
Suppressors of Break-Induced Replication in Human Cells
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
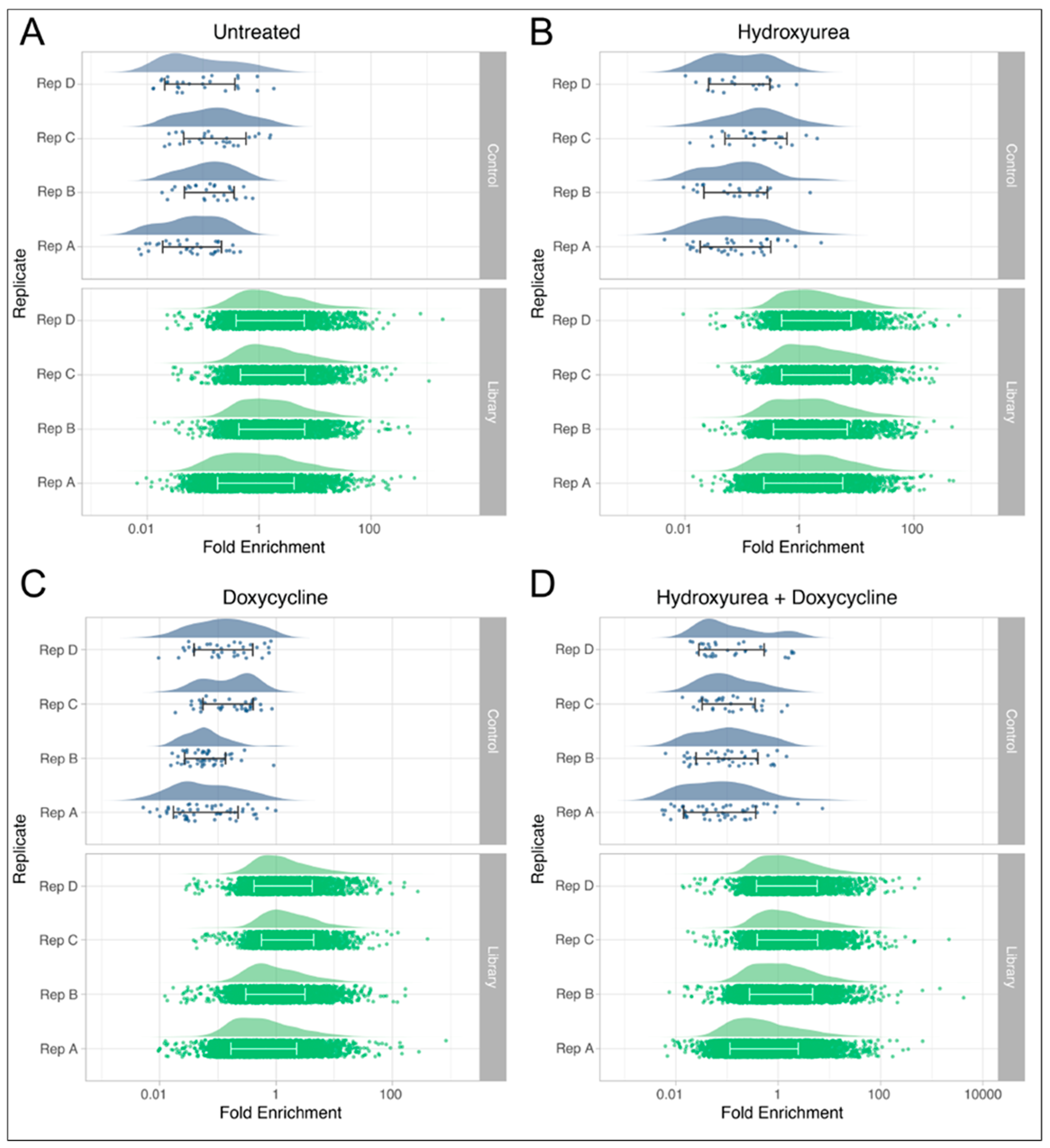
2.2. Library Screens
2.3. DNA Extraction and Next Generation Sequencing (NGS)
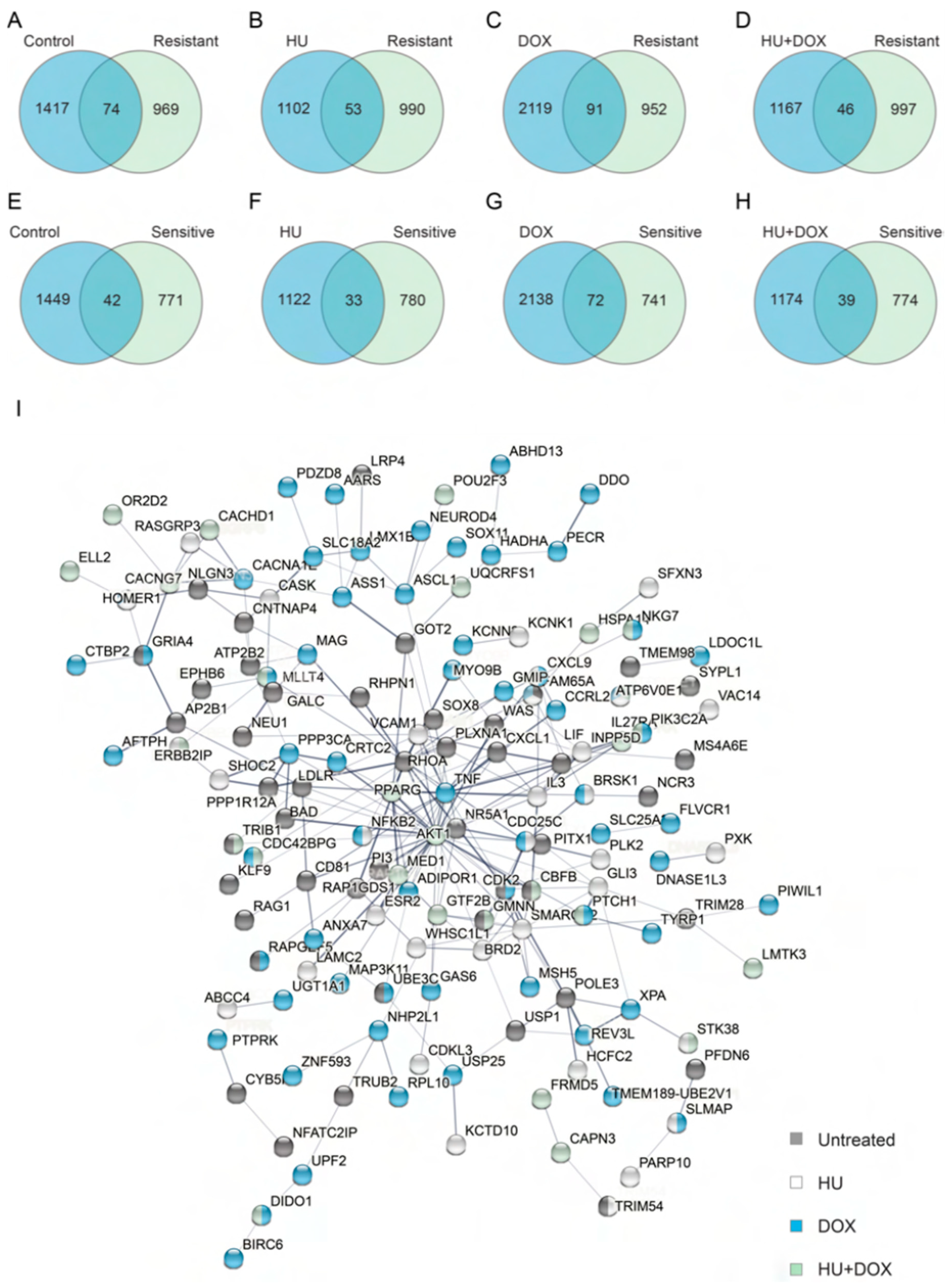
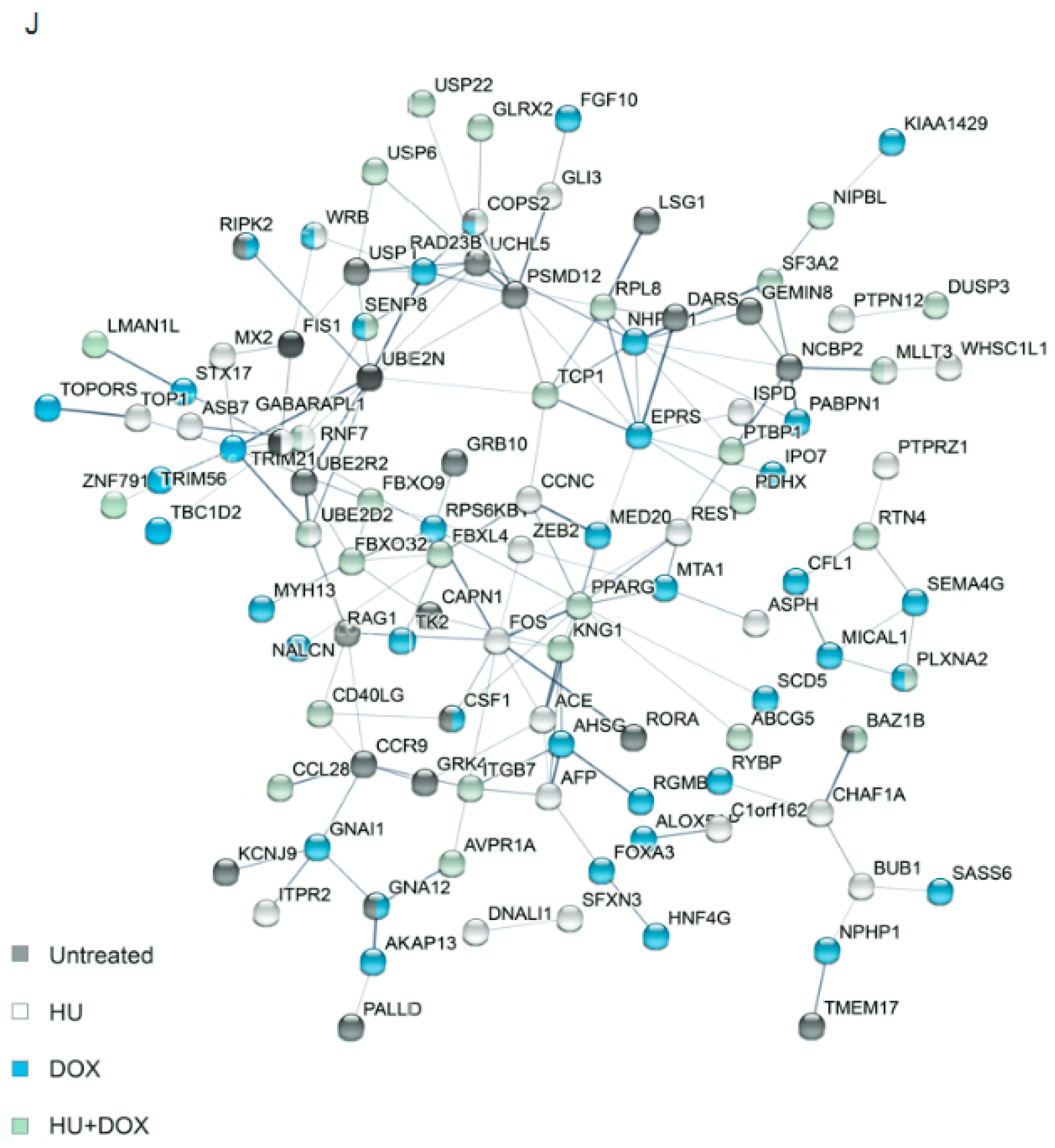
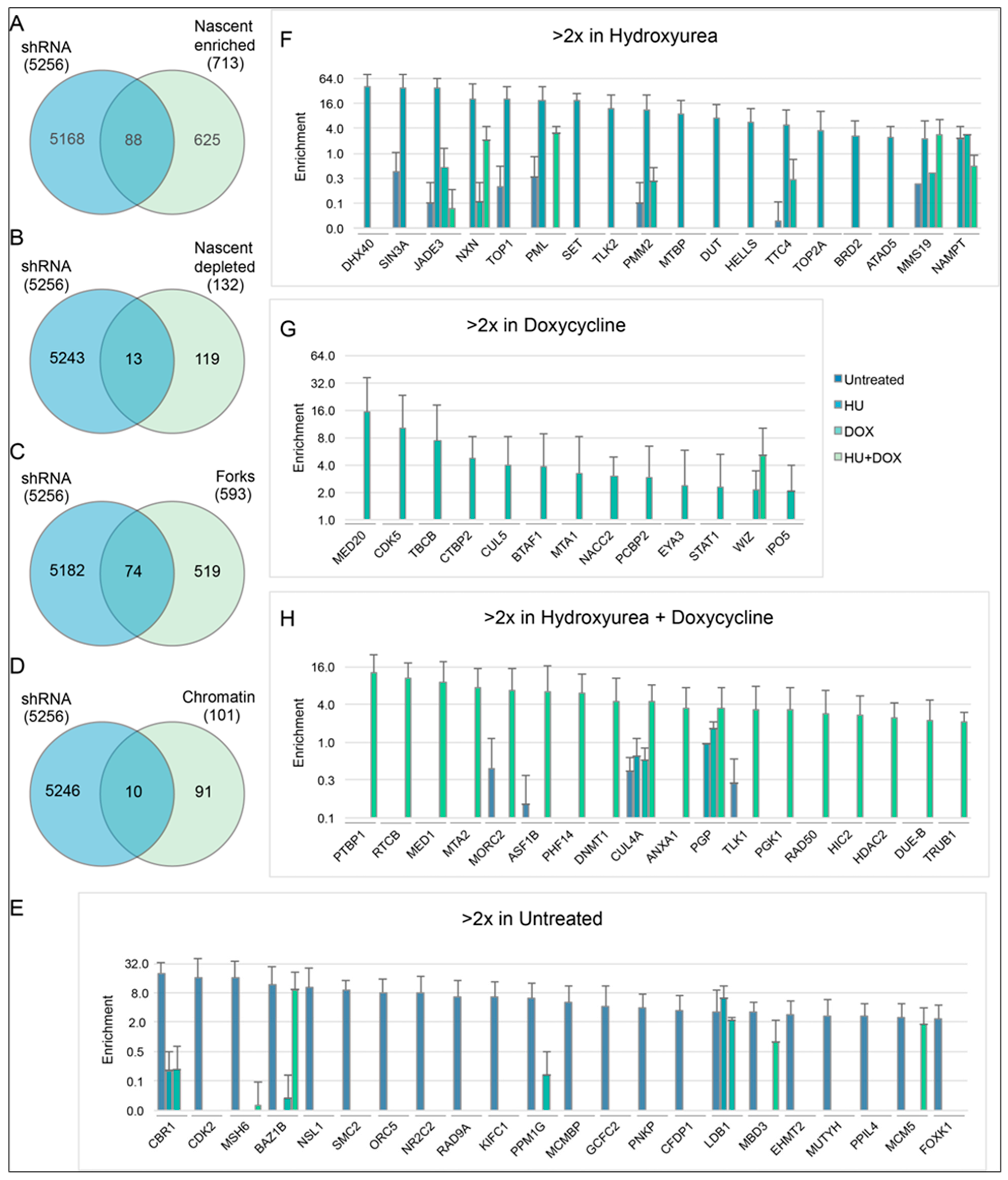
2.4. Bioinformatics
2.5. siRNA Confirmation
2.6. Inverse PCR
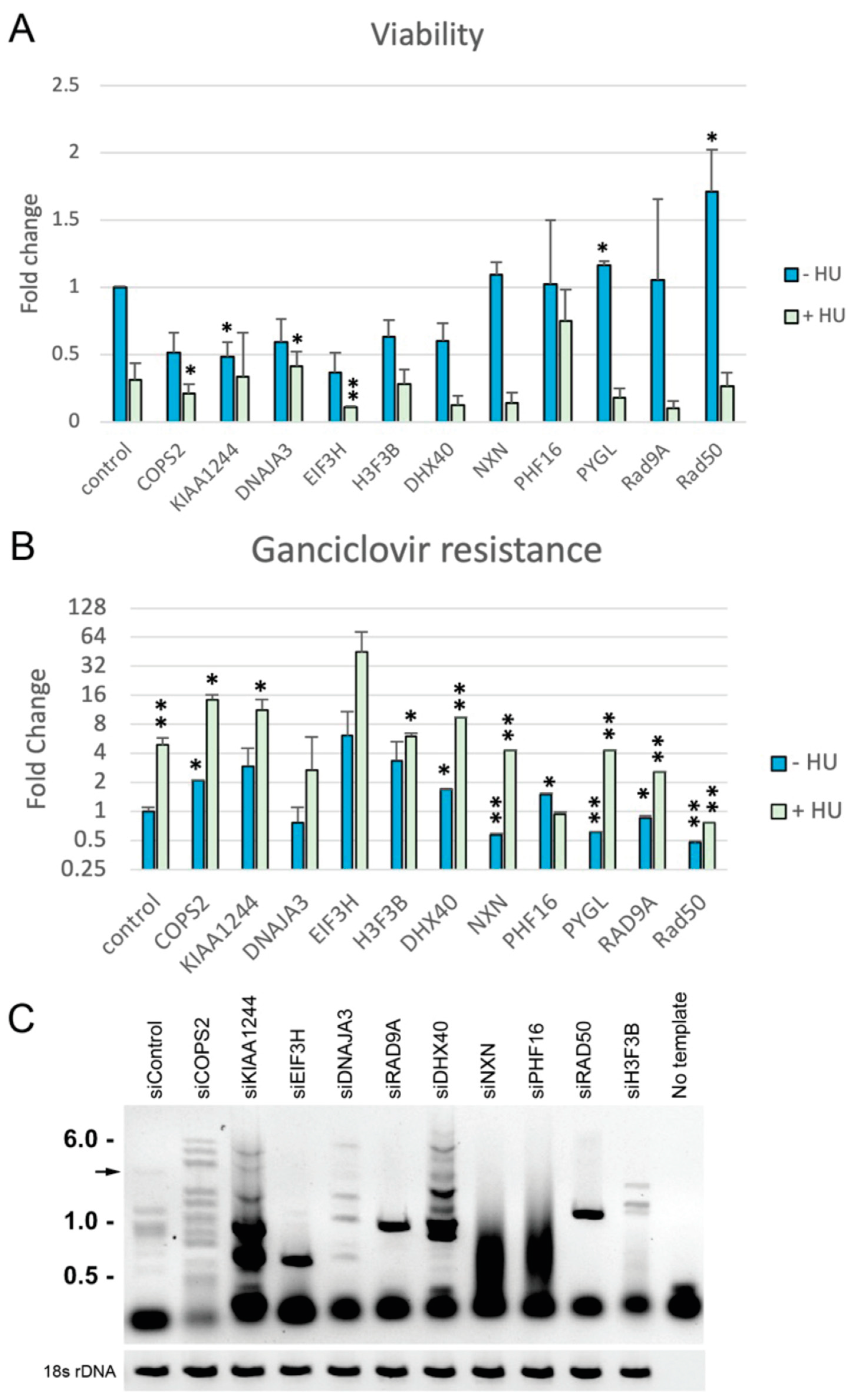
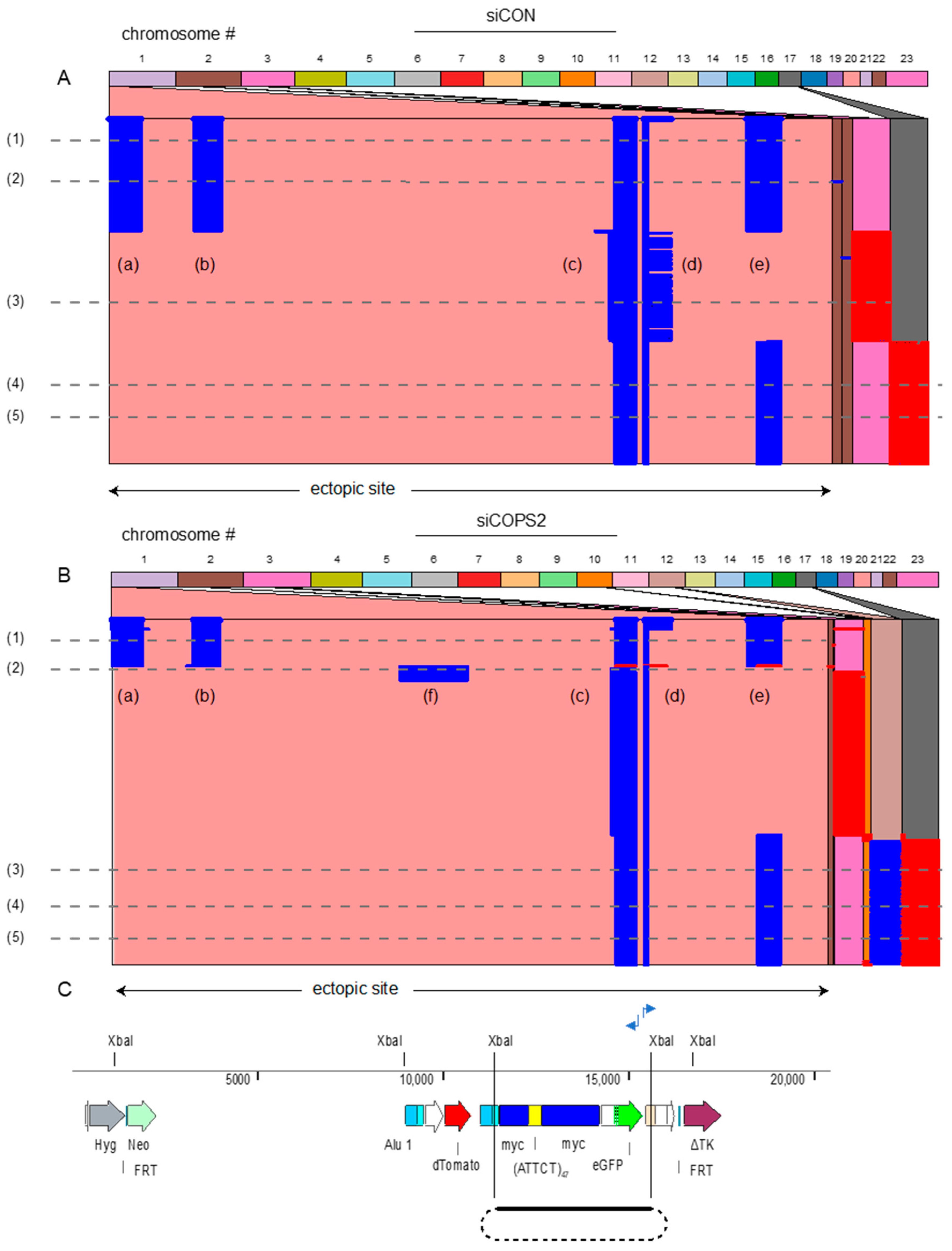
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hile, S.E.; Shabashev, S.; Eckert, K.A. Tumor-specific microsatellite instability: Do distinct mechanisms underlie the MSI-L and EMAST phenotypes? Mutat. Res. 2013, 743–744, 67–77. [Google Scholar]
- Schlotterer, C. Evolutionary dynamics of microsatellite DNA. Chromosoma 2000, 109, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Khristich, A.N.; Mirkin, S.M. On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J. Biol. Chem. 2020, 295, 4134–4170. [Google Scholar]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.A.; Mirkin, S.M. The hidden side of unstable DNA repeats: Mutagenesis at a distance. DNA Repair 2015, 32, 106–112. [Google Scholar] [PubMed]
- Kim, J.C.; Mirkin, S.M. The balancing act of DNA repeat expansions. Curr. Opin. Genet. Dev. 2013, 23, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Polleys, E.J.; Freudenreich, C.H. Homologous recombination within repetitive DNA. Curr. Opin. Genet. Dev. 2021, 71, 143–153. [Google Scholar]
- Koumbaris, G.; Hatzisevastou-Loukidou, H.; Alexandrou, A.; Ioannides, M.; Christodoulou, C.; Fitzgerald, T.; Rajan, D.; Clayton, S.; Kitsiou-Tzeli, S.; Vermeesch, J.R.; et al. FoSTeS, MMBIR and NAHR at the human proximal Xp region and the mechanisms of human Xq isochromosome formation. Hum. Mol. Genet. 2011, 20, 1925–1936. [Google Scholar] [CrossRef]
- Verdin, H.; D’Haene, B.; Beysen, D.; Novikova, Y.; Menten, B.; Sante, T.; Lapunzina, P.; Nevado, J.; Carvalho, C.M.; Lupski, J.R.; et al. Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLoS Genet. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Gene 2016, 17, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Tainer, J.A.; Vasquez, K.M.; Cooper, D.N. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016, 44, 5673–5688. