
Prenatal cfDNA Screening for Emanuel Syndrome and Other Unbalanced Products of Conception in Carriers of the Recurrent Balanced Translocation t(11;22): One Laboratory’s Retrospective Experience


Abstract
:1. Introduction
2. Materials and Methods
3. Results
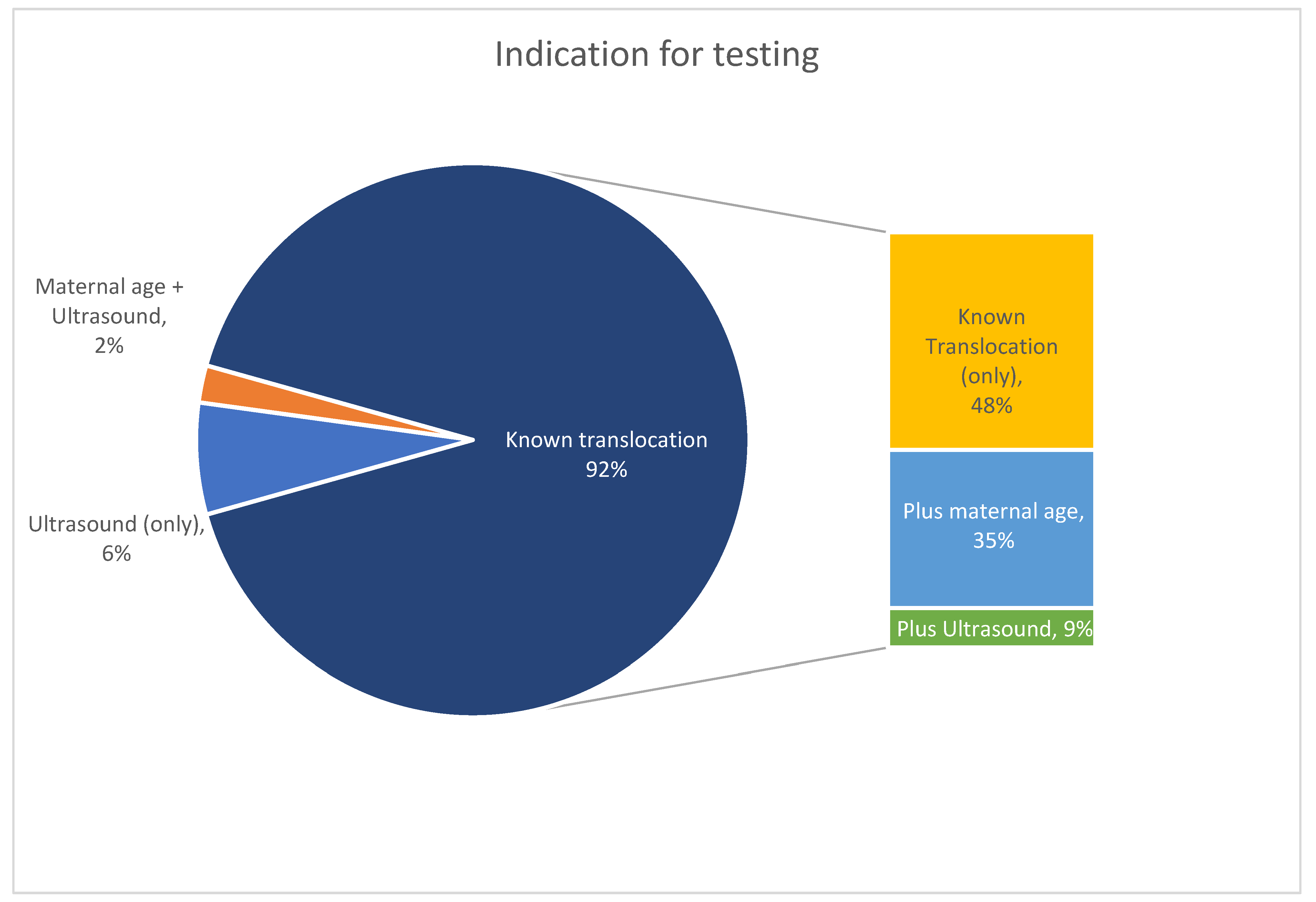
