
Prevalent Pathogenic Variants of ATP7B in Chinese Patients with Wilson’s Disease: Geographical Distribution and Founder Effect







Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Genotype Analysis
2.3. Haplotype Analysis
2.4. Statistical Analysis
3. Results
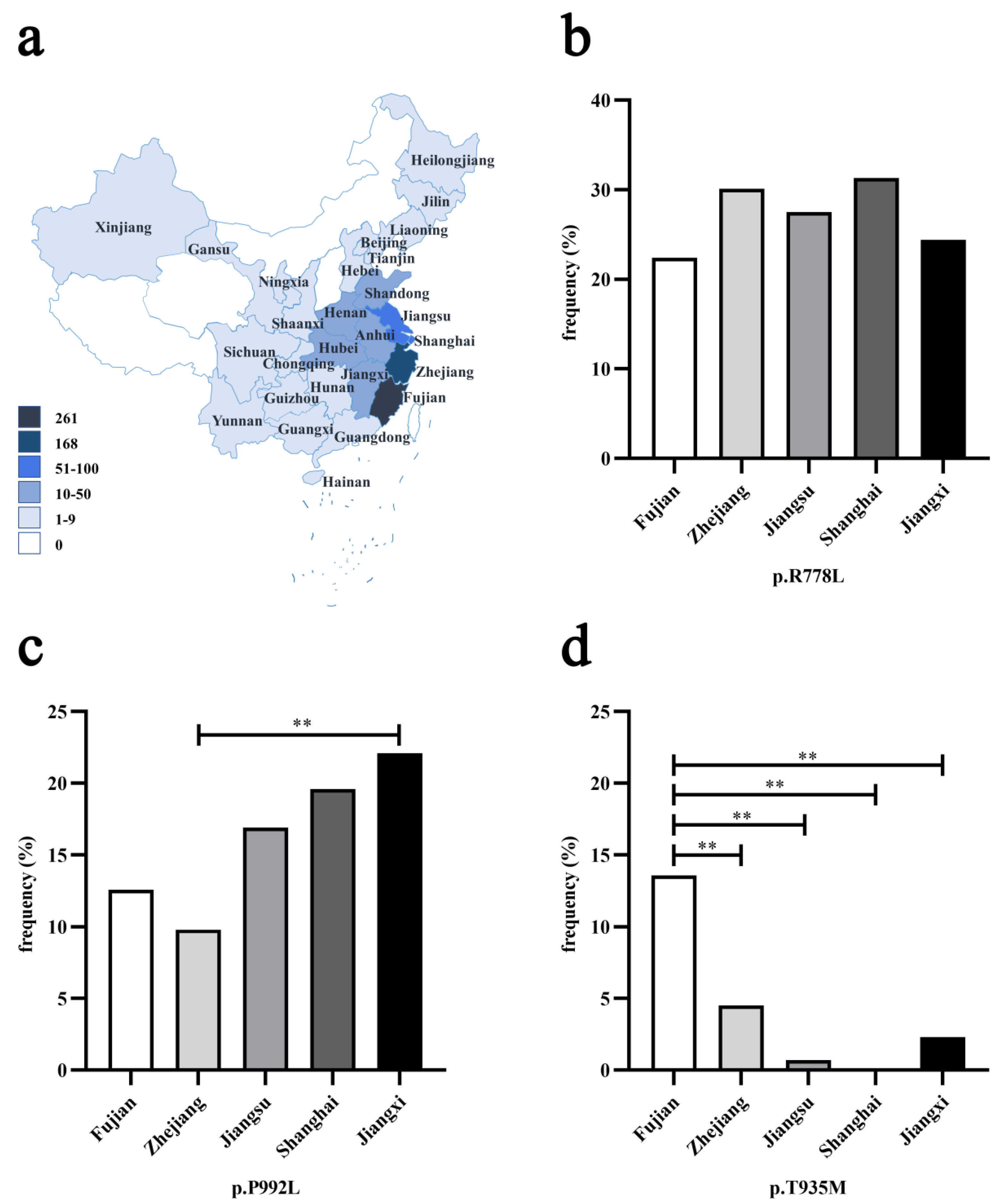
3.1. Geographical Distribution of Three Prevalent Pathogenic Variants
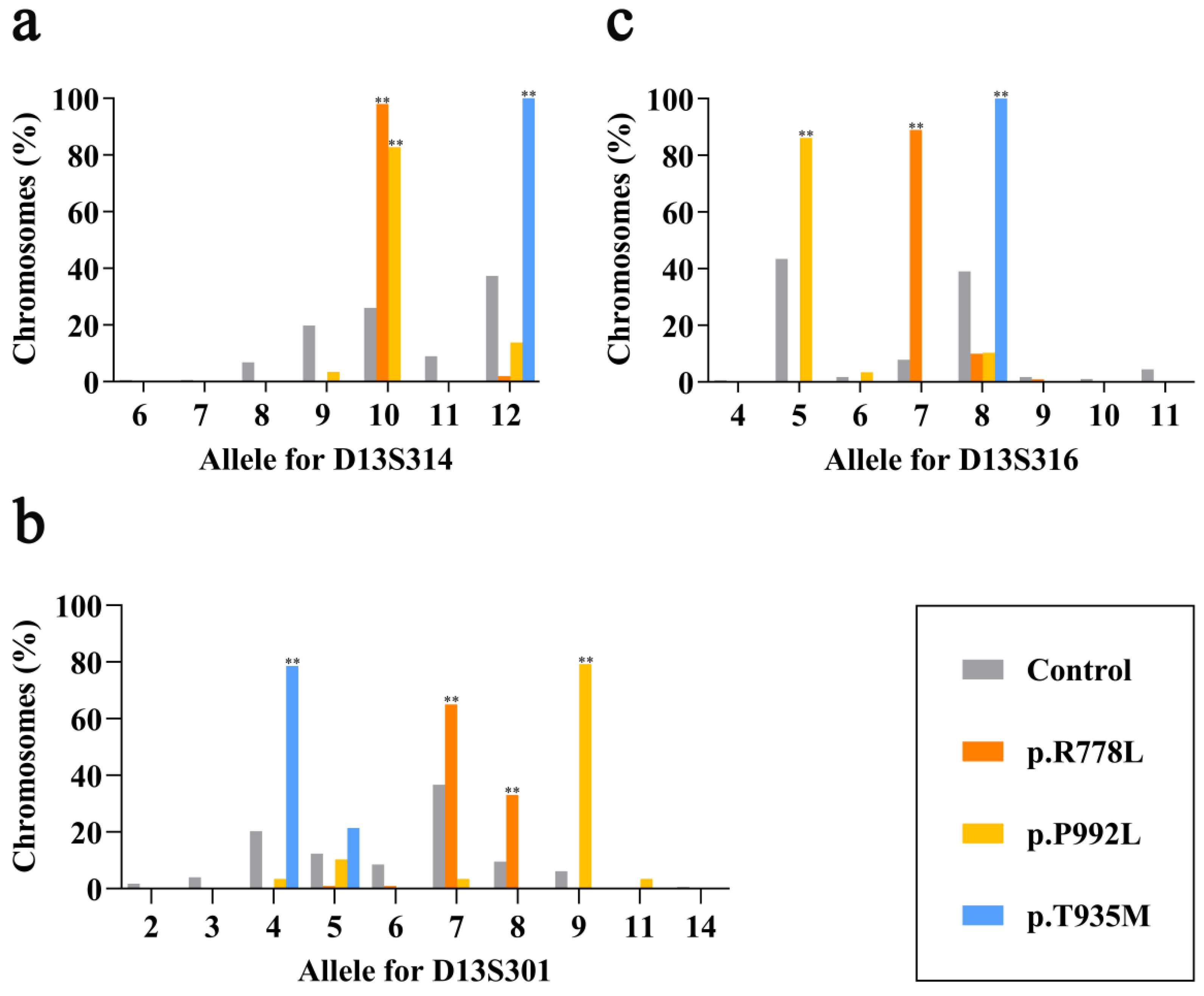
3.2. Linkage Disequilibrium at Three Markers for Three Prevalent Pathogenic Variants
3.3. Haplotype Association of Three Prevalent Pathogenic Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers. 2018, 4, 1–20. [Google Scholar] [CrossRef]
- Ferenci, P.; Caca, K.; Loudianos, G.; Mieli-Vergani, G.; Tanner, S.; Sternlieb, I.; Schilsky, M.; Cox, D.; Berr, F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003, 23, 139–142. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, R.M.; Yang, G.M.; Yu, H.; Xu, W.Q.; Xie, J.J.; Zhang, Y.; Chen, Y.C.; Ni, W.; Wu, Z.Y. Role for Biochemical Assays and Kayser-Fleischer Rings in Diagnosis of Wilson’s Disease. Clin. Gastroenterol. Hepatol. 2021, 19, 590–596. [Google Scholar] [CrossRef]
- Coffey, A.J.; Durkie, M.; Hague, S.; McLay, K.; Emmerson, J.; Lo, C.; Klaffke, S.; Joyce, C.J.; Dhawan, A.; Hadzic, N.; et al. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013, 136, 1476–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gialluisi, A.; Incollu, S.; Pippucci, T.; Lepori, M.B.; Zappu, A.; Loudianos, G.; Romeo, G. The homozygosity index (HI) approach reveals high allele frequency for Wilson disease in the Sardinian population. Eur. J. Hum. Genet. 2013, 21, 1308–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.J.; Wu, Z.Y. Wilson’s Disease in China. Neurosci. Bull. 2017, 33, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 1993, 5, 327–337. