
Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies

 , ,
, , 
 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic DNA Extraction and Quantification
2.2. SNP-Array Analysis
2.3. Whole Exome Sequencing (WES)
3. Results
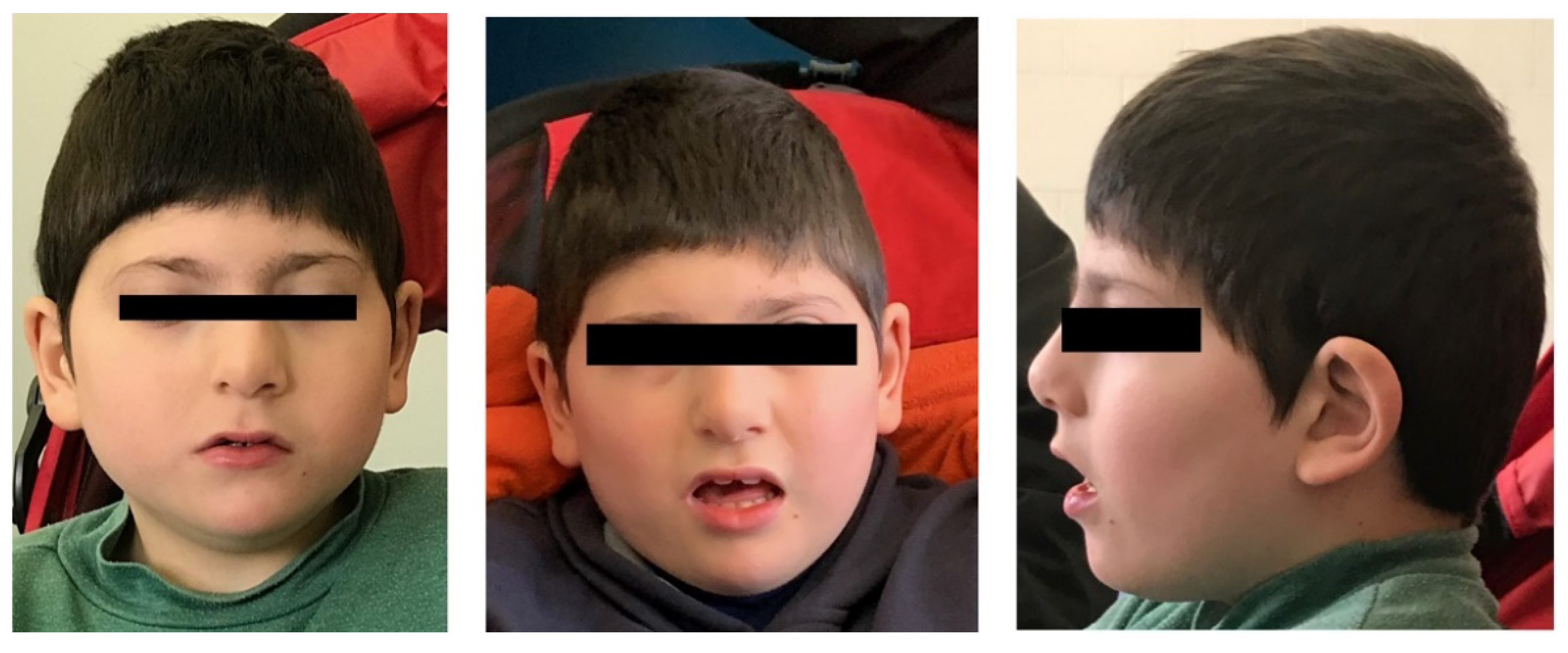
3.1. Clinical Description
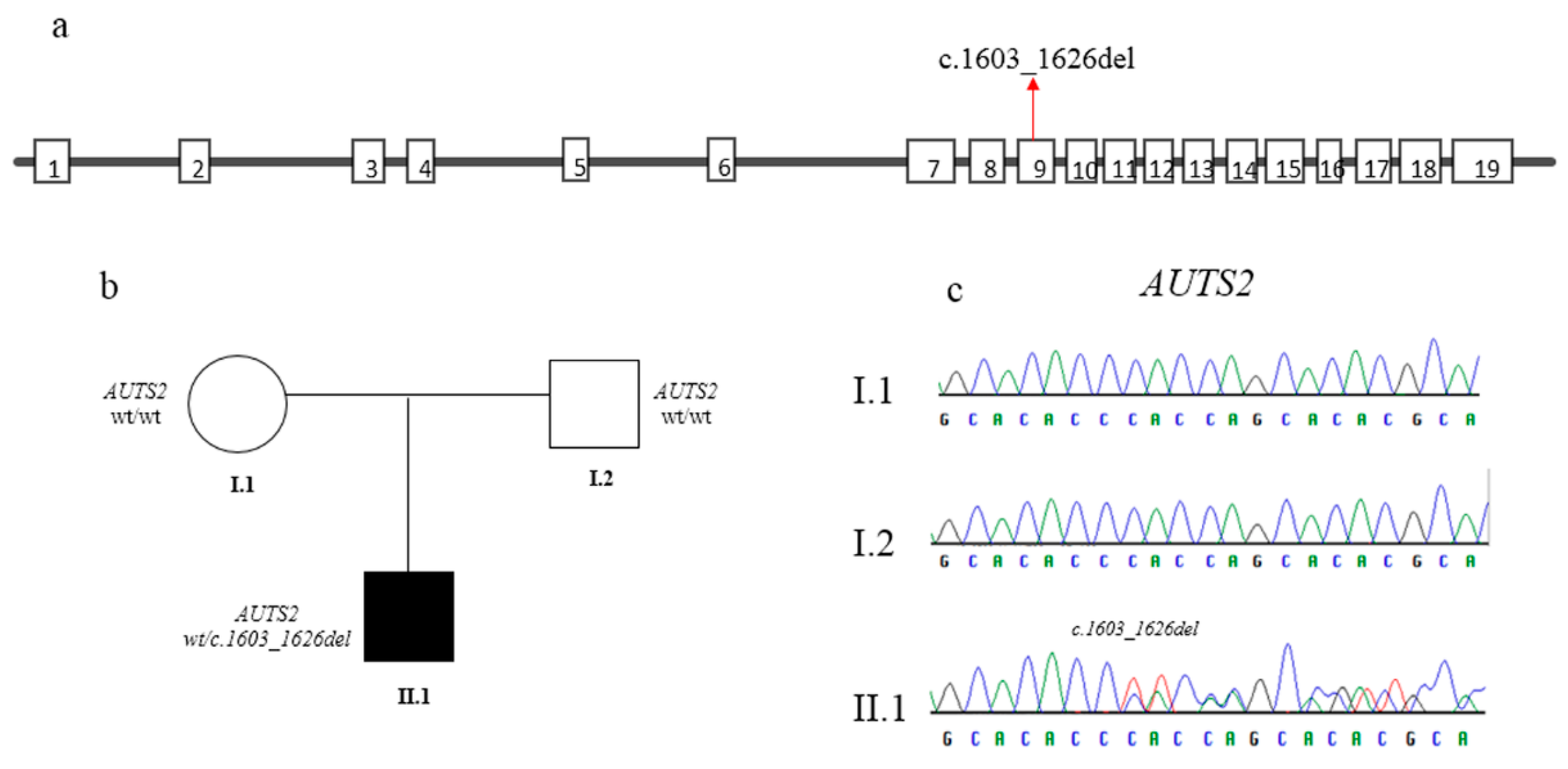
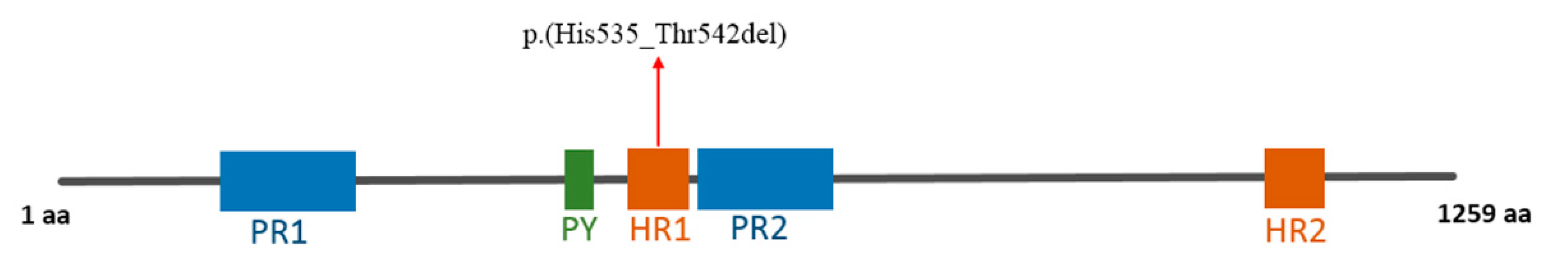
3.2. Molecular Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitchell, K.J. The genetics of neurodevelopmental disease. Curr. Opin. Neurobiol. 2011, 21, 197–203. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Palumbo, O.; Fichera, M.; Palumbo, P.; Rizzo, R.; Mazzolla, E.; Cocuzza, D.M.; Carella, M.; Mattina, T. TBR1 is the candidate gene for intellectual disability in patients with a 2q24.2 interstitial deletion. Am. J. Med. Genet. Part A 2014, 164, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, O.; Palumbo, P.; Di Muro, E.; Cinque, L.; Petracca, A.; Carella, M.; Castori, M. A Private 16q24.2q24.3 Microduplication in a Boy with Intellectual Disability, Speech Delay and Mild Dysmorphic Features. Genes 2020, 11, 707. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Genovesi, M.L.; Guadagnolo, D.; Marchionni, E.; Giovannetti, A.; Traversa, A.; Panzironi, N.; Bernardo, S.; Palumbo, P.; Petrizzelli, F.; Carella, M.; et al. GDF5 mutation case report and a systematic review of molecular and clinical spectrum: Expanding current knowledge on genotype-phenotype correlations. Bone 2021, 12, 115803. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. KAVIAR: An accessible system for testing SNV novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Non-synonymous and Splice Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Kumar, M.; Zeke, A.; Lang, B.; Bely, B.; Chemes, L.B.; Davey, N.E.; Deng, Z.; et al. The eukaryotic linear motif resource—2018 update. Nucleic Acids Res. 2018, 46, D428–D434. [Google Scholar] [CrossRef]