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wojciechowska, M.; Kosmider, B.; Larson, J.E.; Wells, R.D. The involvement of non-B DNA structures in gross chromosomal rearrangements. DNA Repair 2006, 5, 1161–1170. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, H.; Veeraraghavan, J.; Bambara, R.A.; Freudenreich, C.H. Saccharomyces cerevisiae flap endonuclease 1 uses flap equilibration to maintain triplet repeat stability. Mol. Cell. Biol. 2004, 24, 4049–4064. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.; Truong, L.N.; Shi, L.Z.; Hwang, P.Y.; He, J.; Do, J.; Cho, M.J.; Li, H.; Negrete, A.; et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol. Cell 2014, 54, 1012–1021. [Google Scholar] [CrossRef]
- Wang, G.; Carbajal, S.; Vijg, J.; DiGiovanni, J.; Vasquez, K.M. DNA structure-induced genomic instability in vivo. J. Natl. Cancer Inst. 2008, 100, 1815–1817. [Google Scholar] [CrossRef]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair 2014, 19, 143–151. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef]
- Freudenreich, C.H.; Kantrow, S.M.; Zakian, V.A. Expansion and length-dependent fragility of CTG repeats in yeast. Science 1998, 279, 853–856. [Google Scholar] [CrossRef]
- Kumari, D.; Hayward, B.; Nakamura, A.J.; Bonner, W.M.; Usdin, K. Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat. Res. 2015, 781, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Shastri, N.; Tsai, Y.C.; Hile, S.; Jordan, D.; Powell, B.; Chen, J.; Maloney, D.; Dose, M.; Lo, Y.; Anastassiadis, T.; et al. Genome-wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol. Cell 2018, 72, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Myers, S.; Chen, X.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Replication fork stalling and checkpoint activation by a PKD1 locus mirror repeat polypurine-polypyrimidine (Pu-Py) tract. J. Biol. Chem. 2012, 287, 33412–33423. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikova, M.M.; Mirkin, S.M. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Mol. Cell. Biol. 2004, 24, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.C.; Chastain, P.; Lee, J.S.; Hegde, B.G.; Houston, S.; Langen, R.; Hsieh, C.L.; Haworth, I.S.; Lieber, M.R. Evidence for a triplex DNA conformation at the bcl-2 major breakpoint region of the t(14;18) translocation. J. Biol. Chem. 2005, 280, 22749–22760. [Google Scholar] [CrossRef]
