
Long-Term Follow-Up of Three Family Members with a Novel NNT Pathogenic Variant Causing Primary Adrenal Insufficiency


 , , , , , , ,
, , , , , , , 
Abstract
:1. Introduction
2. Materials and Methods
- hypocortisolism (morning serum cortisol levels <140 nmol/L) coupled with elevated ACTH levels (>2-times above the upper limit of normal)
- insufficient response of adrenal glands at ACTH stimulation test (serum cortisol levels <500 nmol/L at 30 or 60 min after intravenous administration of ACTH in a standard dose of 15 μg/kg for infants and 125 μg for children <2 years of age).
3. Results
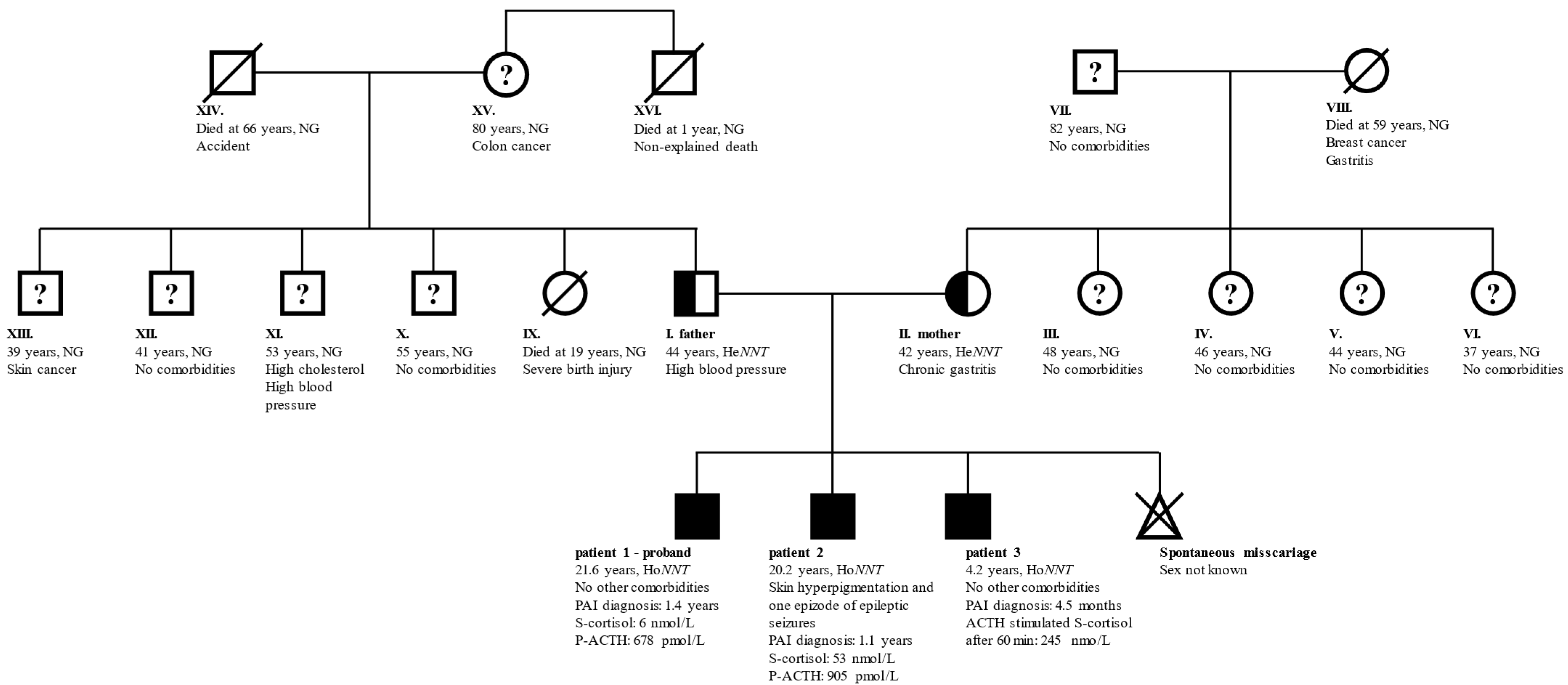
3.1. General Clinical Characteristics
3.2. Genetics
3.3. Hormone Replacement Therapy and Monitoring
3.4. Sex Maturation
3.5. Bone Mineral Density
3.6. Heart Function
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Çamtosun, E.; Dündar, İ.; Akıncı, A.; Kayaş, L.; Çiftçi, N. Pediatric primary adrenal insufficiency: A 21-year single center experience. J. Clin. Res. Pediatr. Endocrinol. 2021, 13, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E. Mechanisms in endocrinology: Update on pathogenesis of primary adrenal insufficiency: Beyond steroid enzyme deficiency and autoimmune adrenal destruction. Eur. J. Endocrinol. 2017, 177, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.; White, P.C. Presentation of primary adrenal insufficiency in childhood. J. Clin. Endocrinol. Metab. 2011, 96, 925–928. [Google Scholar] [CrossRef] [Green Version]
- Kirkgoz, T.; Guran, T. Primary adrenal insufficiency in children: Diagnosis and management. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, M.; Huamei, M.; Jun, Z.; Du, M.; Li, Y.; Chen, Q.; Chen, H.; Song, G. Etiology of primary adrenal insufficiency in children: A 29-year single-center experience. J. Pediatr. Endocrinol. Metab. 2019, 32, 615–622. [Google Scholar] [CrossRef]
- Buonocore, F.; Achermann, J.C. Primary adrenal insufficiency: New genetic causes and their long-term consequences. Clin. Endocrinol. 2020, 92, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Güran, T. Latest insights on the etiology and management of primary adrenal insufficiency in children. J. Clin. Res. Pediatr. Endocrinol. 2018, 9, 9–22. [Google Scholar] [CrossRef]
- Ventura, M.; Serra-Caetano, J.; Cardoso, R.; Dinis, I.; Melo, M.; Carrilho, F.; Mirante, A. The spectrum of pediatric adrenal insufficiency: Insights from 34 years of experience. J. Pediatr. Endocrinol. Metab. 2019, 32, 721–726. [Google Scholar] [CrossRef]
