
Genotype Phenotype Correlation in Dent Disease 2 and Review of the Literature: OCRL Gene Pleiotropism or Extreme Phenotypic Variability of Lowe Syndrome?

 , , and
, , and
Abstract
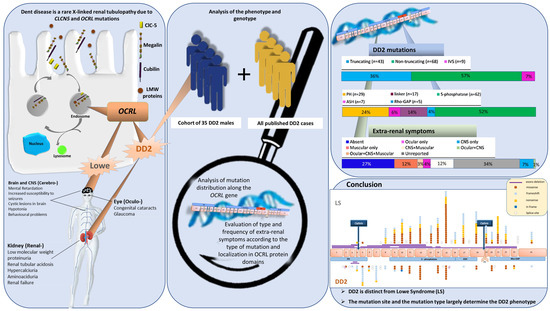
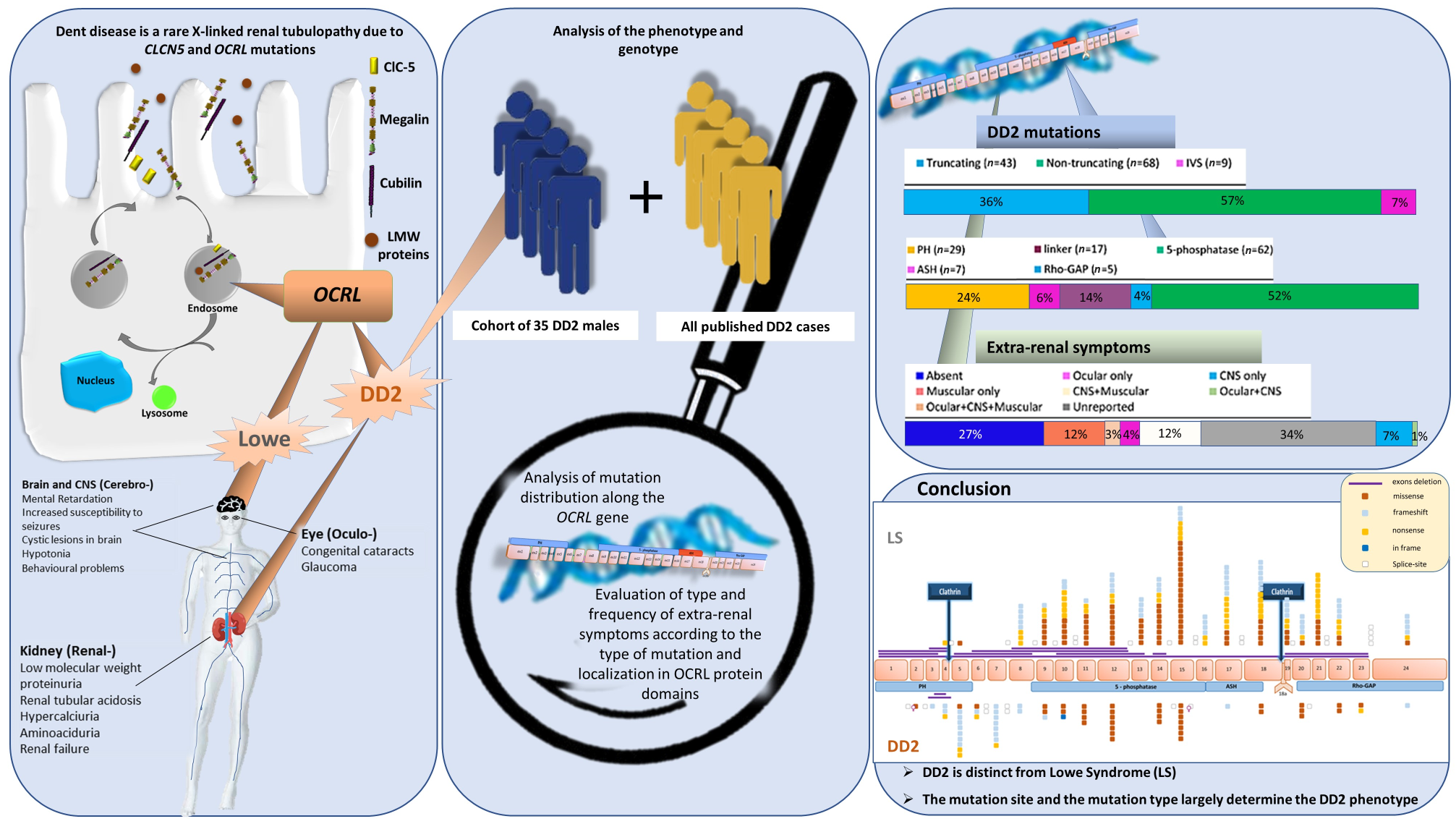
:
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Assessment of Phenotypic and Genetic Features
2.3. Statistical Analysis
3. Results
3.1. Clinical Data of Our Cohort of DD2 Patients
3.2. Histopathological Data of Our Cohort of DD2 Patients
3.3. Genetic Data of Our Cohort of DD2 Patients
3.4. Mapping DD2 Mutations in the OCRL Gene and OCRL Domains
3.5. Genotype–Phenotype Correlation in Dent Disease 2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, S.E.; Black, G.C.; Lloyd, S.E.; Hatchwell, E.; Wrong, O.; Thakker, R.V.; Craig, I.W. Isolation and Partial Characterization of a Chloride Channel Gene Which Is Expressed in Kidney and Is a Candidate for Dent’s Disease (an X-Linked Hereditary Nephrolithiasis). Hum. Mol. Genet. 1994, 3, 2053–2059. [Google Scholar]
- Hoopes, R.R.; Shrimpton, A.E.; Knohl, S.J.; Hueber, P.; Hoppe, B.; Matyus, J.; Simckes, A.; Tasic, V.; Toenshoff, B.; Suchy, S.F.; et al. Dent Disease with Mutations in OCRL1. Am. J. Hum. Genet. 2005, 76, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attree, O.; Olivos, I.M.; Okabe, I.; Bailey, L.C.; Nelson, D.L.; Lewis, R.A.; McInnes, R.R.; Nussbaum, R.L. The Lowe’s Oculocerebrorenal Syndrome Gene Encodes a Protein Highly Homologous to Inositol Polyphosphate-5-Phosphatase. Nature 1992, 358, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Böckenhauer, D.; Bökenkamp, A.; Nuutinen, M.; Unwin, R.; Van’t Hoff, W.; Sirimanna, T.; Vrljicak, K.; Ludwig, M. Novel OCRL Mutations in Patients with Dent-2 Disease. J. Pediatr. Genet. 2012, 1, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Zaniew, M.; Bökenkamp, A.; Kolbuc, M.; La Scola, C.; Baronio, F.; Niemirska, A.; Szczepanska, M.; Bürger, J.; La Manna, A.; Miklaszewska, M.; et al. Long-Term Renal Outcome in Children with OCRL Mutations: Retrospective Analysis of a Large International Cohort. Nephrol. Dial. Transplant. 2018, 33, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hichri, H.; Rendu, J.; Monnier, N.; Coutton, C.; Dorseuil, O.; Poussou, R.V.; Baujat, G.; Blanchard, A.; Nobili, F.; Ranchin, B.; et al. From Lowe Syndrome to Dent Disease: Correlations between Mutations of the OCRL1 Gene and Clinical and Biochemical Phenotypes. Hum. Mutat. 