Fine-Mapping of Sorghum Stay-Green QTL on Chromosome10 Revealed Genes Associated with Delayed Senescence


 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Parents and Fine-Mapping Population Development
2.2. Field Evaluation of Stay-Green
2.3. Estimation of Senescence and Grain Yield
2.4. Statistical Analysis
2.5. Heritability
- S.E. = Standard Error
- N = Number of individuals
- Error MS = Error mean sum of square.
- CV = Coefficient of Variation
- Error MS = Error mean sum of square
- GM = Grand mean.
2.6. Genotyping by Sequencing (GBS), SNP Calling, and Annotation
2.7. Distance Matrix and Principal Coordinate Analysis (PCA)
2.8. SSR-SNP High-Resolution Linkage Map Construction and QTL Analysis
2.9. Stg QTL Cluster Analysis and Fine-Mapping
2.10. Marker Trait Associations (MTAs)
2.11. Candidate Genes Identification
2.12. RNA Extraction, Candidate Gene Primer Designing, and (qRT-PCR) Analysis
2.13. Data Availability Statement
3. Results
3.1. Recombinant Selection for Advancement
3.2. Traits Variation, ANOVA, and Correlation
3.3. Genotyping by Sequencing (GBS) and SNP Annotation
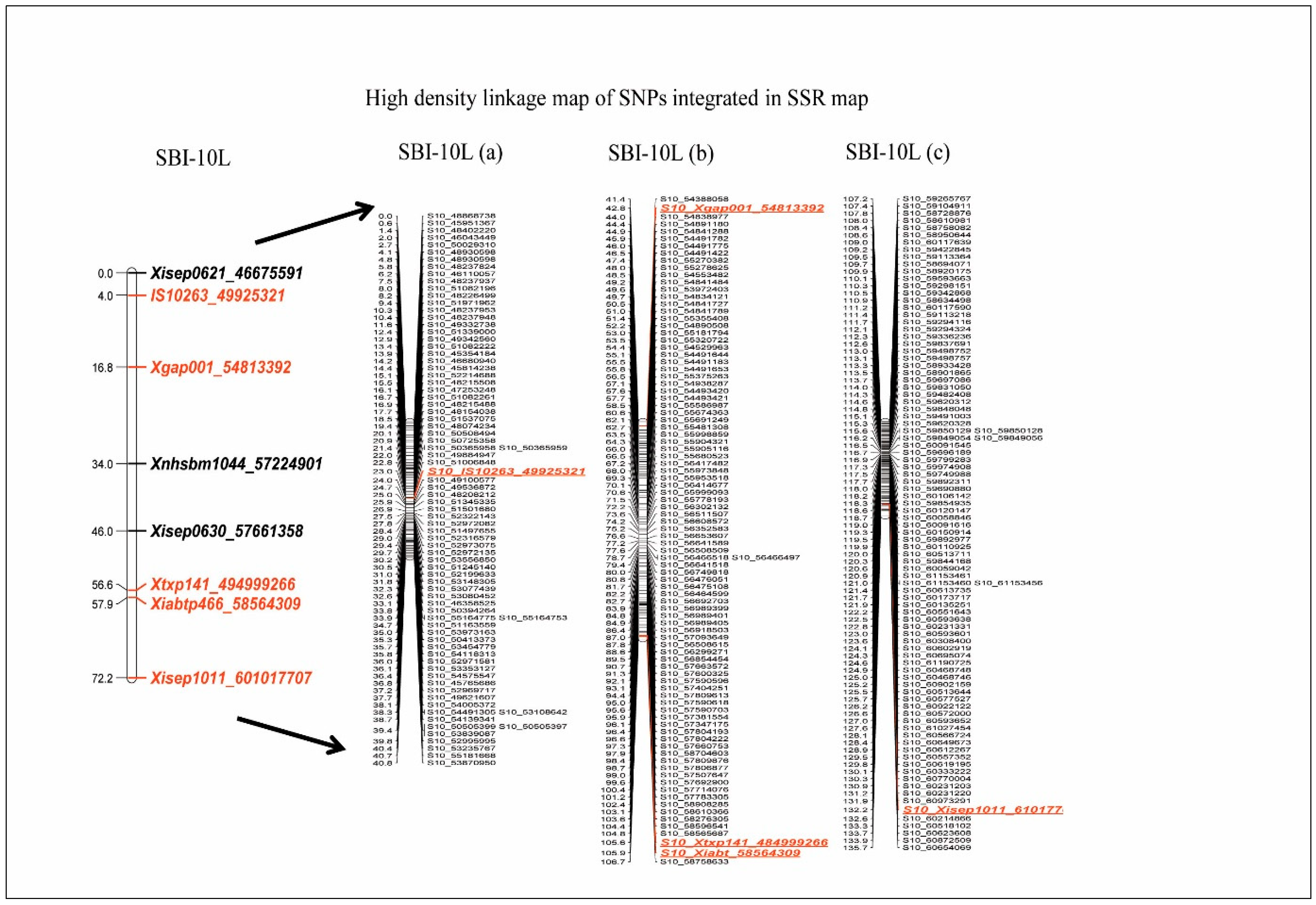
3.4. Development of High-Resolution Genetic Linkage Map by Integrating GBS–SNP into SSR Map of the SBI-10L
3.5. Stay-Green QTL Mapping and QTL Co-Localization
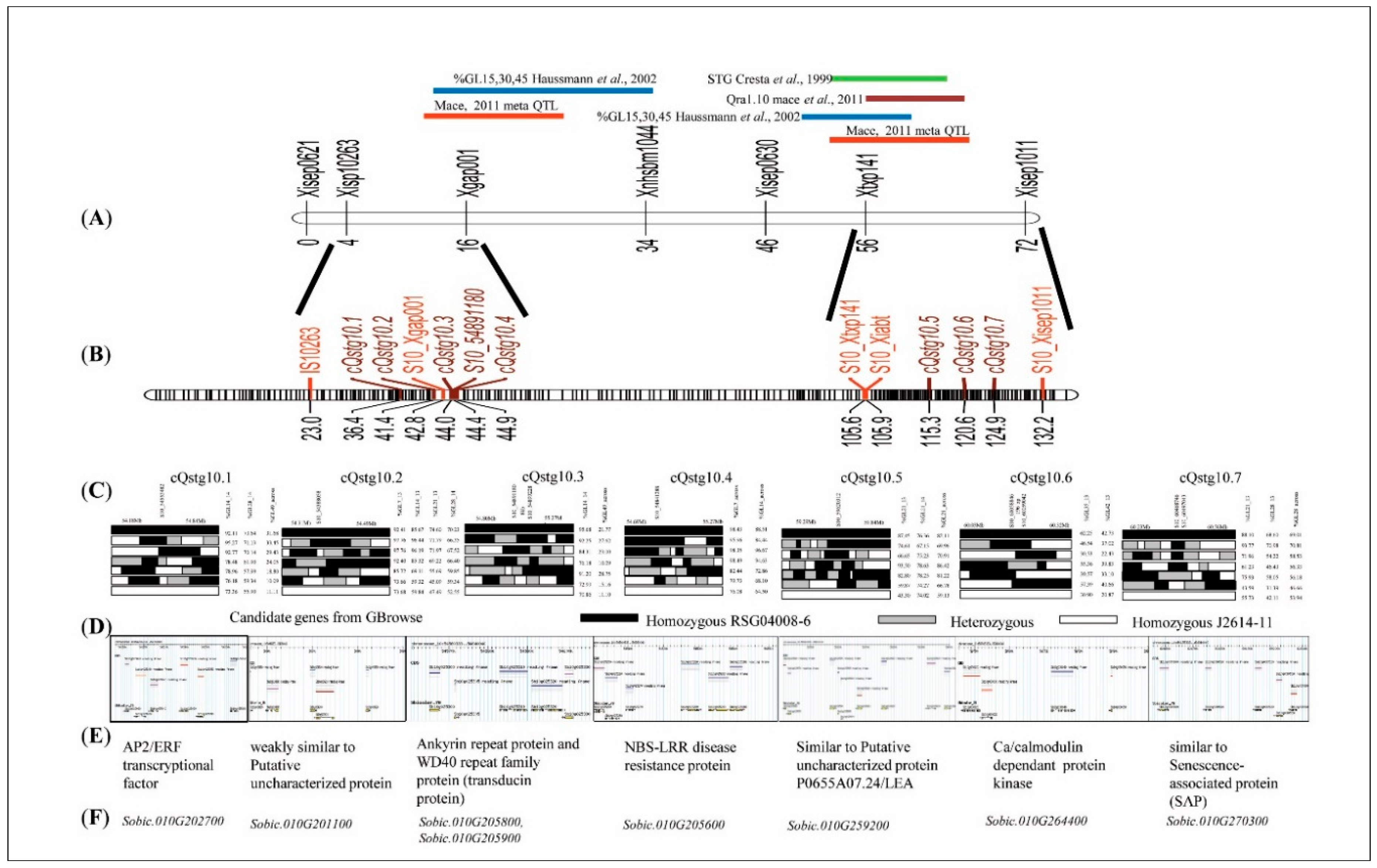
3.6. Fine-Mapping of the Stay-Green QTL Clusters
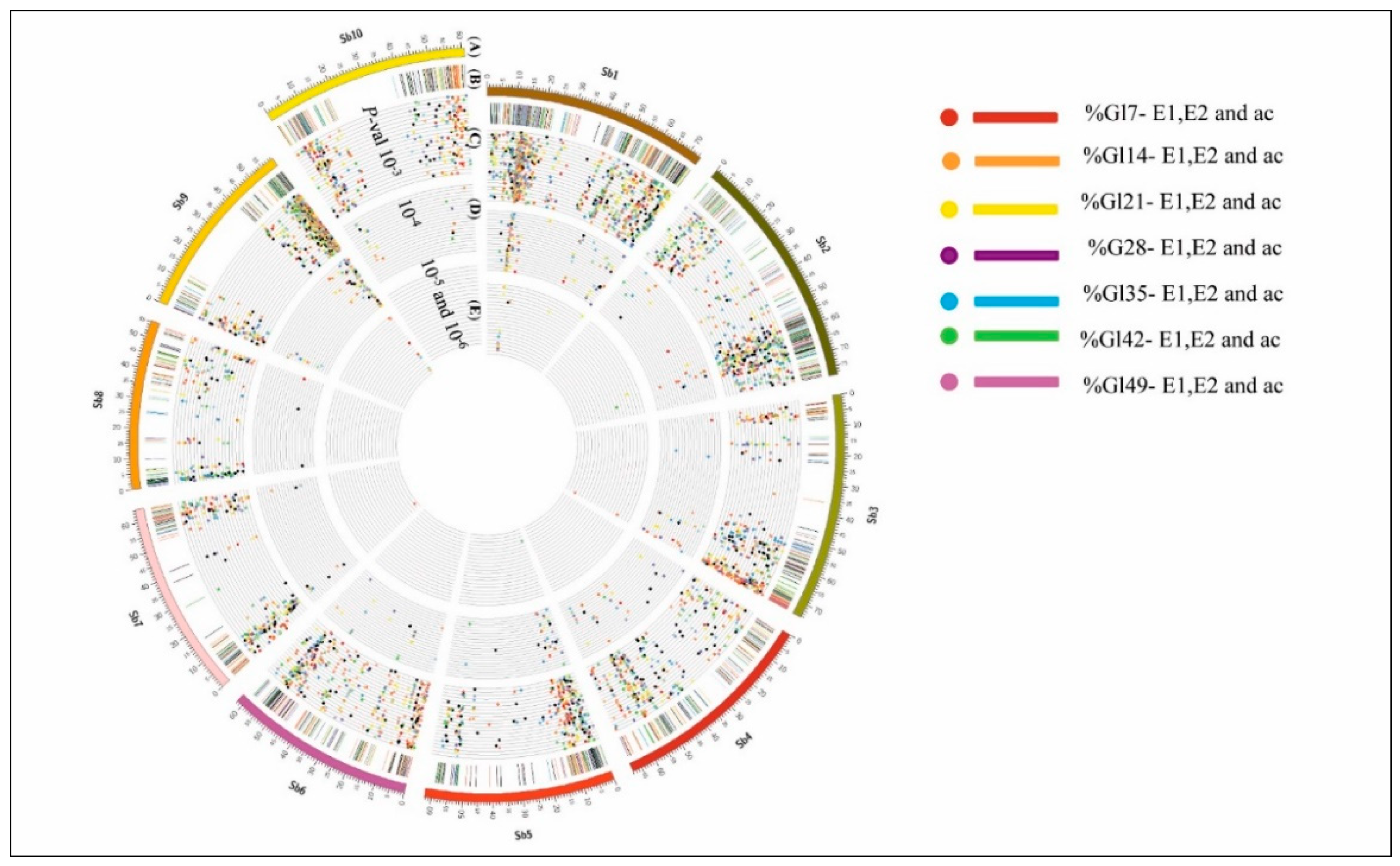
3.7. MTAs for Stay-Green Traits
3.8. Fine-Mapping and MTAs Revealed Putative Candidate Genes for the Stay-Green Trait on SBI-10L
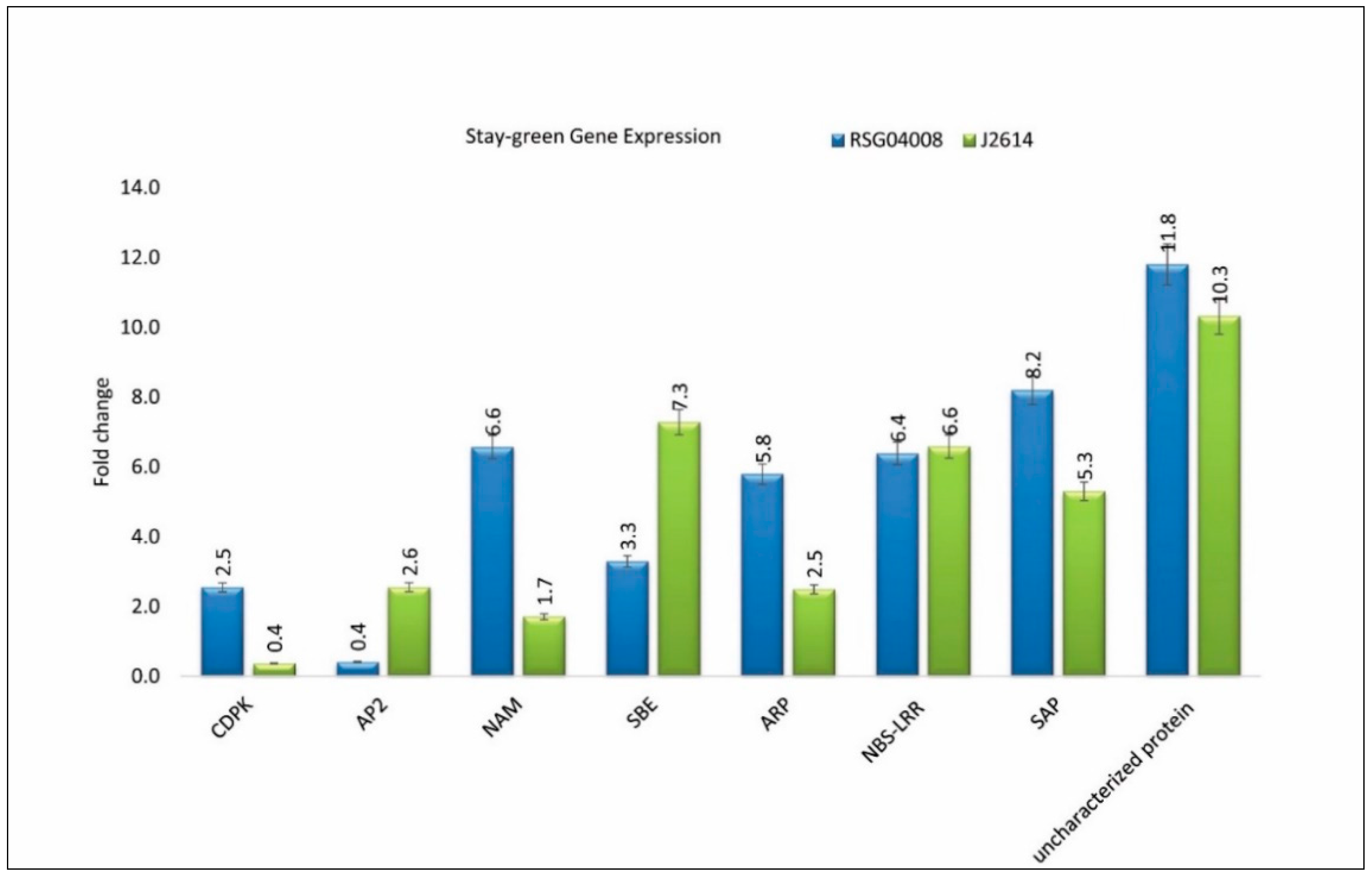
3.9. Stay-Green Candidate Gene Expression Profiling using Quantitative Real-Time PCR (qRT-PCR)
4. Discussion
4.1. Stay-Green Fine-Mapping Population Influenced by the Environment and Its Effect on Yield
4.2. The Significance of High-Resolution Genetic Linkage Mapping and Initial qRT-PCR-based Validation of Candidate Genes Associated with Senescence
4.3. Prioritization of Candidate Genes on SBI-10L