- Sultana, R.; Yu, C.E.; Yu, J.; Munson, J.; Chen, D.; Hua, W.; Estes, A.; Cortes, F.; de la Barra, F.; Yu, D.; et al. Identification of a Novel Gene on Chromosome 7q11.2 Interrupted by a Translocation Breakpoint in a Pair of Autistic Twins. Genomics 2002, 80, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Haliburton, G.D.; Eckalbar, W.L.; Oren, I.; Nishizaki, S.; Murphy, K.; Pollard, K.S.; Birnbaum, R.Y.; Ahituv, N. Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl. Psychiatry 2014, 4, e431. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Lee, P.; Stafford, J.M.; von Schimmelmann, M.; Schaefer, A.; Reinberg, D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 2014, 516, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, K.; Hoshino, M. Neuronal Migration and AUTS2 Syndrome. Brain Sci. 2017, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Quinn, J.F.; Weiss, L.A. An eQTL mapping approach reveals that rare variants in the SEMA5A regulatory network impact autism risk. Hum. Mol. Genet. 2013, 22, 2960–2972. [Google Scholar] [CrossRef] [PubMed]
- Bedogni, F.; Hodge, R.D.; Elsen, G.E.; Nelson, B.R.; Daza, R.A.; Beyer, R.P.; Bammler, T.K.; Rubenstein, J.L.; Hevner, R.F. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. USA 2010, 107, 13129–13134. [Google Scholar] [CrossRef] [Green Version]
- Amarillo, I.E.; Li, W.L.; Li, X.; Vilain, E.; Kantarci, S. De novo single exon deletion of AUTS2 in a patient with speech and language disorder: A review of disrupted AUTS2 and further evidence for its role in neurodevelopmental disorders. Am. J. Med. Genet. A 2014, 164, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Jolley, A.; Corbett, M.; McGregor, L.; Waters, W.; Brown, S.; Nicholl, J.; Yu, S. De novo intragenic deletion of the autism susceptibility candidate 2 (AUTS2) gene in a patient with developmental delay: A case report and literature review. Am. J. Med. Genet. A 2013, 161, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; van de Kamp, J.; Vasudevan, P.; Morton, J.; Smets, K.; Kleefstra, T.; de Munnik, S.A.; Schuurs-Hoeijmakers, J.; Ceulemans, B.; Zollino, M.; et al. A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J. Med. Genet. 2016, 53, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xu, Y.H.; Wei, S.G.; Zhang, H.B.; Fu, D.K.; Feng, Z.F.; Guan, F.L.; Zhu, Y.S.; Li, S.B. Association study identifying a new susceptibility gene (AUTS2) for schizophrenia. Int. J. Mol. Sci. 2014, 15, 19406–19416. [Google Scholar] [CrossRef] [Green Version]