2011, 32, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Matteis, M.A.; Staiano, L.; Emma, F.; Devuyst, O. The 5-Phosphatase OCRL in Lowe Syndrome and Dent Disease 2. Nat. Rev. Nephrol. 2017, 13, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Oltrabella, F.; Pietka, G.; Ramirez, I.B.-R.; Mironov, A.; Starborg, T.; Drummond, I.A.; Hinchliffe, K.A.; Lowe, M. The Lowe Syndrome Protein OCRL1 Is Required for Endocytosis in the Zebrafish Pronephric Tubule. PLoS Genet. 2015, 11, e1005058. [Google Scholar] [CrossRef] [Green Version]
- Gianesello, L.; Del Prete, D.; Anglani, F.; Calò, L.A. Genetics and Phenotypic Heterogeneity of Dent Disease: The Dark Side of the Moon. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Mao, Y.; Balkin, D.M.; Zoncu, R.; Erdmann, K.S.; Tomasini, L.; Hu, F.; Jin, M.M.; Hodsdon, M.E.; De Camilli, P. A PH Domain within OCRL Bridges Clathrin-Mediated Membrane Trafficking to Phosphoinositide Metabolism. EMBO J. 2009, 28, 1831–1842. [Google Scholar] [CrossRef]
- Pirruccello, M.; De Camilli, P. Inositol 5-Phosphatases: Insights from the Lowe Syndrome Protein OCRL. Trends Biochem. Sci. 2012, 37, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Pook, M.A.; Wrong, O.; Wooding, C.; Norden, A.G.; Feest, T.G.; Thakker, R.V. Dent’s Disease, a Renal Fanconi Syndrome with Nephrocalcinosis and Kidney Stones, Is Associated with a Microdeletion Involving DXS255 and Maps to Xp11.22. Hum. Mol. Genet. 1993, 2, 2129–2134. [Google Scholar] [CrossRef]
- Erdmann, K.S.; Mao, Y.; McCrea, H.J.; Zoncu, R.; Lee, S.; Paradise, S.; Modregger, J.; Biemesderfer, D.; Toomre, D.; De Camilli, P. A Role of the Lowe Syndrome Protein OCRL in Early Steps of the Endocytic Pathway. Dev. Cell 2007, 13, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Utsch, B.; Bökenkamp, A.; Benz, M.R.; Besbas, N.; Dötsch, J.; Franke, I.; Fründ, S.; Gok, F.; Hoppe, B.; Karle, S.; et al. Novel OCRL1 Mutations in Patients with the Phenotype of Dent Disease. Am. J. Kidney Dis. 2006, 48, 942–e1. [Google Scholar] [CrossRef]
- Bökenkamp, A.; Ludwig, M. The Oculocerebrorenal Syndrome of Lowe: An Update. Pediatr. Nephrol. 2016, 31, 2201–2212. [Google Scholar] [CrossRef] [Green Version]
- Cogal, A.G.; Arroyo, J.; Shah, R.J.; Reese, K.J.; Walton, B.N.; Reynolds, L.M.; Kennedy, G.N.; Seide, B.M.; Senum, S.R.; Baum, M.; et al. Comprehensive Genetic Analysis Reveals Complexity of Monogenic Urinary Stone Disease. Kidney Int. Rep. 2021. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- National Kidney Foundation K/DOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. Am. J. Kidney Dis. 2002, 39, S1–S266.