- Kim, H.M.; Narayanan, V.; Mieczkowski, P.A.; Petes, T.D.; Krasilnikova, M.M.; Mirkin, S.M.; Lobachev, K.S. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. EMBO J. 2008, 27, 2896–2906. [Google Scholar] [CrossRef] [PubMed]
- Meservy, J.L.; Sargent, R.G.; Iyer, R.R.; Chan, F.; McKenzie, G.J.; Wells, R.D.; Wilson, J.H. Long CTG tracts from the myotonic dystrophy gene induce deletions and rearrangements during recombination at the APRT locus in CHO cells. Mol. Cell. Biol. 2003, 23, 3152–3162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, G.; Del Mundo, I.M.; McKinney, J.A.; Lu, X.; Bacolla, A.; Boulware, S.B.; Zhang, C.; Zhang, H.; Ren, P.; et al. Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep. 2018, 22, 1200–1210. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. MMBR 2007, 71, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. Expandable DNA repeats and human disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Kolinjivadi, A.M.; Sannino, V.; de Antoni, A.; Techer, H.; Baldi, G.; Costanzo, V. Moonlighting at replication forks—a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017, 591, 1083–1100. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short Inverted Repeats Are Hotspots for Genetic Instability: Relevance to Cancer Genomes. Cell Rep. 2015, 10, 674–1680. [Google Scholar] [CrossRef] [PubMed]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 2003, 23, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Gellibolian, R.; Shimizu, M.; Amirhaeri, S.; Kang, S.; Ohshima, K.; Larson, J.E.; Harvey, S.C.; Stollar, B.D.; Wells, R.D. Flexible DNA: Genetically unstable CTG.CAG and CGG.CCG from human hereditary neuromuscular disease genes. J. Biol. Chem. 1997, 272, 16783–16792. [Google Scholar] [CrossRef]
- Bacolla, A.; Jaworski, A.; Connors, T.D.; Wells, R.D. Pkd1 unusual DNA conformations are recognized by nucleotide excision repair. J. Biol. Chem. 2001, 276, 18597–18604. [Google Scholar] [CrossRef]
- Bacolla, A.; Jaworski, A.; Larson, J.E.; Jakupciak, J.P.; Chuzhanova, N.; Abeysinghe, S.S.; O’Connell, C.D.; Cooper, D.N.; Wells, R.D. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc. Natl. Acad. Sci. USA 2004, 101, 14162–14167. [Google Scholar] [CrossRef]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol. Carcinog. 2009, 48, 273–285. [Google Scholar] [CrossRef]