3.1. Study Cohort
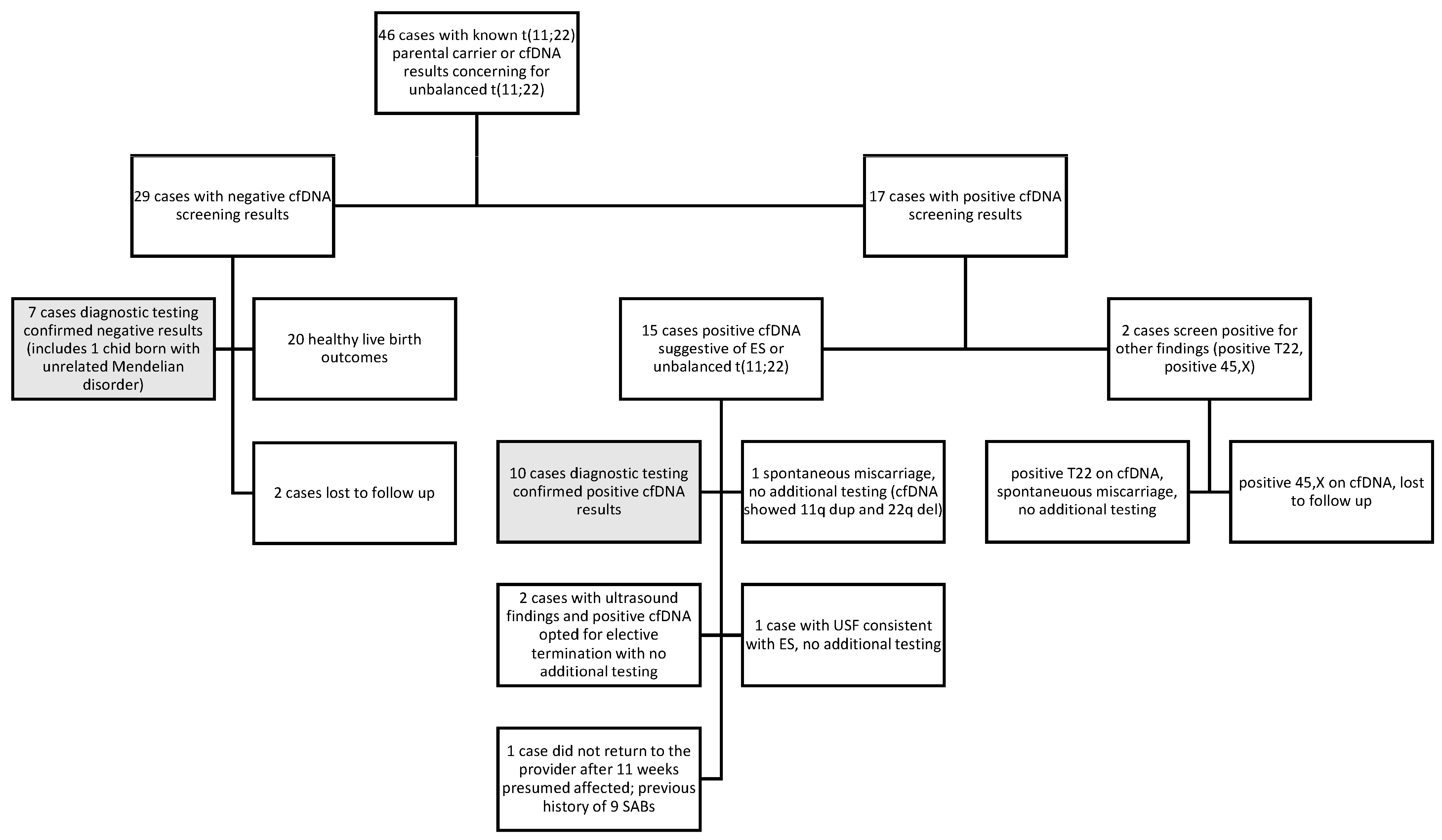
3.2. cfDNA Findings
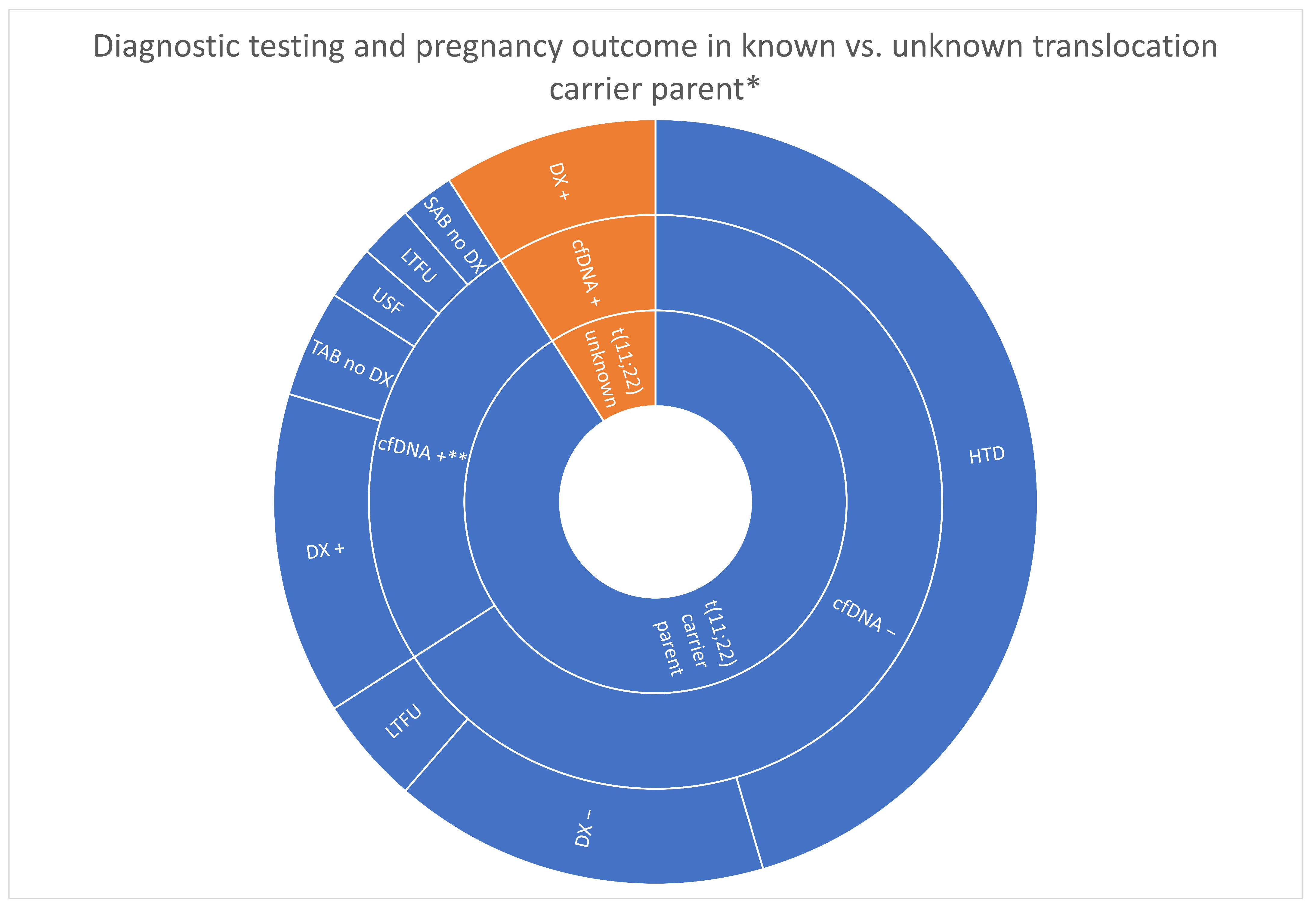
3.3. Diagnostic Testing and Pregnancy Outcome
4. Discussion
4.1. Overview and Comparison to Literature
4.2. Limitations
4.3. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- History of Emanuel Syndrome and the C22C Emanuel Syndrom and the 11/22 Translocation: Emanuelsyndrome.org; Updated July 2023. Available online: https://emanuelsyndrome.org/emanuel-syndrome/history-of-emanuel-syndrome-and-c22c/ (accessed on 22 September 2023).