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Bothwell, S.P.; Chan, E.; Bernardini, I.M.; Kuo, Y.-M.; Gahl, W.A.; Nussbaum, R.L. Mouse Model for Lowe Syndrome/Dent Disease 2 Renal Tubulopathy. J. Am. Soc. Nephrol. 2011, 22, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Loi, M. Lowe Syndrome. Orphanet J. Rare Dis. 2006, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Zhang, H.; Cao, S.; Xiao, H.; Yao, Y. Dent’s Disease Complicated by Nephrotic Syndrome: A Case Report. Intractable Rare Dis. Res. 2016, 5, 297–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, T.; Komoda, F.; Miura, K.; Takita, J.; Shimadzu, M.; Matsuyama, T.; Ashida, A.; Igarashi, T. Japanese Dent Disease Has a Wider Clinical Spectrum than Dent Disease in Europe/USA: Genetic and Clinical Studies of 86 Unrelated Patients with Low-Molecular-Weight Proteinuria. Nephrol. Dial. Transpl. 2014, 29, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, E.; Yoshida, A.; Miyama, Y.; Yabuuchi, T.; Kajiho, Y.; Kanda, S.; Miura, K.; Oka, A.; Harita, Y. Incomplete Cryptic Splicing by an Intronic Mutation of OCRL in Patients with Partial Phenotypes of Lowe Syndrome. J. Hum. Genet. 2020, 65, 831–839. [Google Scholar] [CrossRef]
- Ye, Q.; Shen, Q.; Rao, J.; Zhang, A.; Zheng, B.; Liu, X.; Shen, Y.; Chen, Z.; Wu, Y.; Hou, L.; et al. Multicenter Study of the Clinical Features and Mutation Gene Spectrum of Chinese Children with Dent Disease. Clin. Genet. 2019. [Google Scholar] [CrossRef]
- Cho, H.Y.; Lee, B.H.; Choi, H.J.; Ha, I.S.; Choi, Y.; Cheong, H.I. Renal Manifestations of Dent Disease and Lowe Syndrome. Pediatr. Nephrol. 2008, 23, 243–249. [Google Scholar] [CrossRef]
- Park, E.; Choi, H.J.; Lee, J.M.; Ahn, Y.H.; Kang, H.G.; Choi, Y.M.; Park, S.J.; Cho, H.Y.; Park, Y.-H.; Lee, S.J.; et al. Muscle Involvement in Dent Disease 2. Pediatr. Nephrol. 2014, 29, 2127–2132. [Google Scholar] [CrossRef]
- Recker, F.; Zaniew, M.; Böckenhauer, D.; Miglietti, N.; Bökenkamp, A.; Moczulska, A.; Rogowska-Kalisz, A.; Laube, G.; Said-Conti, V.; Kasap-Demir, B.; et al. Characterization of 28 Novel Patients Expands the Mutational and Phenotypic Spectrum of Lowe Syndrome. Pediatr. Nephrol. 2015, 30, 931–943. [Google Scholar] [CrossRef]