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Heiny, M.E.; Gitlin, J.D. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem. Biophys. Res. Commun. 1993, 197, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yu, L.; Wang, N. Gene diagnosis for hepatolenticular degeneration by genetic linkage analysis with four short tandem repeat polymorphisms. Zhonghua Yi Xue Za Zhi 1996, 76, 578–581. [Google Scholar] [PubMed]
- Thomas, G.R.; Bull, P.C.; Roberts, E.A.; Walshe, J.M.; Cox, D.W. Haplotype studies in Wilson disease. Am. J. Hum. Genet. 1994, 54, 71–78. [Google Scholar] [PubMed]
- Thomas, G.R.; Roberts, E.A.; Walshe, J.M.; Cox, D.W. Haplotypes and mutations in Wilson disease. Am. J. Hum. Genet. 1995, 56, 1315–1319. [Google Scholar] [PubMed]
- Figus, A.; Angius, A.; Loudianos, G.; Bertini, C.; Dessi, V.; Loi, A.; Deiana, M.; Lovicu, M.; Olla, N.; Sole, G.; et al. Molecular pathology and haplotype analysis of Wilson disease in Mediterranean populations. Am. J. Hum. Genet. 1995, 57, 1318–1324. [Google Scholar] [PubMed]
- Loudianos, G.; Dessì, V.; Lovicu, M.; Angius, A.; Kanavakis, E.; Tzetis, M.; Kattamis, C.; Manolaki, N.; Vassiliki, G.; Karpathios, T.; et al. Haplotype and mutation analysis in Greek patients with Wilson disease. Eur. J. Hum. Genet. 1998, 6, 487–491. [Google Scholar] [CrossRef]
- Firneisz, G.; Lakatos, P.L.; Szalay, F.; Polli, C.; Glant, T.T.; Ferenci, P. Common mutations of ATP7B in Wilson disease patients from Hungary. Am. J. Med. Genet. 2002, 108, 23–28. [Google Scholar] [CrossRef]
- Drayna, D. Founder mutations. Sci. Am. 2005, 293, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Dedoussis, G.V. Geographic distribution of ATP7B mutations in Wilson disease. Ann. Hum. Biol. 2016, 43, 1–8. [Google Scholar] [CrossRef]