- Roucher-Boulez, F.; Mallet-Motak, D.; Samara-Boustani, D.; Jilani, H.; Ladjouze, A.; Souchon, P.F.; Simon, D.; Nivot, S.; Heinrichs, C.; Ronze, M.; et al. NNT mutations: A cause of primary adrenal insufficiency, oxidative stress and extra-adrenal defects. Eur. J. Endocrinol. 2016, 175, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Meimaridou, E.; Hughes, C.R.; Kowalczyk, J.; Guasti, L.; Chapple, J.P.; King, P.J.; Chan, L.F.; Clark, A.J.L.; Metherell, L.A. Familial glucocorticoid deficiency: New genes and mechanisms. Mol. Cell. Endocrinol. 2013, 371, 195–200. [Google Scholar] [CrossRef]
- Weinberg-Shukron, A.; Abu-Libdeh, A.; Zhadeh, F.; Carmel, L.; Kogot-Levin, A.; Kamal, L.; Kanaan, M.; Zeligson, S.; Renbaum, P.; Levy-Lahad, E.; et al. Combined mineralocorticoid and glucocorticoid deficiency is caused by a novel founder nicotinamide nucleotide transhydrogenase mutation that alters mitochondrial morphology and increases oxidative stress. J. Med. Genet. 2015, 52, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Meimaridou, E.; Kowalczyk, J.; Guasti, L.; Hughes, C.R.; Wagner, F.; Frommolt, P.; Nürnberg, P.; Mann, N.P.; Banerjee, R.; Saka, H.N.; et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat. Genet. 2012, 44, 740–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haycock, G.B.; Schwartz, G.J.; Wisotsky, D.H. Geometric method for measuring body surface area: A height-weight formula validated in infants, children and adults. J. Pediatr. 1978, 93, 62–66. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Allolio, B.; Arlt, W.; Barthel, A.; Don-Wauchope, A.; Hammer, G.D.; Husebye, E.S.; Merke, D.P.; Murad, M.H.; Stratakis, C.A.; et al. Diagnosis and treatment of primary adrenal insufficiency: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2016, 101, 364–389. [Google Scholar] [CrossRef] [Green Version]
- Resende, E.A.M.R.; Lara, B.H.J.; Reis, J.D.; Ferreira, B.P.; Pereira, G.A.; Borges, M.F. Assessment of basal and gonadotropin-releasing hormone-stimulated gonadotropins by immunochemiluminometric and immunofluorometric assays in normal children. J. Clin. Endocrinol. Metab. 2007, 92, 1424–1429. [Google Scholar] [CrossRef]
- Wudy, S.A.; Hartmann, M.F.; Remer, T. Sexual dimorphism in cortisol secretion starts after age 10 in healthy children: Urinary cortisol metabolite excretion rates during growth. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E970–E976. [Google Scholar] [CrossRef] [Green Version]
- Desvignes, J.P.; Bartoli, M.; Delague, V.; Krahn, M.; Miltgen, M.; Béroud, C.; Salgado, D. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018, 46, 545–553. [Google Scholar] [CrossRef]