- Zheng, B.; Chen, Q.; Wang, C.; Zhou, W.; Chen, Y.; Ding, G.; Jia, Z.; Zhang, A.; Huang, S. Whole-Genome Sequencing Revealed an Interstitial Deletion Encompassing OCRL and SMARCA1 Gene in a Patient with Lowe Syndrome. Mol. Genet Genomic. Med. 2019, 7, e876. [Google Scholar] [CrossRef] [Green Version]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational Readthrough Potential of Natural Termination Codons in Eucaryotes--The Impact of RNA Sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Burge, C.B. Splicing Regulation: From a Parts List of Regulatory Elements to an Integrated Splicing Code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Suchy, S.F.; Bernard, D.; MacDonald, M.; Crawley, J.; Grinberg, A.; Wynshaw-Boris, A.; Westphal, H.; Nussbaum, R.L. Functional Overlap between Murine Inpp5b and Ocrl1 May Explain Why Deficiency of the Murine Ortholog for OCRL1 Does Not Cause Lowe Syndrome in Mice. J. Clin. Investig. 1998, 101, 2042–2053. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Sign | DD2 (n = 35) | |
|---|---|---|
| LMWP | Y | 27 (100.0) |
| N | 0 (0.0) | |
| Albuminuria | Y | 12 (92.3) |
| N | 1 (7.7) | |
| Proteinuria | Y | 29 (96.7) |
| N | 1 (3.3) | |
| Nephrotic syndrome | Y | 8 (29.6) |
| N | 19 (70.4) | |
| eGFR | 90.00 [44.00, 143.00] | |
| CKD | 0 | 1 (4.3) |
| 1 | 13 (56.5) | |
| 2 | 8 (34.8) | |
| 3 | 1 (4.3) | |
| Glucosuria | Y | 8 (30.8) |
| N | 18 (69.2) | |
| Aminoaciduria | Y | 7 (43.8) |
| N | 9 (56.2) | |
| Hematuria | Y | 10 (58.8) |
| N | 7 (41.2) | |
| Hypercalciuria | Y | 20 (95.2) |
| N | 1 (4.8) | |
| TRP | Normal | 7 (63.6) |
| <85% | 4 (36.4) | |
| Hypophosphatemia | Y | 5 (22.7) |
| N | 17 (77.3) | |
| Nephrocalcinosis | Y | 13 (41.9) |
| N | 18 (58.1) | |
| Urolithiasis | Y | 10 (29.4) |
| N | 24 (70.6) | |
| Rickets | Y | 3 (10.3) |
| N | 26 (89.7) | |
| Hypertension | Y | 1 (3.8) |
| N | 25 (96.2) | |
| Family history | Y | 11 (50.0) |
| N | 11 (50.0) | |
| Extrarenal symptoms | Y | 22 (68.8) |
| N | 10 (31.2) | |
| Ocular symptoms | Y | 9 (39.1) |
| N | 14 (12.2) | |
| CNS symptoms | Y | 13 (46.4) |
| N | 15 (53.6) | |
| Muscular abnomalities | Y | 14 (63.6) |
| N | 8 (36.4) | |
| Growth | Normal | 2 (7.1) |
| Below 50 percent | 2 (7.1) | |
| Below 25 percent | 4 (14.3) | |
| Below 10 percent | 2 (7.1) | |
| Below 5 percent | 18 (64.3) |
| DD2 Cohort (n = 7) | Literature (n = 14) | |
|---|---|---|
| Age at Biopsy | 4–11 yr 11 mo | 3–14 yr |
| Glomerular histology | ||
| Number of glomeruli | 8–75 | |
| Normal | 1/7 | 2/14 |
| Unspecified sclerosis | ||
| FGGS | 4/7 | 1/14 |
| FSGS | 0/7 | 3/14 |