- Chang, I.J.; Hahn, S.H. The genetics of Wilson disease. Handb. Clin. Neurol. 2017, 142, 19–34. [Google Scholar] [PubMed] [Green Version]
- Singh, N.; Kallollimath, P.; Shah, M.H.; Kapoor, S.; Bhat, V.K.; Viswanathan, L.G.; Nagappa, M.; Bindu, P.S.; Taly, A.B.; Sinha, S.; et al. Genetic analysis of ATP7B in 102 south Indian families with Wilson disease. PLoS ONE 2019, 14, e0215779. [Google Scholar] [CrossRef]
- Nanji, M.S.; Nguyen, V.T.; Kawasoe, J.H.; Inui, K.; Endo, F.; Nakajima, T.; Anezaki, T.; Cox, D.W. Haplotype and mutation analysis in Japanese patients with Wilson disease. Am. J. Hum. Genet. 1997, 60, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Wu, J.Y.; Tsai, F.J.; Kodama, H.; Abe, T.; Yang, C.F.; Tsai, C.H. Molecular analysis of Wilson disease in Taiwan: Identification of one novel mutation and evidence of haplotype-mutation association. J. Hum. Genet. 2000, 45, 275–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, C.M.; Lam, C.W.; Tam, S.; Lai, C.L.; Chan, L.Y.; Fan, S.T.; Lau, Y.L.; Lai, S.T.; Yuen, P.; Hui, J.; et al. Mutational analysis of 65 Wilson disease patients in Hong Kong Chinese: Identification of 17 novel mutations and its genetic heterogeneity. J. Hum. Genet. 2008, 53, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.H.; Kim, J.W.; Seo, J.K. Haplotype analysis and possible founder effect at the R778L mutation of the ATP7B gene in Korean patients with Wilson’s disease. Korean J. Hepatol. 