- Bacolla, A.; Wang, G.; Vasquez, K.M. New Perspectives on DNA and RNA Triplexes As Effectors of Biological Activity. PLoS Genet. 2015, 11, e1005696. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.P.; Shah, K.A.; Niu, H.; Sung, P.; Mirkin, S.M.; Freudenreich, C.H. Overcoming natural replication barriers: Differential helicase requirements. Nucleic Acids Res. 2012, 40, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callen, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, R.; Elango, R.; Panday, A.; Willis, N.A. Recombination and restart at blocked replication forks. Curr. Opin. Genet. Dev. 2021, 71, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell. Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Kramara, J.; Osia, B.; Malkova, A. Break-Induced Replication: The Where, The Why, and The How. Trends Genet. 2018, 34, 518–531. [Google Scholar] [CrossRef]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Coban-Akdemir, Z.; Hijazi, H.; Yuan, B.; Pendleton, M.; Harrington, E.; Beaulaurier, J.; Juul, S.; Turner, D.J.; Kanchi, R.S.; et al. Interchromosomal template-switching as a novel molecular mechanism for imprinting perturbations associated with Temple syndrome. Genome Med. 2019, 11, 25. [Google Scholar] [CrossRef]
- Bahrambeigi, V.; Song, X.; Sperle, K.; Beck, C.R.; Hijazi, H.; Grochowski, C.M.; Gu, S.; Seeman, P.; Woodward, K.J.; Carvalho, C.M.B.; et al. Distinct patterns of complex rearrangements and a mutational signature of microhomeology are frequently observed in PLP1 copy number gain structural variants. Genome Med. 2019, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Gadgil, R.Y.; Romer, E.J.; Goodman, C.C.; Rider, S.D.; Jr Damewood, F.J.; Barthelemy, J.R.; Shin-Ya, K.; Hanenberg, H.; Leffak, M. Replication stress at microsatellites causes DNA double-strand breaks and break-induced replication. J. Biol. Chem. 2020, 295, 15378–15397. [Google Scholar] [CrossRef] [PubMed]
- Rider, S.D., Jr.; Gadgil, R.Y.; Hitch, D.C.; Damewood, F.J.; Zavada, N.; Shanahan, M.; Alhawach, V.; Shrestha, R.; Shin-ya, K.; Leffak, M. Stable G-quadruplex DNA structures promote replication-dependent genome instability. J. Biol. Chem. 2022, 298, 101947. [Google Scholar] [CrossRef]
