberko et al., 2021 [20] | 2.5 months | 0.5 | F | Germany | Yes | Yes | Lymphadenopathy | No | No | Anemia Neutropenia Thrombocytopenia | No |
| 19 | Del Borrello et al., 2021 [22] | 3 months | 1.5 | M | Negative family history | Yes, recurrent | Yes | Hepatosplenomegaly with lymphadenopathy | Urticarial rash | No | Anemia | Hypotonia, development delay, dystrophic and dysmorphic features, inguinal hernia, axial hypotonia |
| 20 | Borgia et al., 2021 [23] | 4 years | 8 | M | Italian, negative family history | NA | Yes | Hepatosplenomegaly | Pyoderma gangrenosum, cystic acne, poor wound healing | Asymmetrical polyarthritis | Anemia Leucopenia Neutropenia | Kidney involvement with focal segmental glomerulosclerosis, growth hormone deficiency |
| 21 | Resende et al., 2019 [24] | Childhood | 20 | F | NA | NA | Yes | Hepatosplenomegaly | Pyoderma gangrenosum | Arthritis | Pancytopenia | Osteoporosis, portal hypertension with esophageal varices, abdominal pain with recurrent diarrhea in childhood |
| 22 | Hashmi et al., 2019 [25] | 2 years | 12 | F | Grandfather with telomer biology disorder, no PAID in family | Yes, recurrent | NA | Splenomegaly with lymphadenopathy and lymphadenitis | No | Polyarthralgia | Anemia Leucopenia Neutropenia | Chest pain |
| 23 | Hashmi et al., 2019 [25] | At birth | 7 | F | NA | Yes | Yes | Hepatosplenomegaly with lymphadenopathy | No | Arthralgia | Pancytopenia Epistaxis | Born prematurely at 28 W, RSV infection, staph aureus osteomyelitis, periodontal disease, developmental delay |
| 24 | Dai et al., 2019 [26] | 18 years | 56 | F | NA | NA | NA | Splenomegaly | Poor wound healing and wound dehiscence | Symmetrical deforming non-erosive polyarthritis since the age of 18 | Pancytopenia | Macronodular cirrhosis, mild portal hypertension, recurrent childhood chest infections, podocyte effacement and glomerular calprotectin dense deposits |
| 25 | Mejbri et al., 2019 [13]* | 5 months | 2 | F | Caucasian healthy parents, non-consanguineous, same mutation for the father but no symptoms | Yes | No | Hepatosplenomegaly | No | Multifocal osteomyelitis | Anemia Neutropenia Epistaxis | No |
| 26 | Klotgen et al., 2018 [27] | Childhood | 23 | M | NA | Yes | NA | Hepatosplenomegaly | Pyoderma gangrenosum, skin ulceration, nodulocystic acne | Relapsing arthritis and osteomyelitis | Pancytopenia | Recurrent infection |
| 27 | Belelli et al., 2017 [2] | 7 years | 8 | M | mother with PAMI sd | No | No | Hepatosplenomegaly | No | Recurrent right knee swelling | Pancytopenia | No |
| 28 | Belelli et al., 2017 [2] | Childhood | Adulthood | F | Son with PAMI sd | No | No | NA | Psoriasis, acne | Arthralgia | Anemia Leucopenia | No history of infections |
| 29 | Lindwall et al., 2015 [28] | 4 years | 25 | M | Mother with psoriasic arthritis, no other illness | No | No | Hepatosplenomegaly | Cystic acne, scalp pyoderma, recurrent and multiple pyoderma gangrenosum | Recurrent symmetrical arthritis and hyper-mobile joint, osteomyelitis | Anemia Neutropenia Epistaxis | Septic shock with acute renal and respiratory failure, colitis, cellulitis, acute cholecystitis. Allergies to sulfonamides, ciprofloxacin, vancomycin, and Rocephin |
| 30 | Sologuren et al., 2014 [30] | First months | NA | M | NA | Yes | Yes | Hepatosplenomegaly with lymphadenopathy | Abscess | No | Anemia Neutropenia Thrombocytopenia | Hepatic abscess by E. coli |
| 31 | Sampson et al., 2002 [8] | 6 years | Adulthood | M | NA | No | No | Hepatosplenomegaly | PG, ulcerative dermatitis, furunculosis and pustulosis | Arthritis | Pancytopenia | Liver failure with cirrhosis minimal change Glomerulonephritis, deceased post-op complication at 35 y (liver transplantation) |
| 32 | Fessatou et al., 2005 [7] | 14 months | 11 | F | NA | No | Yes | Hepatosplenomegaly with lymphadenopathy | No | No | Anemia Neutropenia Leucopenia | NA |
| 33 | Isidor et al., 2009 [5] | 11 months | 8 | M | Non-consanguineous healthy parents | Yes | Yes | Hepatosplenomegaly | Macular erythematous rash, necrotic palpebral lesions | Arthritis, chronic polyarthralgia | Anemia Neutropenia Leucopenia Epistaxis and hematomas | Generalized muscular atrophy and delayed motor development |