- Chapman, B.; Kirchner, R.; Pantano, L.; Naumenko, S.; Smet, M.D.; Beltrame, L.; Khotiainsteva, T.; Sytchev, I.; Guimera, R.V.; Kern, J.; et al. bcbio/bcbio-nextgen: (v1.2.9). Available online: https://zenodo.org/record/5781867 (accessed on 19 March 2022).
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Hershkovitz, E.; Arafat, M.; Loewenthal, N.; Haim, A.; Parvari, R. Combined adrenal failure and testicular adrenal rest tumor in a patient with nicotinamide nucleotide transhydrogenase deficiency. J. Pediatr. Endocrinol. Metab. 2015, 28, 9–10. [Google Scholar] [CrossRef]
- Jazayeri, O.; Liu, X.; van Diemen, C.C.; Bakker-van Waarde, W.M.; Sikkema-Raddatz, B.; Sinke, R.J.; Zhang, J.; van Ravenswaaij-Arts, C.M.A. A novel homozygous insertion and review of published mutations in the NNT gene causing familial glucocorticoid deficiency (FGD). Eur. J. Med. Genet. 2015, 58, 642–649. [Google Scholar] [CrossRef]
- Winters, S.J.; Vitaz, T.; Nowacki, M.R.; Craddock, D.C.; Silverman, C. Addison’s Disease and pituitary enlargement. Am. J. Med. Sci. 2015, 349, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ruan, L.; Li, H.; Wang, Q.; Zheng, F.; Wu, F. Addison’s disease with pituitary hyperplasia: A case report and review of the literature. Endocrine 2009, 35, 285–289. [Google Scholar] [CrossRef]
- De Menis, E.; Roncaroli, F.; Calvari, V.; Chiarini, V.; Pauletto, P.; Camerino, G.; Cremonini, N. Corticotroph adenoma of the pituitary in a patient with X-linked adrenal hypoplasia congenita due to a novel mutation of the DAX-1 gene. Eur. J. Endocrinol. 2005, 153, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, H.; Ahmed, S.F.; Bensing, S.; Beun, J.G.; Brösamle, M.; Chifu, I.; van der Grinten, H.C.; Clemente, M.; Falhammar, H.; Hahner, S.; et al. Therapy options for adrenal insufficiency and recommendations for the management of adrenal crisis. Endocrine 2021, 71, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Oprea, A.; Bonnet, N.C.G.; Pollé, O.; Lysy, P.A. Novel insights into glucocorticoid replacement therapy for pediatric and adult adrenal insufficiency. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018818821294. [Google Scholar] [CrossRef] [PubMed]
- Jódar, E.; Valdepeñas, M.P.; Martinez, G.; Jara, A.; Hawkins, F. Long-term follow-up of bone mineral density in Addison’s disease. Clin. Endocrinol. 2003, 58, 617–620. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, F.S.; Castell, E.C.; Marco, F.C.; Ruiz, M.J.; Rico, J.A.Q.; Roca, A.P.N. Influence of weight status on bone mineral content measured by DXA in children. BMC Pediatr. 2021, 21, 185. [Google Scholar] [CrossRef]
- Domazetovic, V. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209. [Google Scholar] [CrossRef]