- Carter, M.T.; St. Pierre, S.A.; Zackai, E.H.; Emanuel, B.S.; Boycott, K.M. Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): Clinical features of 63 individuals. Am. J. Med. Genet. Part A 2009, 149A, 1712–1721. [Google Scholar] [CrossRef]
- Zackai, E.H.; Emanuel, B.S. Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am. J. Med. Genet. 1980, 7, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H.; Budarf, M.L.; Celle, L.; Zackai, E.H.; Emanuel, B.S. Clustered 11q23 and 22q11 Breakpoints and 3:1 Meiotic Malsegregation in Multiple Unrelated t(11;22) Families. Am. J. Hum. Genet. 1999, 65, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, B.S.; Zackai, E.H.; Medne, L. Emanuel Syndrome. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2007. [Google Scholar]
- Ohye, T.; Inagaki, H.; Kato, T.; Tsutsumi, M.; Kurahashi, H. Prevalence of Emanuel syndrome: Theoretical frequency and surveillance result. Pediatr. Int. 2014, 56, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Fraccaro, M.; Lindsten, J.; Ford, C.E.; Iselius, L. The 11q;22q translocation: A European collaborative analysis of 43 cases. Hum. Genet. 1980, 56, 21–51. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lin, J.; Sun, Y.; Qian, Y.; Wang, L.; Chen, M.; Dong, M.; Jin, F. Non-invasive prenatal screening for Emanuel syndrome. Mol. Cytogenet. 2020, 13, 9. [Google Scholar] [CrossRef]
- Hao, X.; Wu, J.; Fu, W.; Zhang, R.; Zhong, S.; Deng, Y.; Zhu, Y.; Ye, Y.; Fang, Q. Prenatal diagnosis of fetuses with Emanuel syndrome: Results of ultrasound examination and invasive genetic testing. Prenat. Diagn. 2022, 42, 469–477. [Google Scholar] [CrossRef]
- Zaki, M.S.; Mohamed, A.M.; Kamel, A.K.; El-Gerzawy, A.M.; El-Ruby, M.O. Emanuel syndrome due to unusual segregation of paternal origin. Genet. Couns. 2012, 23, 319–328. [Google Scholar]
- Walfisch, A.; Mills, K.E.; Chodirker, B.N.; Berger, H. Prenatal screening characteristics in Emanuel syndrome: A case series and review of the literature. Arch. Gynecol. Obstet. 2012, 286, 299–302. [Google Scholar] [CrossRef]
- Flowers, N.J.; Burgess, T.; Giouzeppos, O.; Shi, G.; Love, C.J.; Hunt, C.E.; Scarff, K.L.; Archibald, A.D.; Pertile, M.D. Genome-wide noninvasive prenatal screening for carriers of balanced reciprocal translocations. Genet. Med. 2020, 22, 1944–1955. [Google Scholar] [CrossRef]
- Dungan, J.S.; Klugman, S.; Darilek, S.; Malinowski, J.; Akkari, Y.M.N.; Monaghan, K.G.; Erwin, A.; Best, R.G. Noninvasive prenatal screening (NIPS) for fetal chromosome abnormalities in a general-risk population: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2022, 25, 100336. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Ellis, K.; Mayen, D.; Pertile, M.D.; Riemers, R.; Sun, L.; Vermeesch, J.; Vora, N.L.; Chitty, L.S. Position statement from the International Society for Prenatal Diagnosis (ISPD) on the use of non-invasive prenatal testing (NIPT) for the detection of fetal chromosomal conditions in singleton pregnancies. Prenat. Diagn. 2023, 43, 814–828. [Google Scholar] [CrossRef]
- Jensen, T.J.; Zwiefelhofer, T.; Tim, R.C.; Dzakula, Z.; Kim, S.K.; Mazloom, A.R.; Zhu, Z.; Tynan, J.; Lu, T.; McLennan, G.; et al. High-throughput massively parallel sequencing for fetal aneuploidy detection from maternal plasma. PLoS ONE 2013, 8, e57381. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Tynan, J.; Ehrich, M.; Hannum, G.; McCullough, R.; Saldivar, J.-S.; Oeth, P.; Van Den Boom, D.; Deciu, C. Detection of Fetal Subchromosomal Abnormalities by Sequencing Circulating Cell-Free DNA from Maternal Plasma. Clin. Chem. 2015, 61, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.B.; Tynan, J.A.; Liu, T.; Wu, Y.; Mazloom, A.R.; Almasri, E.; Hogg, G.; Angkachatchai, V.; Zhao, C.; Grosu, D.S.; et al. Clinical validation of a noninvasive prenatal test for genomewide detection of fetal copy number variants. Am. J. Obstet. Gynecol. 2016, 215, 227.e1–227.e16. [Google Scholar] [CrossRef]