- Lambert, S.; Mizuno, K.; Blaisonneau, J.; Martineau, S.; Chanet, R.; Freon, K.; Murray, J.M.; Carr, A.M.; Baldacci, G. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 2010, 39, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Lambert, S. Replication stress-induced genome instability: The dark side of replication maintenance by homologous recombination. J. Mol. Biol. 2013, 425, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kononenko, A.V.; Ebersole, T.; Vasquez, K.M.; Mirkin, S.M. Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat. Struct. Mol. Biol. 2018, 25, 669–676. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approcah to Multiple Testing. J. R. Statist. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Klipper-Aurbach, Y.; Wasserman, M.; Braunspiegel-Weintrob, N.; Borstein, D.; Peleg, S.; Assa, S.; Karp, M.; Benjamini, Y.; Hochberg, Y.; Laron, Z. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med. Hypotheses 1995, 45, 486–490. [Google Scholar] [CrossRef]
- Barthelemy, J.; Hanenberg, H.; Leffak, M. FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 2016, 44, 6803–6816. [Google Scholar] [CrossRef]
- Liu, G.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Unstable spinocerebellar ataxia type 10 (ATTCT) × (AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol. Cell. Biol. 2007, 27, 7828–7838. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Liu, G.; Kemp, M.; Chen, X.; Katrangi, N.; Myers, S.; Ghosh, M.; Yao, J.; Gao, Y.; Bubulya, P.; et al. The DNA unwinding element binding protein DUE-B interacts with Cdc45 in preinitiation complex formation. Mol. Cell. Biol. 2010, 30, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, X.; Bissler, J.J.; Sinden, R.R.; Leffak, M. Replication-dependent instability at (CTG) × (CAG) repeat hairpins in human cells. Nat. Chem. Biol. 2010, 6, 652–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Liu, G.; Leffak, M. Activation of a human chromosomal replication origin by protein tethering. Nucleic Acids Res. 2013, 41, 6460–6474. [Google Scholar] [CrossRef]
- Liu, G.; Chen, X.; Leffak, M. Oligodeoxynucleotide binding to (CTG) × (CAG) microsatellite repeats inhibits replication fork stalling, hairpin formation, and genome instability. Mol. Cell. Biol. 2013, 33, 571–581. [Google Scholar] [CrossRef]
- Guo, J.; Gu, L.; Leffak, M.; Li, G.M. MutSbeta promotes trinucleotide repeat expansion by recruiting DNA polymerase beta to nascent (CAG)n or (CTG)n hairpins for error-prone DNA synthesis. Cell Res. 2016, 26, 775–786. [Google Scholar] [CrossRef]
- Waltz, S.E.; Trivedi, A.A.; Leffak, M. DNA replication initiates non-randomly at multiple sites near the c-myc gene in HeLa cells. Nucleic Acids Res. 1996, 24, 1887–1894. [Google Scholar] [CrossRef]