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Timu, A.; Mulwa, R.; Okello, J.; Kamau, M. The role of varietal attributes on adoption of improved seed varieties: The case of Sorghum in Kenya. Agric. Food Secur. 2014, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H.; Ougham, H. The stay-green trait. J. Exp. Bot. 2014, 65, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Haussmann, B.I.G.; Mahalakshmi, V.; Reddy, B.V.S.; Seetharama, N.; Hash, C.T.; Geiger, H.H. QTL mapping of stay-green in two sorghum recombinant inbred populations. Theor. Appl. Genet. 2002, 106, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belicuas, P.R.; Aguiar, A.M.; Bento, D.A.V.; Câmara, T.M.M.; de Souza Junior, C.L. Inheritance of the stay-green trait in tropical maize. Euphytica 2014, 198, 163–173. [Google Scholar] [CrossRef]
- Chen, J.; Liang, Y.; Hu, X.; Wang, X.; Tan, F.; Zhang, H.; Ren, Z.; Luo, P.G. Physiological characterization of ‘stay green’ wheat cultivars during the grain filling stage under field growing conditions. Acta Physiol. Plant. 2010, 32, 875–882. [Google Scholar] [CrossRef]
- Gous, P.W.; Hasjim, J.; Franckowiak, J.; Fox, G.P.; Gilbert, R.G. Barley genotype expressing ‘stay-green’-like characteristics maintains starch quality of the grain during water stress condition. J. Cereal Sci. 2013, 58, 414–419. [Google Scholar] [CrossRef]
- Huang, L.; Dai, L.; Wang, L.; Leng, Y.; Yang, Y.; Xu, J.; Hu, J.; Rao, Y.; Zhang, G.; Zhu, L.; et al. Genetic dissection for chlorophyll content of the top three leaves during grain filling in rice (Oryza sativa L.). J. Plant Growth Regul. 2015, 34, 381–391. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Park, S.-Y.; Kim, Y.-S.; Wang, S.-H.; Yoo, S.-C.; Hörtensteiner, S.; Paek, N.-C. Arabidopsis STAY-GREEN2 Is a Negative Regulator of Chlorophyll Degradation during Leaf Senescence. Mol. Plant Pathol. 2014, 7, 1288–1302. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.M.; Cummins, I.; Lim, F.L.; Slabas, A.R.; Knight, M.R. Transcriptomic analysis comparing stay-green and senescent Sorghum bicolor lines identifies a role for proline biosynthesis in the stay-green trait. J. Exp. Bot. 2015, 22, 7061–7073. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, G.N.; Fakrudin, B. Candidate gene prediction and expression profiling of near isogenic lines (NILs) carrying stay-green QTLs in rabi sorghum. J. Plant Biochem. Biotechnol. 2017, 26, 64–72. [Google Scholar] [CrossRef]