- Kim, S.K.; Hannum, G.; Geis, J.; Tynan, J.; Hogg, G.; Zhao, C.; Jensen, T.J.; Mazloom, A.R.; Oeth, P.; Ehrich, M.; et al. Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenat. Diagn. 2015, 35, 810–815. [Google Scholar] [CrossRef]
- Mazloom, A.R.; Koch, A.; Sun, Y.; Geis, J.; Whidden, M.; Almasri, E.; Tynan, J.; Burcham, T.; Ehrich, M. Sample specific fetal fraction threshold for non-invasive prenatal testing. In Proceedings of the American College of Medical Genetics Annual Clinical Genetics Meeting, Phoenix, AZ, USA, 21–25 March 2017. [Google Scholar]
- Palomaki, G.E.; Deciu, C.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; Van Den Boom, D.; Bombard, A.T.; Grody, W.W.; et al. DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: An international collaborative study. Genet. Med. 2012, 14, 296–305. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Kloza, E.M.; Lambert-Messerlian, G.M.; Haddow, J.E.; Neveux, L.M.; Ehrich, M.; Van Den Boom, D.; Bombard, A.T.; Deciu, C.; Grody, W.W.; et al. DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet. Med. 2011, 13, 913–920. [Google Scholar] [CrossRef]
- Soster, E.; Boomer, T.; Hicks, S.; Caldwell, S.; Dyr, B.; Chibuk, J.; Almasri, E. Three years of clinical experience with a genome-wide cfDNA screening test for aneuploidies and copy-number variants. Genet. Med. 2021, 23, 1349–1355. [Google Scholar] [CrossRef]
- Gremeau, A.S.; Coste, K.; Blanc, P.; Goumy, C.; Francannet, C.; Dechelotte, P.J.; Vago, P.; Laurichesse-Delmas, H.; Labbe, A.; Lemery, D.; et al. Congenital diaphragmatic hernia and genital anomalies: Emanuel syndrome. Prenat. Diagn. 2009, 29, 816–818. [Google Scholar] [CrossRef]
- Cifuentes Ochoa, M.; Flowers, N.J.; Pertile, M.D.; Archibald, A.D. “It becomes your whole life”—Exploring experiences of reciprocal translocation carriers and their partners. J. Genet. Couns. 2023, 32, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Srebniak, M.I.; Vogel, I.; Van Opstal, D. Is carriership of a balanced translocation or inversion an indication for non-invasive prenatal testing? Expert Rev. Mol. Diagn. 2018, 18, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Mazloom, A.R.; Dzakula, Z.; Oeth, P.; Wang, H.; Jensen, T.; Tynan, J.; McCullough, R.; Saldivar, J.S.; Ehrich, M.; van den Boom, D.; et al. Noninvasive prenatal detection of sex chromosomal aneuploidies by sequencing circulating cell-free DNA from maternal plasma. Prenat. Diagn. 2013, 33, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Piwowarczyk, P.; Massalska, D.; Obodzinska, I.; Gawlik Zawislak, S.; Bijok, J.; Kucinska-Chahwan, A.; Roszkowski, T. Prenatal diagnosis of Emanuel syndrome—Case series and review of the literature. J. Obstet. Gynaecol. 2022, 42, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.E.; Chapman, A.; Cormack, C.L.; Campbell, K.; Ebanks, A.H.; Annibale, D.J.; Hollinger, L.E. Emanuel syndrome and congenital diaphragmatic hernia: A systematic review. J. Pediatr. Surg. 2022, 57, 24–28. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patient # | Case | GA | cfDNA Indication | Known Translocation? (Origin) | cfDNA Assay Type | cfDNA Result (FF) | Diagnostic Testing + Pregnancy Outcome | Category |
|---|---|---|---|---|---|---|---|---|
| 1 * | - | 9.00 | AMA, Tx Hx | Yes (Paternal) | Genome-wide | Negative (8.57%) | CVS karyotype: one twin WNL and one twin balanced carrier | Concordant negative |
| 2 | A | 10.14 | Tx Hx | Yes (Paternal) | Genome-wide | Positive (11 and 22) (9.94%) | SAB; No testing. | Likely concordant positive |