- Trivedi, A.; Waltz, S.E.; Kamath, S.; Leffak, M. Multiple initiations in the c-myc replication origin independent of chromosomal location. DNA Cell Biol. 1998, 17, 885–896. [Google Scholar] [CrossRef]
- Malott, M.; Leffak, M. Activity of the c-myc replicator at an ectopic chromosomal location. Mol. Cell. Biol. 1999, 19, 5685–5695. [Google Scholar] [CrossRef]
- Berberich, S.; Trivedi, A.; Daniel, D.C.; Johnson, E.M.; Leffak, M. In vitro replication of plasmids containing human c-myc DNA. J. Mol. Biol. 1995, 245, 92–109. [Google Scholar] [CrossRef]
- McWhinney, C.; Waltz, S.E.; Leffak, M. Cis-acting effects of sequences within 2.4-kb upstream of the human c-myc gene on autonomous plasmid replication in HeLa cells. DNA Cell Biol. 1995, 14, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Dong, Z.; Leffak, M.; Zannis-Hadjopoulos, M.; Price, G. Major DNA replication initiation sites in the c-myc locus in human cells. J. Cell. Biochem. 2000, 78, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Malott, M.; Leffak, M. Multiple functional elements comprise a Mammalian chromosomal replicator. Mol. Cell. Biol. 2003, 23, 1832–1842. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Liu, G.; Randall, G.; Bevington, J.; Leffak, M. Transcription factor binding and induced transcription alter chromosomal c-myc replicator activity. Mol. Cell. Biol. 2004, 24, 10193–10207. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Kemp, M.; Liu, G.; Ritzi, M.; Schepers, A.; Leffak, M. Differential binding of replication proteins across the human c-myc replicator. Mol. Cell. Biol. 2006, 26, 5270–5283. [Google Scholar] [CrossRef] [PubMed]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef]
- Lewis, T.W.; Barthelemy, J.R.; Virts, E.L.; Kennedy, F.M.; Gadgil, R.Y.; Wiek, C.; Linka, R.M.; Zhang, F.; Andreassen, P.R.; Hanenberg, H.; et al. Deficiency of the Fanconi anemia E2 ubiqitin conjugase UBE2T only partially abrogates Alu-mediated recombination in a new model of homology dependent recombination. Nucleic Acids Res. 2019, 47, 3503–3520. [Google Scholar] [CrossRef]
- Wessel, S.R.; Mohni, K.N.; Luzwick, J.W.; Dungrawala, H.; Cortez, D. Functional Analysis of the Replication Fork Proteome Identifies BET Proteins as PCNA Regulators. Cell Rep. 2019, 28, 3497–3509. [Google Scholar] [CrossRef]
- Dungrawala, H.; Cortez, D. Purification of proteins on newly synthesized DNA using iPOND. Methods Mol. Biol. 2015, 1228, 123–131. [Google Scholar]