- Abdelrahman, M.; Ei-Sayed, M.; Jogaiah, S.; Burritt, D.J.; Tran, L.-S.T. The “STAY-GREEN” trait and phytohormone signaling networks in plants under heat stress. Plant Cell Rep. 2017, 36, 1009–1025. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.R.; Upadhyaya, H.D.; Ramu, P.; Deshpande, S.; Hash, C.T.; Bonnette, J.; Juenger, T.E.; Hyma, K.; Acharya, C.; Mitchell, S.E.; et al. Genome-environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 2015, 1, e1400218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadez, V.; Deshpande, S.P.; Kholova, J.; Hammer, G.L.; Borrell, A.; Talwar, H.; Hash, C.T., Jr. Stay-green quantitative trait loci’s effects on water extraction, transpiration efficiency and seed yield depend on recipient parent background. Funct. Plant Biol. 2011, 38, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrell, A.K.; Mullet, J.E.; George-Jaeggli, B.; van Oosterom, E.J.; Hammer, G.L.; Klein, P.E. Drought adaptation of stay-green sorghum is associated with canopy development, leaf anatomy, root growth, and water uptake. J. Exp. Bot. 2014, 65, 6251–6263. [Google Scholar] [CrossRef]
- Kiranmayee, K.N.S.U.; Hash, C.T.; Deshpande, S.P.; Varaprasad, K.V.G.K.; Kishor, P.B.K. Biotechnological Approaches to Evolve Sorghum (Sorghum bicolor (L.) Moench) for Drought Stress Tolerance and Shoot fly Resistance. Curr. Trends Biotechnol. Pharm. 2015, 9, 281–292. [Google Scholar]
- Hash, C.T.; Raj, A.G.B.; Lindup, S.; Sharma, A.; Beniwal, C.R.; Folkertsma, R.T.; Mahalakshmi, V.; Zerbini, E.; Blümmeld, M. Opportunities for marker-assisted selection (MAS) to improve the feed quality of crop residues in pearl millet and sorghum. Field Crop. Res. 2003, 84, 79–88. [Google Scholar] [CrossRef]
- Kassahun, B.; Bidinger, F.R.; Hash, C.T.; Kuruvinashetti, M.S. Stay-green expression in early generation sorghum [Sorghum bicolor (L.) Moench]. Euphytica 2010, 172, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Harris, K.; Subudhi, P.K.; Borrell, A.; Jordan, D.; Rosenow, D.; Nguyen, H.; Klein, P.; Klein, R.; Mullet, J. Sorghum stay-green QTL individually reduce post-flowering drought-induced leaf senescence. J. Exp. Bot. 2007, 58, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Gan, S. Convergence and divergence in gene expression profiles induced by leaf senescence and 27 senescence-promoting hormonal, pathological and environmental stress treatments. Plant. Cell Environ. 2012, 35, 644–655. [Google Scholar] [CrossRef]