| B | 10.86 | Tx Hx | Yes (Paternal) | Genome-wide | Negative (7.48%) | Healthy, term delivery. No testing. | Concordant negative | |
| 3 | - | 9.00 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (12.51%) | CVS karyotype: WNL | Concordant negative |
| 4 | - | 9.00 | Tx Hx | Yes (Paternal) | ‘Traditional’ | Negative (verbal requested) (6.68%) | Postnatal karyotype: balanced carrier | Concordant negative |
| 5 * | - | 10.00 | Tx Hx | Yes (Maternal) | ‘Traditional’ | Negative (verbal requested) (6.57%) | Healthy, term delivery. No testing. | Concordant negative |
| 6 | - | 11.57 | Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (6.50%) | CVS karyotype and array: 47,XY,+der(22)t(11;22)(q23.3;q11.21); 18.26 Mb terminal duplication of 11q23.3->q25; 3.84 Mb proximal duplication of 22q11.1->q11.21 | Concordant positive |
| 7 | - | 11.71 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (7.42%) | LTFU | LTFU |
| 8 | - | 12.14 | Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 only) (4.54%) | Amniocentesis karyotype: 47,XY,+der(22)t(11;22)(q23.3;q11.2) Patient is gravida 11—all early SABs | Concordant positive |
| 9 * | A | 10.14 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (15.27%) | Multiple anomalies; TAB; No testing. | Likely concordant positive |
| B | 9.00 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (9.46%) | Egg donor/IVF; Monochorionic/diamniotic twins; CVS karyotype: WNL | Concordant negative | |
| 10 | - | 11.71 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (10.91%) | Healthy, term delivery. No testing. | Concordant negative |
| 11 | A | 10.00 | AMA, Tx Hx | Yes (Paternal) | Genome-wide | Negative (10.08%) | PGT-SR with presumably balanced/normal embryo; Healthy, term delivery. No testing | Concordant negative |
| B | 9.71 | AMA, Tx Hx | Yes (Paternal) | Genome-wide | Negative (10.95%) | Amniocentesis karyotype and array: balanced carrier | Concordant negative | |
| 12 | - | 21.57 | USF | No | Genome-wide | Positive (11 and 22) (7.04%) | Diaphragmatic hernia on ultrasound. Previous pregnancy also had diaphragmatic hernia, neonatal death with no testing. Postnatal array: 18.2 Mb terminal duplication of 11q23.3->q25; 3.4 Mb proximal duplication of 22q11.1->q11.21 | Concordant positive |
| 13 | A | 9.00 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (15.75%) | Healthy, term delivery. No testing. Reports previous child with ES. | Concordant negative |
| B | 10.29 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (10.76%) | Healthy, term delivery. No testing. | Concordant negative | |
| 14 | A | 13.57 | USF, Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (10.81%) | Maternal family history of ES with history of 2 prior SABs; Maternal karyotype concurrent to cfDNA confirmed carrier status; Amniocentesis: FISH showed 3 copies of 11q23 and 22q11.2; Products of conception karyotype: 47,XX,+der(22)t(11;22)(q23.3;q11.2); TAB | Concordant positive |
| B | 11.00 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (6.31%) | Healthy, term delivery. No testing. | Concordant negative | |
| 15 | A | 12.00 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (10.20%) | Healthy, term delivery. No testing. | Concordant negative |
| B | 12.29 | Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (9.95%) | SAB; CVS karyotype: 47,XX,+der(22)t(11;22)(q23.3;q11.2) | Concordant positive | |
| C | 9.00 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (7.79%) | CVS karyotype: balanced carrier | Concordant negative | |
| D | 10.71 | USF, Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 only) (8.33%) | Patient did not return for care; History of 9+ SABs and reports previous children affected; 22q dup noted on data review | Likely concordant positive | |