2009, 15, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Ni, W.; Chen, W.J.; Wan, B.; Zhao, G.X.; Shi, Z.Q.; Zhang, Y.; Wang, N.; Yu, L.; Xu, J.F.; et al. Spectrum and Classification of ATP7B Variants in a Large Cohort of Chinese Patients with Wilson’s Disease Guides Genetic Diagnosis. Theranostics 2016, 6, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Yu, H.; Wang, R.M.; Xie, J.J.; Ni, W.; Zhang, Y.; Dong, Y.; Wu, Z.Y. Contribution of intragenic deletions to mutation spectrum in Chinese patients with Wilson’s disease and possible mechanism underlying ATP7B gross deletions. Parkinsonism Relat. Disord. 2019, 62, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [PubMed] [Green Version]
- Li, K.; Zhang, W.M.; Lin, S.; Wen, L.; Wang, Z.F.; Xie, D.; Wei, M.; Qiu, Z.Q.; Dai, Y.; Lin, M.C.; et al. Mutational analysis of ATP7B in north Chinese patients with Wilson disease. J. Hum. Genet. 2013, 58, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Huang, Y.; Liu, A.; Diao, S.; Yu, Q.; Peng, Z.; Hong, M. Mutational characterization of ATP7B gene in 103 Wilson’s disease patients from Southern China: Identification of three novel mutations. Neuroreport 2014, 25, 1075–1080. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Variant | Group 1 | Haplotype | No. of Chromosomes (%) | ||||
|---|---|---|---|---|---|---|---|
| D13S314 | D13S301 | D13S316 | WD | Control (n = 177) | |||
| p.R778L (n = 100) | A | A1 | 10 | 7 | 7 | 57(57.0) | 4(2.3) |
| A2 | 10 | 7 | 8 | 7(7.0) | 3(1.7) | ||
| A3 | 10 | 8 | 7 | 30(30.0) | 8(4.5) | ||
| A4 | 10 | 8 | 8 | 3(3.0) | 2(1.1) | ||
| Subtotal | 97(97.0) ** | 17(9.6) | |||||
| B | 10 | 7 | 9 | 1(1.0) | 0(0.0) | ||
| C | C1 | 12 | 5 | 7 | 1(1.0) | 0(0.0) | |
| C2 | 12 | 6 | 7 | 1(1.0) | 0(0.0) | ||
| p.P992L (n = 29) | D | 9 | 7 | 5 | 1(3.4) | 13(7.3) | |
| E | E1 | 10 | 9 | 5 | 20(69.0) | 6(3.4) | |
| E2 | 10 | 9 | 6 | 1(3.4) | 0(0.0) | ||
| Subtotal | 21(72.4) ** | 6(3.4) | |||||
| F | 10 | 9 | 8 | 2(6.9) | 0(0.0) | ||
| G | 10 | 11 | 5 | 1(3.4) | 0(0.0) | ||