- Fujisawa, Y.; Napoli, E.; Wong, S.; Song, G.; Yamaguchi, R.; Matsui, T.; Nagasaki, K.; Ogata, T.; Giulivi, C. Impact of a novel homozygous mutation in nicotinamide nucleotide transhydrogenase on mitochondrial DNA integrity in a case of familial glucocorticoid deficiency. BBA Clin. 2015, 3, 70–78. [Google Scholar] [CrossRef] [Green Version]
- El Amrousy, D.; El-Afify, D.; Shabana, A. Relationship between bone turnover markers and oxidative stress in children with type 1 diabetes mellitus. Pediatr. Res. 2021, 89, 878–881. [Google Scholar] [CrossRef]
- Williams, J.L.; Hall, C.L.; Meimaridou, E.; Metherell, L.A. Loss of Nnt increases expression of oxidative phosphorylation complexes in C57BL/6J hearts. Int. J. Mol. Sci. 2021, 22, 6101. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.L.; Paudyal, A.; Awad, S.; Nicholson, J.; Grzesik, D.; Botta, J.; Meimaridou, E.; Maharaj, A.V.; Stewart, M.; Tinker, A.; et al. Mylk3 null C57BL/6N mice develop cardiomyopathy, whereas Nnt null C57BL/6J mice do not. Life Sci. Alliance 2020, 3, e201900593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.; Chen, C.H.; Ursell, P.; Huang, T.T. Genetic modifier of mitochondrial superoxide dismutase-deficient mice delays heart failure and prolongs survival. Mamm. Genome 2010, 21, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, M.N.; Davis, E.E.; Choi, W.Y.; Dickson, A.; Martinez, H.R.; Wang, M.; Dinh, H.; Muzny, D.; Pignatelli, R.; Katsanis, N.; et al. Loss of function mutations in NNT are associated with left ventricular noncompaction. Circ. Cardiovasc. Genet. 2015, 8, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Sheeran, F.L.; Rydström, J.; Shakhparonov, M.I.; Pestov, N.B.; Pepe, S. Diminished NADPH transhydrogenase activity and mitochondrial redox regulation in human failing myocardium. Biochim. Biophys. Acta 2010, 1797, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Steroid Abbreviation | Steroid Name | Value (μg/L) | Reference Range (μg/L) |
|---|---|---|---|
| A5-3b,17a | 5-androstene-3β,17α-diol | 13 | 0–40 |
| DHEA | 5-androstene-3β-ol-17-on (dehydroepiandrosterone) | 0 | 0–0 |
| A5-3b,17b | 5-androstene-3β,17β-diol | 0 | 0–0 |
| 11-O-An | 5α-androstane-3α-ol-11,17-dione (11-oxo-androsterone) | 20 | 0–40 |
| Po-5b,3a | 5β-pregnane-3α,17α-diol-20-one (17a-OH-pregnanolone) | 11 | 10–80 |
| 11-OH-An | 5α-androstane-3α,11β-diol-17-one (11-hydroxy-androsterone) | 22 | 20–70 |
| 11-OH-Et | 5β-androstane-3α,11β-diol-17-one (11-hydroxy-etiocholanolone) | 0 | 0–40 |
| Po-5a,3a | 5α-pregnane-3α,17α-diol-20-one | 8 | 5–50 |
| 16a-OH-DHEA | 5-androstene-3β,16α-diol-17-one | 548 | 0–750 |
| PD | 5β-pregnane-3α,20α-diol (pregnanediol) | 0 | 0–250 |