- Mace, E.; Singh, V.; van Oosterom, E.; Hamer, G.; Hunt, C.; Jordan, D. QTL for nodal root angle in sorghum (Sorghum bicolor L. Moench) co-locate with QTL for traits associated with drought adaptation. Theor. Appl. Genet. 2012, 124, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Kiranmayee, K.N.S.U.; Kishor, P.B.K.; Hash, C.T.; Deshpande, S.P. Evaluation of QTLs for Shoot Fly (Atherigona soccata) Resistance Component Traits of Seedling Leaf Blade Glossiness and Trichome Density on Sorghum (Sorghum bicolor) Chromosome SBI-10L. Trop. Plant. Biol. 2016, 9, 12–28. [Google Scholar] [CrossRef] [Green Version]
- Van Berloo, R. GGT 2.0: Versatile software for visualization and analysis of genetic data. J. Hered. 2008, 99, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, H.D.; Williams, E.R. A new class of resolvable incomplete block designs. Biometrika 1976, 63, 83–92. [Google Scholar] [CrossRef]
- Mahalakshmi, V.; Bidinger, F.R. Evaluation of stay-green sorghum germplasm lines at ICRISAT. Crop. Sci. 2002, 42, 965–974. [Google Scholar]
- Patterson, H.D.; Thompson, R. Recovery of inter-block information when block sizes are unequal. Biometrika 1971, 58, 545–554. [Google Scholar] [CrossRef]
- Falconer, D.S. Introduction to Quantitative Genetics, 3rd ed.; Longman Scientific & Technical: New York, NY, USA, 1989. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant. J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [Green Version]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wanq le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118, iso-2, iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Cheema, J.; Ellis, T.N.; Dicks, J. THREaD Mapper Studio: A novel, visual web server for the estimation of genetic linkage maps. Nucleic Acids Res. 2010, 38, 188–193. [Google Scholar] [CrossRef]
- Van Ooijen, J.W.; Voorrips, R.E. JoinMap 3.0 Software for the Calculation of Genetic Linkage Maps; Plant Researchs International: Wageningen, The Netherlands, 2001. [Google Scholar]
- Kosambi, D.D. The estimation of map distances from recombination values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Wang, S.; Basten, C.J.; Zeng, Z.B. Windows QTL Cartographer 2.5; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2010; Available online: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm (accessed on 3 November 2015).