| H | H1 | 12 | 4 | 5 | 1(3.4) | 5(2.8) | |
| H2 | 12 | 5 | 5 | 2(6.9) | 8(4.5) | ||
| I | 12 | 5 | 8 | 1(3.4) | 9(5.1) | ||
| p.T935M (n = 14) | J | J1 | 12 | 4 | 8 | 11(78.6) | 18(10.2) |
| J2 | 12 | 5 | 8 | 3(21.4) | 9(5.1) | ||
| Subtotal | 14(100.0) ** | 27(15.3) | |||||
| Study | Variant | Haplotype | No. of Chromosomes | ||
|---|---|---|---|---|---|
| D13S314 | D13S301 | D13S316 | |||
| Japan [20] | p.R778L | 5 | 5 | 6 | 1 |
| 5 | 7 | 4 | 2 | ||
| 5 | 7 | 5 | 1 | ||
| 5 | 7 | 5.5 | 4 | ||
| 5 | 7 | 7 | 1 | ||
| p.P992L | 6 | 9 | 4 | 3 | |
| Taiwan, China [21] | p.R778L | 8 | 4 | 4 | 4 |
| 8 | 4 | 5.5 | 4 | ||
| p.P992L | 8.5 | 6.5 | 2 | 2 | |
| 8.5 | 6.5 | 5.5 | 3 | ||
| Hong Kong, China [22] | p.R778L | 10 | 4 | 7 | 5 |
| 10 | 5 | 7 | 2 | ||
| 10 | 6 | 7 | 5 | ||
| 10 | 9 | 7 | 1 | ||
| 13 | 3 | 7 | 1 | ||
| p.P992L | 10 | 7 | 4 | 7 | |
| 10 | 7 | 8 | 1 | ||
| p.T935M | 13 | 1 | 8 | 1 | |
| 13 | 3 | 8 | 1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.-M.; Wang, R.-M.; Xia, N.; Zheng, Z.-W.; Dong, Y.; Wu, Z.-Y. Prevalent Pathogenic Variants of ATP7B in Chinese Patients with Wilson’s Disease: Geographical Distribution and Founder Effect. Genes 2021, 12, 336. https://doi.org/10.3390/genes12030336
Yang G-M, Wang R-M, Xia N, Zheng Z-W, Dong Y, Wu Z-Y. Prevalent Pathogenic Variants of ATP7B in Chinese Patients with Wilson’s Disease: Geographical Distribution and Founder Effect. Genes. 2021; 12(3):336. https://doi.org/10.3390/genes12030336
Chicago/Turabian StyleYang, Guo-Min, Rou-Min Wang, Nan Xia, Zi-Wei Zheng, Yi Dong, and Zhi-Ying Wu. 2021. "Prevalent Pathogenic Variants of ATP7B in Chinese Patients with Wilson’s Disease: Geographical Distribution and Founder Effect" Genes 12, no. 3: 336. https://doi.org/10.3390/genes12030336