| 16 | A | 12.43 | AMA, Tx Hx | Yes (Paternal) | Genome-wide | Negative (10.51%) | Healthy, term delivery. No testing. | Concordant negative |
| B | 10.14 | AMA, Tx Hx | Yes (Paternal) | Genome-wide | Negative (7.10%) | Healthy, term delivery. No testing. | Concordant negative | |
| 17 | - | 26.14 | USF | No | Genome-wide | Positive (11 and 22) (15.41%) | Dandy–Walker malformation on ultrasound; Amniocentesis karyotype: 47,XX,+der(22)t(11;22)(q23.3;q11.2). Delivered affected infant; no additional outcome available. | Concordant positive |
| 18 | A | 12.29 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (14.35%) | Healthy, term delivery. No testing. | Concordant negative |
| B | 12.00 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (13.76%) | Healthy, term delivery. No testing. | Concordant negative | |
| 19 | - | 12.86 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (11.43%) | Healthy, term delivery. No testing. | Concordant negative |
| 20 | - | 19.86 | AMA, USF | No | Genome-wide | Positive (11 and 22) (9.76%) | Diaphragmatic hernia and mild ventriculomegaly on ultrasound; Amniocentesis array: 18.3 Mb duplication of 11q23.3->11qter; 3.42 terminal duplication of 22qter->22q11.21 | Concordant positive |
| 21 | - | 11.43 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (11.24%) | LTFU | LTFU |
| 22 | - | 23.14 | USF, Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (7.54%) | Dandy–Walker malformation on ultrasound; history of several SABs and one healthy child; Postnatal karyotype: 47,XY,+der(22)t(11;22)(q23.3;q11.2) | Concordant positive |
| 23 | - | 11.00 | Tx Hx | Yes (Paternal) | Genome-wide | Negative (6.46%) | Healthy, term delivery. No testing. | Concordant negative |
| 24 | A | 22.29 | USF | No | ‘Traditional’ | Positive (verbal requested) (14.35%) | Multiple anomalies including micrognathia, growth restriction, and possible heart defect. Amniocentesis karyotype showed ‘derivative chromosome 22′ per verbal report from genetic counselor | Concordant positive |
| B | 9.29 | Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (8.49%) | Multiple anomalies including micrognathia, growth restriction, and possible heart defect; No testing; TAB | Likely concordant positive | |
| C | 9.14 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (12.24%) | PGT-SR, Anomalies/complication related to Mendelian disorder diagnosed via WES. Amniocentesis karyotype: WNL. | Concordant negative | |
| 25 | - | 11.57 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (5.50%) | Healthy, term delivery. No testing. Previous child with ES. | Concordant negative |
| 26 | A | 10.00 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Positive (trisomy 22) (5.88%) | SAB at 11 weeks; No testing. | Likely concordant positive |
| B | 10.43 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (5.94%) | Healthy, term delivery. No testing. | Concordant negative | |
| C | 10.29 | AMA, Tx Hx | Yes (Maternal) | Genome-wide | Negative (5.57%) | Healthy, term delivery. No testing. | Concordant negative | |
| 27 | - | 10.86 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (11.24%) | Healthy, term delivery. No testing. Previous child with another chromosome disorder which prompted maternal karyotype. | Concordant negative |
| 28 | - | 11.57 | Tx Hx | Yes (Maternal) | Genome-wide | Negative (6.53%) | Healthy, term delivery. No testing. | Concordant negative |
| 29 | - | 18.86 | USF, Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (7.43%) | Multiple anomalies including micrognathia, diaphragmatic hernia, Dandy–Walker malformation, and two vessel cord; Amniocentesis karyotype and array: 47,XX,+der(22)t(11;22)(q23; q11.2); TAB | Concordant positive |