- Li, J.; Thomson, M.; Mc Couch, S.R. Fine mapping of a grain-weight quantitative trait locus in the pericentromeric region of rice chromosome 3. Genetics 2004, 168, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryzwinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos an information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Hospital, F. Size of donor chromosome segments around introgressed loci and reduction of linkage drag in marker-assisted backcross programs. Genetics 2001, 158, 1363–1379. [Google Scholar]
- Jordan, D.R.; Hunt, C.H.; Cruickshank, A.W.; Borrell, A.K.; Henzell, R.G. The relationship between the stay-green trait and grain yield in elite sorghum hybrids grown in a range of environments. Crop. Sci. 2012, 52, 1153–1161. [Google Scholar] [CrossRef]
- Fakrudin, B.R.; Kavil, S.P.; Girma, Y.; Arun, S.S.; Dadakhalandar, D.; Gurusiddesh, B.H.; Patil, A.M.; Thudi, M.; Bhairappanavar, S.B.; Narayana, Y.D.; et al. Molecular mapping of genomic regions harbouring QTLs for root and yield traits in sorghum (Sorghum bicolor L. Moench). Physiol. Mol. Biol. Plants 2013, 19, 409–419. [Google Scholar]
- Xiao, B.; Huang, Y.; Tang, N.; Xiong, L. Over-expression of a LEA gene in rice improves drought resistance under the field conditions. Theor. Appl. Genet. 2007, 115, 35–46. [Google Scholar] [CrossRef]
- Dai, C.; Lee, Y.; Lee, I.C.; Nam, H.G.; Kwak, J.M. Calmodulin 1 Regulates Senescence and ABA Response in Arabidopsis. Front. Plant. Sci. 2018, 9, 803. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.J.; Franks, C.D.; Burow, G.; Xin, Z. Selection System for the Stay-Green Drought Tolerance Trait in Sorghum Germplasm. Agron. J. 2010, 102, 1118. [Google Scholar] [CrossRef] [Green Version]
- Cai, R.; Zhao, Y.; Wang, Y.; Lin, Y.; Peng, X.; Li, Q.; Chang, Y.; Jiang, H.; Xiang, Y.; Cheng, B. Over expression of a maize WRKY58 gene enhances drought and salt tolerance in transgenic rice. Plant Cell Tissue Organ. Cult. 2014, 119, 565–577. [Google Scholar] [CrossRef]
- Shore, P.; Sharrocks, A.D. The MADS-box family of transcription factors. Eur. J. Biochem. 1995, 229, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.O.; Kim, H.J.; Nam, H.G. Leaf senescence. Annu. Rev. Plant Biol. 2007, 58, 115–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cui, X.; Meng, Z.; Huang, X.; Xie, Q.; Wu, H.; Jin, H.; Zhang, D.; Liang, W. Transcriptional Regulation of Arabidopsis MIR168a and ARGONAUTE1 Homeostasis in Abscisic Acid and Abiotic Stress Responses. Plant Physiol. 2012, 158, 1279–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Jiang, C.Z.; Donnelly, L.; Reid, M.S. Functional analysis of a RING domain ankyrin repeat protein that is highly expressed during flower senescence. J. Exp. Bot. 2007, 58, 3623–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, S. An overview of pentatricopeptide repeat proteins and their applications. Biochimie 2015, 113, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Preston, J.C.; Hileman, L.C. Functional Evolution in the Plant SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) Gene Family. Front. Plant Sci. 2013, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Post Rainy 2012–2013/E1 | Post Rainy 2013–2014/E2 | Across Season | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Range F4 Progeny | Mean | Range F4 Progeny | Mean | Range F4 Progeny | ||||||||
| Trait | RSG04008-6 | F4 Progeny | J2614-11 | RSG04008-6 | F4 Progeny | J2614-11 | RSG04008-6 | F4 Progeny | J2614-11 | ||||
| 1 | %GL 7 DAF | 88.38 | 88.67 | 83.68 | 72.33–97.78 | 99.03 | 96.05 | 95.24 | 79.14–99.26 | 93.86 | 92.36 | 89.56 | 76.31–98.65 |
| 2 | %GL14 DAF | 79.18 | 79.58 | 72.06 | 58.73–96.64 | 84.22 | 84.29 | 85.26 | 68.60–96.93 | 81.71 | 81.94 | 78.65 | 63.34–96.69 |
| 3 | %GL21 DAF | 62.52 | 65.96 | 61.88 | 43.29–93.77 | 74.39 | 73.73 | 75.05 | 63.34–84.91 | 68.43 | 69.85 | 68.48 | 54.12–90.37 |
| 4 | %GL28 DAF | 47.00 | 50.72 | 47.68 | 29.92–78.11 | 66.49 | 65.03 | 64.04 | 51.36–77.49 | 56.83 | 57.88 | 55.78 | 42.44–77.36 |
| 5 | %GL35 DAF | 34.46 | 39.49 | 37.72 | 22.90–62.25 | 54.17 | 51.57 | 50.54 | 19.89–66.05 | 44.39 | 45.53 | 44.19 | 19.74–61.07 |
| 6 | %GL42 DAF | 23.44 | 29.41 | 30.73 | 17.19–45.88 | 25.96 | 39.73 | 40.1 | 6.09–53.89 | 25.28 | 34.71 | 35.25 | 13.73–48.77 |
| 7 | %GL 49 DAF | 17.12 | 18.56 | 20.33 | 9.90–31.90 | 11.68 | 29.07 | 28.4 | 4.73–44.61 | 14.67 | 24.01 | 24.94 | 8.72–38.12 |