- Yim, C.Y.; Bikorimana, E.; Khan, E.; Warzecha, J.M.; Shin, L.; Rodriguez, J.; Dmitrovsky, E.; Freemantle, S.J.; Spinella, M.J. G0S2 represses PI3K/mTOR signaling and increases sensitivity to PI3K/mTOR pathway inhibitors in breast cancer. Cell Cycle 2017, 16, 2146–2155. [Google Scholar] [CrossRef]
- Yim, C.Y.; Sekula, D.J.; Hever-Jardine, M.P.; Liu, X.; Warzecha, J.M.; Tam, J.; Freemantle, S.J.; Dmitrovsky, E.; Spinella, M.J. G0S2 Suppresses Oncogenic Transformation by Repressing a MYC-Regulated Transcriptional Program. Cancer Res. 2016, 76, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.T.; Sardiu, M.E.; Martin-Brown, S.A.; Seidel, C.; Mushegian, A.; Egidy, R.; Florens, L.; Washburn, M.P.; Workman, J.L. Human family with sequence similarity 60 member A (FAM60A) protein: A new subunit of the Sin3 deacetylase complex. Mol. Cell. Proteomics 2012, 11, 1815–1828. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.; Dellaire, G. The Role of the COP9 Signalosome and Neddylation in DNA Damage Signaling and Repair. Biomolecules 2015, 5, 2388–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavadini, S.; Fischer, E.S.; Bunker, R.D.; Potenza, A.; Lingaraju, G.M.; Goldie, K.N.; Mohamed, W.I.; Faty, M.; Petzold, G.; Beckwith, R.E.; et al. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature 2016, 531, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Dubiel, W.; Chaithongyot, S.; Dubiel, D.; Naumann, M. The COP9 Signalosome: A Multi-DUB Complex. Biomolecules 2020, 10, 1082. [Google Scholar] [CrossRef]
- Qin, N.; Xu, D.; Li, J.; Deng, X.W. COP9 signalosome: Discovery, conservation, activity, and function. J. Integr. Plant Biol. 2020, 62, 90–103. [Google Scholar] [CrossRef]
- Rao, F.; Lin, H.; Su, Y. Cullin-RING Ligase Regulation by the COP9 Signalosome: Structural Mechanisms and New Physiologic Players. Adv. Exp. Med. Biol. 2020, 1217, 47–60. [Google Scholar]
- Nattestad, M.; Aboukhalil, R.; Chin, C.S.; Schatz, M.C. Ribbon: Intuitive visualization for complex genomic variation. Bioinformatics 2021, 37, 413–415. [Google Scholar] [CrossRef]
- Kusakabe, M.; Kutomi, T.; Watanabe, K.; Emoto, N.; Aki, N.; Kage, H.; Hamano, E.; Kitagawa, H.; Nagase, T.; Sano, A.; et al. Identification of G0S2 as a gene frequently methylated in squamous lung cancer by combination of in silico and experimental approaches. Int. J. Cancer 2010, 126, 1895–1902. [Google Scholar] [CrossRef]