- Elia, J.; Gai, X.; Xie, H.M.; Perin, J.C.; Geiger, E.; Glessner, J.T.; D’Arcy, M.; deBerardinis, R.; Frackelton, E.; Kim, C.; et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 2010, 15, 637–646. [Google Scholar] [CrossRef]
- Girirajan, S.; Brkanac, Z.; Coe, B.P.; Baker, C.; Vives, L.; Vu, T.H.; Shafer, N.; Bernier, R.; Ferrero, G.B.; Silengo, M.; et al. Relative burden of large cnvs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011, 7, e1002334. [Google Scholar] [CrossRef]
- Mefford, H.C.; Muhle, H.; Ostertag, P.; von Spiczak, S.; Buysse, K.; Baker, C.; Franke, A.; Malafosse, A.; Genton, P.; Thomas, P.; et al. Genome-wide copy number variation in epilepsy: Novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010, 6, e1000962. [Google Scholar] [CrossRef] [PubMed]
- Myung, W.; Kim, J.; Lim, S.W.; Shim, S.; Won, H.H.; Kim, S.; Kim, S.; Lee, M.S.; Chang, H.S.; Kim, J.W.; et al. A genome-wide association study of antidepressant response in koreans. Transl. Psychiatry 2015, 5, e633. [Google Scholar] [CrossRef] [Green Version]
- Schumann, G.; Coin, L.J.; Lourdusamy, A.; Charoen, P.; Berger, K.H.; Stacey, D.; Desrivieres, S.; Aliev, F.A.; Khan, A.A.; Amin, N.; et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc. Natl. Acad. Sci. USA 2011, 108, 7119–7124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, M.; Wang, J.C.; Wetherill, L.; Le, N.; Bertelsen, S.; Hinrichs, A.L.; Budde, J.; Agrawal, A.; Bucholz, K.; Dick, D.; et al. A meta-analysis of two genome-wide association studies to identify novel loci for maximum number of alcoholic drinks. Hum. Genet. 2013, 132, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Dang, W.; Zhang, Q.; Zhu, Y.S.; Lu, X.Y. The evidence for the contribution of the autism susceptibility candidate 2 (auts2) gene in heroin dependence susceptibility. J. Mol. Neurosci. 2014, 54, 811–819. [Google Scholar] [CrossRef]