| 8 | PnDW/plot | 598.1 | 577.49 | 372.53 | 416.17–779.52 | 945.95 | 935.88 | 1016.03 | 763.31–1102.78 | 768.65 | 756.15 | 738.23 | 668.70–887.70 |
| 9 | GDW/plot | 392.72 | 397.96 | 233.42 | 267.61–539.88 | 632.53 | 650.60 | 732.21 | 531.50–777.47 | 513.47 | 523.70 | 518.19 | 461.19–599.05 |
| 10 | PHI | 66.9 | 68.4 | 62.88 | 63.08–78.21 | 68.63 | 70.14 | 71.69 | 66.35–74.21 | 67.48 | 69.27 | 67.72 | 65.00–73.43 |
| 11 | HGM | 2.14 | 1.94 | 2.06 | 1.28–2.44 | 3.28 | 2.95 | 2.71 | 2.27–3.73 | 2.70 | 2.45 | 2.38 | 1.79–3.07 |
| 12 | GNP/plot | 20,456.12 | 21,232.45 | 17,807.87 | 18,453.82–43,294.30 | 20,212.25 | 22,191.42 | 26,891.54 | 17,204.13–27,250.23 | 20,746.04 | 21,720.92 | 21,254.12 | 19,611.06–33,652.47 |
| 13 | GNPP | 786.78 | 816.64 | 684.92 | 709.77–1665.17 | 777.40 | 853.52 | 1034.29 | 661.70–1048.09 | 797.92 | 835.42 | 817.47 | 754.27–1294.33 |
| Post Rainy 2012–2013 | Post Rainy 2013–2014 | Across Season | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S. No. | Trait | σ2g | ±SE | h2 | σ2g | ±SE | h2 | σ2g | GхE | ±SE | h2 |
| 1 | %GL7 DAF | 64.34 | 7.941 | 75.38 | 26.42 | 5.92 | 69.37 | 73.44 | 100.99 | 7.00 | 81.80 |
| 2 | %GL14 DAF | 183.30 | 13.8 | 74.27 | 42.63 | 7.65 | 68.64 | 181.90 | 256.50 | 11.16 | 81.42 |
| 3 | %GL21 DAF | 198.33 | 13.85 | 75.63 | 17.13 | 6.54 | 54.59 | 160.13 | 283.30 | 10.83 | 80.39 |
| 4 | %GL28 DAF | 121.70 | 11.13 | 74.65 | 30.21 | 7.31 | 62.93 | 120.54 | 182.82 | 9.42 | 80.31 |
| 5 | %GL35 DAF | 63.26 | 8.775 | 71.14 | 62.33 | 9.48 | 67.52 | 98.61 | 164.42 | 9.14 | 77.99 |
| 6 | %GL42 DAF | 29.74 | 8.04 | 57.98 | 97.99 | 8.39 | 80.70 | 80.03 | 210.67 | 8.21 | 78.08 |
| 7 | %GL49 DAF | 28.20 | 8.719 | 52.67 | 104.33 | 10.13 | 75.31 | 79.26 | 249.26 | 9.44 | 72.73 |
| 8 | PnDW/plot | 4307.00 | 4562.00 | 57.94 | 8033.33 | 208.50 | 35.66 | 7717.67 | 40,344.00 | 162.40 | 46.74 |
| 9 | GDW/plot | 3341.67 | 88.51 | 56.13 | 3906.67 | 139.50 | 37.59 | 3610.33 | 21672.00 | 113.40 | 45.70 |
| 10 | PHI | 9.48 | 7.61 | 32.96 | 1.73 | 8.02 | 7.45 | 8.75 | 69.66 | 7.65 | 30.99 |
| 11 | HGM | 0.04 | 0.12 | 87.95 | 0.07 | 0.26 | 75.91 | 0.08 | 0.11 | 0.19 | 86.91 |
| 12 | GNP/plot | 12,233,333.00 | 1,282,200 | 18.25 | 6,710,000.00 | 5443.00 | 40.45 | 12,460,000.00 | 116,400,000.00 ns | 9850 | 27.81 |
| 13 | GNPP | 18,072.00 | 493.20 | 18.23 | 9924.00 | 209.40 | 40.45 | 18417.00 | 172259.00 ns | 378.80 | 27.78 |
| Trait | %GL_7 | %GL_14 | %GL_21 | %GL_28 | %GL_35 | %GL_42 | %GL_49 | GDW | GDW/Plot | GNPP | GNP/Plot | HGM | PHI | PnDW | PnDW/Plot |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| %GL13_7 | 1 | ||||||||||||||
| %GL14_7 | 1 | ||||||||||||||
| %GL13_14 | 0.98 ** | 1 | |||||||||||||
| %GL14_14 | 0.68 ** | 1 | |||||||||||||
| %GL13_21 | 0.93 ** | 0.94** | 1 | ||||||||||||
| %GL14_21 | 0.65 ** | 0.88 ** | 1 | ||||||||||||
| %GL13_28 | 0.89 ** | 0.9 ** | 0.99 ** | 1 | |||||||||||
| %GL14_28 | 0.72 ** | 0.82 ** | 0.88 ** | 1 | |||||||||||
| %GL13_35 | 0.82 ** | 0.82 ** | 0.93 ** | 0.97 ** | 1 | ||||||||||
| %GL14_35 | 0.67 ** | 0.81 ** | 0.76 ** | 0.75 ** | 1 | ||||||||||
| %GL13_42 | 0.68 ** | 0.66 ** | 0.81 ** | 0.87 ** | 0.94 ** | 1 | |||||||||
| %GL14_42 | 0.5 ** | 0.59 ** | 0.56 ** | 0.53 ** | 0.77 ** | 1 | |||||||||
| %GL13_49 | 0.81 ** | 0.83 ** | 0.85 ** | 0.87 ** | 0.87 ** | 0.85 ** | 1 | ||||||||
| %GL14_49 | 0.65 ** | 0.61 ** | 0.54 ** | 0.6 ** | 0.7 ** | 0.82 ** | 1 | ||||||||
| GDW_13 | 0.08 | 0.1 | −0.02 | −0.06 | −0.13 | −0.22 | −0.08 | 1 | |||||||
| GDW_14 | 0.12 | 0.01 | −0.1 | −0.09 | 0.06 | 0.12 | 0.15 | 1 | |||||||
| GDW_plot_13 | 0.01 | −0.02 | −0.08 | −0.08 | −0.09 | −0.11 | −0.07 | 0.75 ** | 1 | ||||||
| GDW_plot_14 | 0.09 | 0.06 | 0.01 | 0.01 | 0.06 | 0.09 | 0.1 | 0.57 ** | 1 | ||||||
| GNPP_13 | 0.1 | 0.07 | 0.06 | 0.05 | 0.05 | 0.05 | 0.01 | 0.01 | 0.05 | 1 | |||||
| GNPP_14 | 0.02 | 0.06 | 0.01 | 0.03 | 0.09 | −0.02 | −0.04 | 0.04 | 0.02 | 1 | |||||
| GNP_plot_13 | 0.07 | 0.07 | 0.06 | 0.05 | 0.05 | 0.05 | 0.01 | 0.01 | 0.05 | 1 | 1 | ||||
| GNP_plot_14 | 0.09 | 0.06 | 0.01 | 0.01 | 0.06 | 0.09 | 0.1 | 0.57 ** | 1 | 0.02 | 1 | ||||
| HGM_13 | −0.16 | −0.18 | −0.17 | −0.11 | −0.07 | 0.05 | −0.01 | −0.04 | 0.15 | −0.13 | −0.13 | 1 | |||
| HGM_14 | −0.09 | −0.03 | −0.09 | −0.13 | 0.01 | −0.01 | −0.08 | −0.12 | 0.06 | 0.01 | 0.06 | 1 | |||
| PHI_13 | −0.05 | −0.07 | −0.11 | −0.12 | −0.12 | −0.13 | −0.07 | 0.62 ** | 0.62 ** | −0.02 | −0.02 | −0.15 | 1 | ||