| Mesangial proliferation | 0/7 | 6/14 |
| Minor glomerular abnormalities | 0/7 | 1/14 |
| Periglomerular fibrosis | 0/7 | |
| Expansion of mesangial matrix | 2/7 | 1/14 |
| Immature glomeruli | 1/7 | |
| Adherence to Bowman capsule | 0/7 | 1/14 |
| Other (Perihyliar hyalinosis, ECM hyperplasia, Collapsed tuft, Podocytes’ hypertrophy, glom lesion) | 2/7 | 2/14 |
| Tubular histology | ||
| Normal | 2/7 | 2/6 |
| Tubular atrophy | 4/7 | 1/6 |
| Interstitial fibrosis | 4/7 | 1/6 |
| Calcification | 0/7 | |
| Tubulointerstitial lesions | 0/7 | 2/6 |
| Calcium deposits | 0/7 | |
| Intratubular proteinaceous casts | 0/7 | 1/6 |
| Interstitial inflammation | 1/7 | |
| Nephrocalcinosis | 0/7 | |
| Vascular degeneration | 1/7 | |
| Interstitial mononuclear cells infiltrate | 0/7 | |
| Interstitial lymphocytes infiltrate | 0/7 | 1/6 |
| Acute tubular necrosis | 0/7 | 1/6 |
| Other (Cortical fibrosis, Interstitial chronic inflammation, chronic tubulointerstitial nephropathy with ischemic renal damage) | 1/7 | |
| Immunofluorescence | ||
| Negative | 3/7 | 2/2 |
| IgM deposits | 1/7 | |
| C3 deposits | 1/7 | |
| OTHER | 3/7 | |
| TEM | ||
| Normal | 0/6 | |
| Foot process effacement | 5/6 | 2/2 |
| Electrondense deposits | 1/6 | |
| Mesangial proliferation | 2/6 | |
| Global sclerosis | 2/6 | |
| Irregular GBM folding | 2/6 | 1/2 |
| Other (Expansion of mesangial matrix, Microvillarization of podocytes) | 4/6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianesello, L.; Arroyo, J.; Del Prete, D.; Priante, G.; Ceol, M.; Harris, P.C.; Lieske, J.C.; Anglani, F. Genotype Phenotype Correlation in Dent Disease 2 and Review of the Literature: OCRL Gene Pleiotropism or Extreme Phenotypic Variability of Lowe Syndrome? Genes 2021, 12, 1597. https://doi.org/10.3390/genes12101597
Gianesello L, Arroyo J, Del Prete D, Priante G, Ceol M, Harris PC, Lieske JC, Anglani F. Genotype Phenotype Correlation in Dent Disease 2 and Review of the Literature: OCRL Gene Pleiotropism or Extreme Phenotypic Variability of Lowe Syndrome? Genes. 2021; 12(10):1597. https://doi.org/10.3390/genes12101597
Chicago/Turabian StyleGianesello, Lisa, Jennifer Arroyo, Dorella Del Prete, Giovanna Priante, Monica Ceol, Peter C. Harris, John C. Lieske, and Franca Anglani. 2021. "Genotype Phenotype Correlation in Dent Disease 2 and Review of the Literature: OCRL Gene Pleiotropism or Extreme Phenotypic Variability of Lowe Syndrome?" Genes 12, no. 10: 1597. https://doi.org/10.3390/genes12101597