| 34 | Sugiura et al., 2006 [6] | 4 years | 20 | F | NA | NA | Yes | Hepatosplenomegaly | Pyoderma gangrenosum, ulcerative dermatitis | Arthritis | Pancytopenia | Portal hypertension with ascites and esophageal varix, moderately impaired mental and motor development |
| 35 | Holzinger et al., 2015 [3] | First month | 5 | M | NA | Yes | Yes | Hepatosplenomegaly with lymphadenopathy | Recurrent perianal and gluteal abscesses | No | Anemia Neutropenia Thrombocytopenia | NA |
| 36 | Holzinger et al., 2015 [3] | 7 months | 9 | F | Paternal family history of multiple members with early gout, father at 14 y | No | Yes | Hepatosplenomegaly with lymphadenopathy | Pustular lesions | Arthralgia | Pancytopenia | Ig A nephropathy with hematuria and proteinuria (7 y), recurrent infection with cellulitis, conjunctivitis, pneumonia and central line infections, poor weight gain and requirement of gastrotomy tube feeding and growth retardation |
| 37 | Holzinger et al., 2015 [3] | 4 years | 5 | M | Father with PAMI sd | No | NA | No | No | No | Anemia Neutropenia | No recurrent infection |
| 38 | Holzinger et al., 2015 [3] | 18 years | 24 | M | NA | No | NA | No | Skin abscesses, Acne | Morning stiffness | Anemia Neutropenia/Leucopenia | History of salmonella meningitis at age of 16 m, no recurrent infection |
| 39 | Holzinger et al., 2015 [3] | 10 weeks | 7 | F | NA | Yes, recurrent fever | Yes | Splenomegaly | Erythema multiforme rash | Arthritis, transient non infectious osteitis | Pancytopenia | No |
| 40 | Demidowich et al., 2012 [9] | 1 month | Deceased (36) Sepsis-associated MOF | F | NA | NA | No | Splenomegaly with lymphadenopathy | Pyoderma gangrenosum, bullae, ulcerations, erythematous lesions | Recurrent sterile arthritis | Anemia Neutropeia Leucopenia Bleeding diasthesis | Recurrent upper respiratory infections and pneumonias, episodic lymphangitis and cellulitis, saddle nose deformity following spontaneous septal perforation, pharyngeal papillomatosis, large granular lymphocytosis of the T cells |
| 41 | Holzinger et al., 2015 [3] | At birth | 5 | F | Mother with SLE/RA overlap with Ro/La antibodies | Yes, recurrent | Yes | Hepatosplenomegaly with lymphadenopathy | Eczematous rash, photosensitivity, scalp hair heterochromia | Dactylitis, arthralgia, aseptic osteomyelitis | Pancytopenia | Viral infections, scoliosis, delayed motor and communication development, chronic diarrhea |
| 42 | Holzinger et al., 2015 [3] | 18 months | 16 | M | NA | No | Yes | Hepatosplenomegaly with lymphadenopathy | Pyoderma gangrenosum, furunculosis | Symmetric aseptic polyarthritis and aseptic necrosis of the right femoral head | Pancytopenia | Glomerulonephritis (hematuria + Proteinuria) |
| 43 | Holzinger et al., 2015 [3] | 1 year | 17 | M | NA | No | No | Hepatosplenomegaly | Acne vulgaris, pyoderma gangrenosum and pustular lesions | Osteomyelitis | Pancytopenia | Bilateral hearing loss after multiple middle ear infections, hepatic noncaseating granulomas, mild steatosis, and mild inflammation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejbri, M.; Renella, R.; Candotti, F.; Jaques, C.; Holzinger, D.; Hofer, M.; Theodoropoulou, K. PSTPIP1-Associated Myeloid-Related Proteinemia Inflammatory (PAMI) Syndrome: A Systematic Review. Genes 2023, 14, 1655. https://doi.org/10.3390/genes14081655
Mejbri M, Renella R, Candotti F, Jaques C, Holzinger D, Hofer M, Theodoropoulou K. PSTPIP1-Associated Myeloid-Related Proteinemia Inflammatory (PAMI) Syndrome: A Systematic Review. Genes. 2023; 14(8):1655. https://doi.org/10.3390/genes14081655
Chicago/Turabian StyleMejbri, Manel, Raffaele Renella, Fabio Candotti, Cecile Jaques, Dirk Holzinger, Michael Hofer, and Katerina Theodoropoulou. 2023. "PSTPIP1-Associated Myeloid-Related Proteinemia Inflammatory (PAMI) Syndrome: A Systematic Review" Genes 14, no. 8: 1655. https://doi.org/10.3390/genes14081655