| PT | 5β-pregnane-3α,17α,20α-triol (pregnanetriol) | 6 | 0–105 |
| P5D | 5-pregnene-3β,20α-diol (pregnenediol) | 62 | 0–75 |
| A5T-16a | 5-androstene-3β,16α,17β-triol (androstenetriol-16α) | 439 | 0–480 |
| THS | 5β-pregnane-3α,17α,21-triol-20-one (tetrahydro-11-deoxycortisol) | 168 | 33–280 |
| 11-O-PT | 5β-pregnane-3α,17α,20α-triol-11-one (11-oxo-pregnanetriol) | 0 | 0–0 |
| P5T-17a | 5-pregnene-3β,17α,20α-triol (pregnenetriol-17α) | 40 | 0–140 |
| THE | 5β-pregnane-3α,17 α,21-triol-11,20-dione (tetrahydro-cortisone) | 869 | 465–1570 |
| THA | 5β-pregnane-3α,21-diol-11,20-dione | 36 | 0–230 |
| THB | 5β-pregnane-3α,11β,21-triol-20-one (tetrahydro-corticosteron) | 0 | 0–250 |
| a-THB | 5α-pregnane-3α,11β,21-triol-20-one (allo-tetrahydro-corticosteron) | 92 | 0–100 |
| THF | 5β-pregnane-3α,11β,17α,21-tetrol-20-one (tetrahydro-cortisol) | 122 | 10–200 |
| a-THF | 5α-pregnane-3α,11β,17α,21-tetrol-20-one (allo-tetrahydro-cortisol) | 629 | 10–1000 |
| a-Cl | 5β-pregnane-3α,17α,20α,21-tetrol-11-one (α-Cortolone) | 184 | 113–350 |
| b-C | 5β-pregnane-3a,11b,17a,20β,21-pentol (β-Cortol) | 40 | 10–100 |
| b-Cl | 5β-pregnane-3α,17α,20β,21-tetrol-11-one (β-Cortolone) | 182 | 30–800 |
| a-C | 5β-pregnane-3α,11β,17α,20α,21-pentol (α-Cortol) | 0 * | 150–525 |
| F | 4-pregnene-11β,17α,21-triol-3,20-dione (cortisol) | 58 | 20–100 |
| 6b-OH F | 4-pregnene-6β,11β,17α,21-tetrol-3,20-dione (6β-hydroxycortisol) | 110 | 0–660 |
| 20a-DHF | 4-pregnene-11β,17α,20α,21-tetrol-3-one (20α-dihydrocortisol) | 28 | 0–100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasovec, T.; Sikonja, J.; Zerjav Tansek, M.; Debeljak, M.; Ilovar, S.; Trebusak Podkrajsek, K.; Bertok, S.; Tesovnik, T.; Kovac, J.; Suput Omladic, J.; et al. Long-Term Follow-Up of Three Family Members with a Novel NNT Pathogenic Variant Causing Primary Adrenal Insufficiency. Genes 2022, 13, 717. https://doi.org/10.3390/genes13050717
Krasovec T, Sikonja J, Zerjav Tansek M, Debeljak M, Ilovar S, Trebusak Podkrajsek K, Bertok S, Tesovnik T, Kovac J, Suput Omladic J, et al. Long-Term Follow-Up of Three Family Members with a Novel NNT Pathogenic Variant Causing Primary Adrenal Insufficiency. Genes. 2022; 13(5):717. https://doi.org/10.3390/genes13050717
Chicago/Turabian StyleKrasovec, Tjasa, Jaka Sikonja, Mojca Zerjav Tansek, Marusa Debeljak, Sasa Ilovar, Katarina Trebusak Podkrajsek, Sara Bertok, Tine Tesovnik, Jernej Kovac, Jasna Suput Omladic, and et al. 2022. "Long-Term Follow-Up of Three Family Members with a Novel NNT Pathogenic Variant Causing Primary Adrenal Insufficiency" Genes 13, no. 5: 717. https://doi.org/10.3390/genes13050717