- McCarthy, S.E.; Gillis, J.; Kramer, M.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-David, E.; Granot-Hershkovitz, E.; Monderer-Rothkoff, G.; Lerer, E.; Levi, S.; Yaari, M.; Ebstein, R.P.; Yirmiya, N.; Shifman, S. Identification of a functional rare variant in autism using genome-wide screen for monoallelic expression. Hum. Mol. Genet. 2011, 20, 3632–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.L.; Zou, Y.S.; Maher, T.A.; Newton, S.; Milunsky, J.M. A de novo balanced translocation breakpoint truncating the autism susceptibility candidate 2 (AUTS2) gene in a patient with autism. Am. J. Med. Genet. A 2010, 152A, 2112–2114. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Bakkaloglu, B.; O’Roak, B.J.; Louvi, A.; Gupta, A.R.; Abelson, J.F.; Morgan, T.M.; Chawarska, K.; Klin, A.; Ercan-Sencicek, A.G.; Stillman, A.A.; et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2008, 82, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012, 27, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Chromosome | Start | End | Reference Allele | Alternative Allele | Genotype | Gene | Nucleotide Change | Amino Acid Change | dbSNP ID | gnomAD_exome Allele Count | TOPMED Allele Count | ExAC_ALL Allele Count |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 70231220 | 70231244 | AGCACCAGCACACCCACCAGCACAC | A | Heterozygous | AUTS2 NM_015570 | c.1603_1626del | p.(His535_Thr542del) | N.A. | N.A. | N.A. | N.A. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, P.; Di Muro, E.; Accadia, M.; Benvenuto, M.; Di Giacomo, M.C.; Castellana, S.; Mazza, T.; Castori, M.; Palumbo, O.; Carella, M. Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies. Genes 2021, 12, 229. https://doi.org/10.3390/genes12020229
Palumbo P, Di Muro E, Accadia M, Benvenuto M, Di Giacomo MC, Castellana S, Mazza T, Castori M, Palumbo O, Carella M. Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies. Genes. 2021; 12(2):229. https://doi.org/10.3390/genes12020229
Chicago/Turabian StylePalumbo, Pietro, Ester Di Muro, Maria Accadia, Mario Benvenuto, Marilena Carmela Di Giacomo, Stefano Castellana, Tommaso Mazza, Marco Castori, Orazio Palumbo, and Massimo Carella. 2021. "Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies" Genes 12, no. 2: 229. https://doi.org/10.3390/genes12020229