- Cho, E.; Kwon, Y.J.; Ye, D.J.; Baek, H.S.; Kwon, T.U.; Choi, H.K.; Chun, Y.J. G0/G1 Switch 2 Induces Cell Survival and Metastasis through Integrin-Mediated Signal Transduction in Human Invasive Breast Cancer Cells. Biomol. Ther. 2019, 27, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.; Santra, M.K.; El-Assaad, W.; Zhu, X.; Huber, W.E.; Keys, R.A.; Teodoro, J.G.; Green, M.R. Identification of a protein, G0S2, that lacks Bcl-2 homology domains and interacts with and antagonizes Bcl-2. Cancer Res. 2009, 69, 6782–6789. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Alland, L.; David, G.; Shen-Li, H.; Potes, J.; Muhle, R.; Lee, H.C.; Hou, H.; Jr Chen, K.; DePinho, R.A. Identification of mammalian Sds3 as an integral component of the Sin3/histone deacetylase corepressor complex. Mol. Cell. Biol. 2002, 22, 2743–2750. [Google Scholar] [CrossRef]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef]
- Naiman, K.; Campillo-Funollet, E.; Watson, A.T.; Budden, A.; Miyabe, I.; Carr, A.M. Replication dynamics of recombination-dependent replication forks. Nat. Commun. 2021, 12, 923. [Google Scholar] [CrossRef]
- Nagpal, A.; Raja, S.; Van Houten, B. The role of UV-DDB in processing 8-oxoguanine during base excision repair. Biochem. Soc. Trans. 2022, 50, 1481–1488. [Google Scholar] [CrossRef]
- Dorn, J.; Ferrari, E.; Imhof, R.; Ziegler, N.; Hubscher, U. Regulation of human MutYH DNA glycosylase by the E3 ubiquitin ligase mule. J. Biol. Chem. 2014, 289, 7049–7058. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rider, S.D., Jr.; Damewood, F.J., IV; Gadgil, R.Y.; Hitch, D.C.; Alhawach, V.; Shrestha, R.; Shanahan, M.; Zavada, N.; Leffak, M. Suppressors of Break-Induced Replication in Human Cells. Genes 2023, 14, 398. https://doi.org/10.3390/genes14020398
Rider SD Jr., Damewood FJ IV, Gadgil RY, Hitch DC, Alhawach V, Shrestha R, Shanahan M, Zavada N, Leffak M. Suppressors of Break-Induced Replication in Human Cells. Genes. 2023; 14(2):398. https://doi.org/10.3390/genes14020398
Chicago/Turabian StyleRider, Stanley Dean, Jr., French J. Damewood, IV, Rujuta Yashodhan Gadgil, David C. Hitch, Venicia Alhawach, Resha Shrestha, Matilyn Shanahan, Nathen Zavada, and Michael Leffak. 2023. "Suppressors of Break-Induced Replication in Human Cells" Genes 14, no. 2: 398. https://doi.org/10.3390/genes14020398