| PHI_14 | −0.02 | 0.05 | 0.03 | 0.03 | 0.04 | 0.05 | 0.07 | 0.33 ** | 0.22 ** | −0.12 | 0.22 ** | −0.22 ** | 1 | ||
| PnDW_13 | 0.11 | 0.15 | 0.03 | −0.02 | −0.11 | −0.22 | −0.07 | 0.95 ** | 0.65 ** | 0.02 | 0.02 | 0.01 | 0.36 ** | 1 | |
| PnDW_14 | 0.12 | −0.02 | −0.12 | −0.12 | 0.05 | 0.11 | 0.12 | 0.91 ** | 0.51 ** | 0.09 | 0.51 ** | −0.05 | −0.1 | 1 | |
| PnDW_plot_13 | 0.04 | 0.01 | −0.05 | −0.05 | −0.07 | −0.08 | −0.06 | 0.65 ** | 0.95 ** | 0.08 | 0.08 | 0.22 ** | 0.35 ** | 0.65 ** | 1 |
| PnDW_plot_14 | 0.08 | 0.03 | −0.01 | −0.01 | 0.04 | 0.07 | 0.05 | 0.38 ** | 0.88 ** | 0.07 | 0.88 ** | 0.17 | −0.28 | 0.53 ** | 1 |
| QTLs on SBI-10 | Pos cM | Nearest Marker | Marker Interval | Support Interval | LOD | %R2 (PVE) | Additive | Dominant |
|---|---|---|---|---|---|---|---|---|
| Q10GL7a_across | 44.41 | S10_54891180 | Xgap001_54813392–S10_54491782 | 42.8–45.9/44.9 | 2.76 | 6.75 | 2.2331 | 0.7273 |
| Q10GL7a_13 | 41.41 | S10_54388058 | S10_55181668–Xgap001_54813392 | 40.6–42.4 | 2.93 | 8.86 | 2.8201 | −2.6178 |
| Q10GL7a_14 | 104.81 | S10_58565687 | S10_58276305–Xiabt_58564309 | 103.7–105.9 | 3.40 | 6.98 | −1.5675 | −3.095 |
| Q10GL7b_14 | 112.11 | S10_59294324 | S10_58634498–S10_59498752 | 110.9–113.1 | 3.11 | 9.43 | −1.5537 | 1.6478 |
| Q10GL7combined r2 | 32.02 | |||||||
| Q10GL14d_14 | 29.01 | S10_52316579 | S10_51497655‒S10_52973075 | 28.4‒29.4 | 2.21 | 0.09 | 0.5767 | 5.886 |
| Q10GL14a_14 | 36.41 | S10_54575547 | S10_54575547–S10_52969717 | 36.3–37.2 | 2.59 | 5.03 | 2.0097 | 2.8007 |
| Q10GL14b_14 | 45.01 | S10_54841288 | Xgap001_54813392–S10_55270382 | 42.8–47.4 | 2.55 | 6.49 | 2.7621 | 0.5624 |
| Q10GL14e_14 | 123.61 | S10_60308400 | S10_60231331‒S10_60602919 | 122.9‒124.1 | 2.37 | 9.70 | −2.3214 | 3.225 |
| Q10GL14c_14 | 129.51 | S10_60557352 | S10_60612267–S10_60333222 | 129–130.0 | 3.38 | 8.94 | −2.7106 | 6.1702 |
| Q10GL14a_13 | 41.41 | S10_54388058 | S10_55181668–Xgap001_54813392 | 40.7–42.8 | 2.86 | 9.00 | 4.9036 | −4.2892 |
| Q10GL14a_across | 44.41 | S10_54891180 | Xgap001_54813392–S10_54491782 | 42.8–45.6 | 3.73 | 10.20 | 4.5978 | 0.0381 |
| Q10GL14b_13 | 82.71 | S10_56692703 | S10_56476051‒S10_56989405 | 82.6‒85 | 2.18 | 4.24 | −5.2408 | −4.2484 |
| Q10GL14combined r2 | 53.68 | |||||||
| Q10GL21a_across | 115.31 | S10_59620328 | S10_59620312–S10_59850129 | 114.5–115.6 | 2.91 | 10.14 | −5.0744 | 2.4482 |
| Q10GL21b_13 | 41.41 | S10_54388058 | S10_55181668‒Xgap001_54813392 | 40.5‒42.8 | 2.18 | 6.91 | 4.9353 | −3.5171 |
| Q10GL21a_13 | 115.31 | S10_59620328 | S10_59620312–S10_59849054 | 114.5–116.2 | 2.76 | 9.07 | −6.0311 | 6.0466 |
| Q10GL21c_13 | 125.01 | S10_60468748 | S10_60308400–S10_60513644 | 123.6–125.5 | 2.33 | 1.41 | −0.3211 | 13.282 |
| Q10GL21b_14 | 79.41 | S10_56641518 | S10_56608572‒S10_56692703 | 74.3‒82.7 | 2.12 | 7.04 | −1.4956 | 2.3253 |
| Q10GL21c_14 | 99.01 | S10_57507647 | S10_58704603‒S10_57714076 | 97.9‒100.4 | 2.16 | 5.31 | 1.055 | −4.1377 |
| Q10GL21a_14 | 115.31 | S10_59620328 | S10_59620312–S10_59850129 | 114.5–115.6 | 2.74 | 9.20 | −2.2148 | 1.2957 |
| Q10GL21combined r2 | 49.08 | |||||||
| Q10GL28a_14 | 36.41 | S10_54575547 | S10_54575547–S10_52969717 | 36.3–37.2 | 3.67 | 4.96 | 2.044 | 3.4449 |
| Q10GL28b_14 | 41.41 | S10_54388058 | S10_52995995–Xgap001_54813392 | 39.8–42.8 | 2.77 | 6.62 | 2.1001 | 0.7883 |
| Q10GL28a_across | 124.91 | S10_60468748 | S10_60308400–S10_60513644 | 123.6–125.5 | 2.74 | 2.85 | −0.6203 | 7.2213 |
| Q10GL28a_13 | 125.01 | S10_60468748 | S10_60308400–S10_60513644 | 123.6–125.5 | 2.52 | 1.20 | −0.1975 | 11.128 |
| Q10GL28combined r2 | 15.62 | |||||||
| Q10GL35a_13 | 121.91 | S10_60135251 | S10_60613735‒S10_60593638 | 121.4‒122.5 | 2.36 | 2.06 | −0.6135 | 8.0863 |
| Q10GL42a_13 | 121.91 | S10_60135251 | S10_60173717–S10_60593638 | 121.7–122.5 | 2.59 | 4.54 | −0.5686 | 5.187 |
| Q10GL42b_13 | 131.91 | S10_60973291 | S10_60231220‒Xisep1011_61017707 | 131.2–132 | 2.01 | 0.52 | 2.4162 | 3.149 |
| Q10GL42d_14 | 32.31 | S10_53077439 | S10_52199633‒S10_55164775 | 30.8‒33.9 | 2.47 | 6.66 | 2.0509 | −6.9547 |
| Q10GL42b_14 | 38.41 | S10_53108642 | S10_52969717‒S10_53839087 | 37.7‒39.6 | 2.33 | 2.28 | 3.5114 | 4.965 |
| Q10GL42a_14 | 102.31 | S10_57783305 | S10_57714076–S10_58610366 | 100.4–103.6 | 2.60 | 2.43 | 1.7652 | −29.678 |
| Q10GL42c_14 | 107.81 | S10_58728876 | S10_58758633‒S10_58758082 | 106.7‒108.5 | 2.44 | 5.75 | 2.0457 | −7.1224 |
| Q10GL42combined r2 | 22.18 | |||||||