| 30 | - | 12.14 | Tx Hx | Yes (Maternal) | Genome-wide | Positive (11 and 22) (6.83%) | Ultrasound anomalies including recessed chin and growth restriction; ‘partial T11′ in previous pregnancy | Likely concordant positive |
| 31 | - | 12.00 | AMA, Tx Hx | Yes (Maternal) | ‘Traditional’ | Negative (Verbal requested) (10.03%) | PGT-SR: carrier male; healthy, term delivery. No additional testing. | Concordant negative |
| 32 | - | 10.00 | Tx Hx | Yes (Maternal) | Genome-wide | Positive (45,X) (5.09%) | Normal nuchal translucency on ultrasound; LTFU. | LTFU |
| Study Identifier | cfDNA Positive Reported | Z-Scores | Details of cfDNA Findings |
|---|---|---|---|
| Patient 2-A | Positive (11 and 22) | 11p: 45.3 22q: −4.88 | 116.65 Mb gain of 11p15.5-q23.3 2.75 Mb loss of 22q11.2-q11.2 (DiGeorge region) |
| Patient 6 | Positive (11 and verbal 22) | 11q: 11.1 22q: 5.23 | 18.35 Mb gain of 11q23.3-q25 Verbal report of small ~3 Mb dup on 22q |
| Patient 8 | Positive (11) | 11q: 8.86 | 20.6 Mb gain of 11q23.2-q25 |
| Patient 9-A | Positive (11 and 22) | 11q: 33.16 22q: 8.76 | 18.25 Mb gain of 11q23.3-q25 3.0 Mb gain of 22q11.1-q11.21 |
| Patient 12 | Positive (11 and 22) | 11q: 15.34 22q: 6.37 | 18.1 Mb gain of 11q23.3-q25 2.85 Mb gain of 22q11.1-q11.21 |
| Patient 14-A | Positive (11 and 22) | 11q: 22.82 22q: 6.89 | 18.40 Mb gain of 11q23.3-q25 3.0 Mb gain of 22q11.1-q11.21 |
| Patient 15-B | Positive (11 and 22) | 11q: 18.27 22q: 6.34 | 18.5 Mb gain of 11q23.3-q25 2.8 Mb gain of 22q11.1-q11.2 |
| Patient 15-D | Positive (11) | 11q: 23.56 22q: 8.69 | 18.3 Mb gain of 11q23.3-q25 2.05 Mb gain of 22q11.21-q11.21 noted on retrospective data review |
| Patient 17 | Positive (11 and 22) | 11q: 37.21 22q: 14.32 | 18.25 Mb gain of 11q23.3-q25 3.0 Mb gain of 22q11.1-q11.21 |
| Patient 20 | Positive (11 and 22) | 11q: 19.92 22q: 7.77 | 18.3 Mb gain of 11q23.3-q25 3.55 Mb gain of 22q11.1-q11.21 |
| Patient 22 | Positive (11 and 22) | 11q: 13.76 22q: 4.54 | 18.3 Mb gain of 11q23.3-q25 3.65 Mb gain of 22q11.1-q11.21 |
| Patient 24-A | Positive-verbal review requested of 11/22 | 11q: 22.18 22q: 6.79 | Verbal report of duplications on 11q and 22q in cfDNA sequencing data |
| Patient 24 B | Positive (11 and 22) | 11q: 14.22 22q: 7.68 | 17.75 Mb gain of 11q23.3-q25 4.75 Mb gain of 22q11.1-q11.21 |
| Patient 29 | Positive (11 and 22) | 11q: 20.91 22q: 8.00 | 18.25 Mb gain of 11q23.3-q25 3.55 Mb gain of 22q11.1-q11.21 |
| Patient 30 | Positive (11 and 22) | 11q: 14.25 22q: 4.65 | 18.05 Mb gain of 11q23.3-q25 3.0 Mb gain of 22q11.1-q11.21 |
| Patient 32 | Positive (45,X) | NA | cfDNA data consistent with monosomy X Chromosome 11 and 22 data unremarkable |
| Patient 26-A | Positive (T22) | NA | cfDNA data consistent with full trisomy 22 Chromosome 11 data unremarkable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soster, E.; Dyr, B.; Caldwell, S.; Sussman, A.; Magharyous, H. Prenatal cfDNA Screening for Emanuel Syndrome and Other Unbalanced Products of Conception in Carriers of the Recurrent Balanced Translocation t(11;22): One Laboratory’s Retrospective Experience. Genes 2023, 14, 1924. https://doi.org/10.3390/genes14101924
Soster E, Dyr B, Caldwell S, Sussman A, Magharyous H. Prenatal cfDNA Screening for Emanuel Syndrome and Other Unbalanced Products of Conception in Carriers of the Recurrent Balanced Translocation t(11;22): One Laboratory’s Retrospective Experience. Genes. 2023; 14(10):1924. https://doi.org/10.3390/genes14101924
Chicago/Turabian StyleSoster, Erica, Brittany Dyr, Samantha Caldwell, Amanda Sussman, and Hany Magharyous. 2023. "Prenatal cfDNA Screening for Emanuel Syndrome and Other Unbalanced Products of Conception in Carriers of the Recurrent Balanced Translocation t(11;22): One Laboratory’s Retrospective Experience" Genes 14, no. 10: 1924. https://doi.org/10.3390/genes14101924