| Q10GL49a_13 | 34.71 | S10_51163559 | S10_50394264‒S10_54118313 | 33.9–35.8 | 2.37 | 2.17 | 1.4224 | 3.2659 |
| Q10GL49a_across | 36.41 | S10_54575547 | S10_54575547–S10_52969712 | 36.4–37.2 | 2.54 | 2.45 | 2.1417 | 3.2671 |
| Q10GL49b_across | 45.01 | S10_54841288 | Xgap001_54813392–S10_54491782 | 42.8–45.9 | 3.04 | 4.08 | 2.6273 | 2.1788 |
| Q10GL49combined r2 | 8.71 |
| cQTL | Nearest Marker | Position cM | Marker Intervals | No.of QTLs | Individual QTLs Co-Localized | Gene ID/MTAs | Combined r2 | Previous Studies Reporting stg QTL | Candidate Genes | SNP Effect |
|---|---|---|---|---|---|---|---|---|---|---|
| cQstg10.1 | S10_54553482 | 36.4 | 36.4 | 3 | Q10GL14a_14, Q10GL28a_14, Q10GL49a_across | Sobic.010G202700 | 12.4 | Haussmann et al. 2002 [3] | AP2/ERF transcriptional factor | Splice_site_ region + Intron |
| cQstg10.2 | S10_54388058 | 41.4 | 39.8–42.8 | 4 | Q10GL7a_13, Q10GL14a_13, Q10GL21b_13, Q10GL28b_14 | Sobic.010G201100 | 31.3 | Haussmann et al. 2002 [3] | weakly similar to Putative uncharacterized protein | Non synonymous |
| cQstg10.3 | S10_54891180S10_54899228 | 44.4 | 54.6–54.6 | 2 | Q10GL14b_14, Q10GL49b_across | Sobic.010G205800, Sobic.010G205900 | 10.6 | Haussmann et al. 2002 [3] | Ankyrin repeat protein and WD40 repeat family protein (transducin protein) | Intron and synonymous |
| cQstg10.4 | S10_54841288 | 45.0 | 42.8–47.4 | 2 | Q10GL7a_across, Q10GL14a_across | Sobic.010G205600 | 16.9 | Haussmann et al. 2002 [3] | NBS-LRR disease resistance protein | synonymous |
| cQstg10.5 | S10_59620328 | 115.3 | 114.5–115.6 | 3 | Q10GL21a_13, Q10GL21a_14, Q10GL21a_across | Sobic.010G259200 | 28 | Haussmann et al. 2002 [3] | similar to Putative uncharacterized protein P0655A07.24/LEA | Nonsynonymous |
| cQstg10.6 | S10_60059042 | 121.9 | 121.4‒122.5 | 2 | Q10GL35a_13, Q10GL42a_13 | Sobic.010G264400 | 6.6 | Haussmann et al. 2002 [3] | Ca/calmodulin dependent protein kinase | Intron |
| cQstg10.7 | S10_60468746 | 125.0 | 124.7–125.5 | 3 | Q10GL21c_13, Q10GL28a_13, Q10GL28a_across | Sobic.010G270300 | 5.5 | Haussmann et al. 2002 [3] | similar to Senescence-associated protein | synonymous |
| SNP ID | Gene ID 3.1V | Gene Abbrevation | Gene Function | Forward Primer Seq | Reverse Primer Seq | Product | Allele | |
|---|---|---|---|---|---|---|---|---|
| 1 | S10_54553482 | Sobic.010G202700 | AP2 | similar to Apetala 2 | attcgacactgctcatgctg | gtacttggagctgcctctgg | 196 | T/A |
| 2 | S10_54841288 | Sobic.010G205600 | NBS-LRR | NBS-LRR disease resistance protein | ggagtgcagcattgttcaga | caatgagctcaggggcttag | 184 | A/G |
| 3 | S10_54891180 | Sobic.010G205800 | ARP | similar to ankyrin repeat-containing protein-like | cgagcatggagcagacataa | tgtgtcgcctgatacccata | 153 | A/T |
| 4 | S10_59620328 | Sobic.010G259200 | Uncharacterised | similar to Putative uncharacterized protein P0655A07.24 | acctgctgtacaagcccaag | tcggtcttggagttggagtt | 142 | A/T |
| 5 | S10_60059042 | Sobic.010G264400 | CAMK | Calcium/calmodulin-dependent protein kinase | gtgtggcagtaggccatttt | tgcttggacagcagtcattc | 175 | A/C |
| 6 | S10_60468746 | Sobic.010G270300 | SAP | similar to Senescence-associated protein | ggctcaggaatgacgaaaaa | cagctcattctccctccaag | 192 | C/A |
| 7 | S10_60684319 | Sobic.010G273700 | NAM | No apical meristem (NAM) protein | ggagtggagaccatgacgat | gtcagaaatgggtcctgcat | 182 | T/A |
| 8 | S10_60695074 | Sobic.010G273800 | SBE | Starch branching enzyme I precursor | attgggatcctcctgcttct | ccatcaactgaacggtgttg | 192 | A/G |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiranmayee, K.N.S.U.; Hash, C.T.; Sivasubramani, S.; Ramu, P.; Amindala, B.P.; Rathore, A.; Kishor, P.B.K.; Gupta, R.; Deshpande, S.P. Fine-Mapping of Sorghum Stay-Green QTL on Chromosome10 Revealed Genes Associated with Delayed Senescence. Genes 2020, 11, 1026. https://doi.org/10.3390/genes11091026
Kiranmayee KNSU, Hash CT, Sivasubramani S, Ramu P, Amindala BP, Rathore A, Kishor PBK, Gupta R, Deshpande SP. Fine-Mapping of Sorghum Stay-Green QTL on Chromosome10 Revealed Genes Associated with Delayed Senescence. Genes. 2020; 11(9):1026. https://doi.org/10.3390/genes11091026
Chicago/Turabian StyleKiranmayee, K. N. S. Usha, C. Tom Hash, S. Sivasubramani, P. Ramu, Bhanu Prakash Amindala, Abhishek Rathore, P. B. Kavi Kishor, Rajeev Gupta, and Santosh P. Deshpande. 2020. "Fine-Mapping of Sorghum Stay-Green QTL on Chromosome10 Revealed Genes Associated with Delayed Senescence" Genes 11, no. 9: 1026. https://doi.org/10.3390/genes11091026