The Acidic Stress Response of the Intracellular Pathogen Brucella melitensis: New Insights from a Comparative, Genome-Wide Transcriptome Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Public Data Sets
2.2. Data Analysis
2.3. Statistical Analysis
2.4. Reverse-Transcriptase PCR (RT-PCR)
3. Results
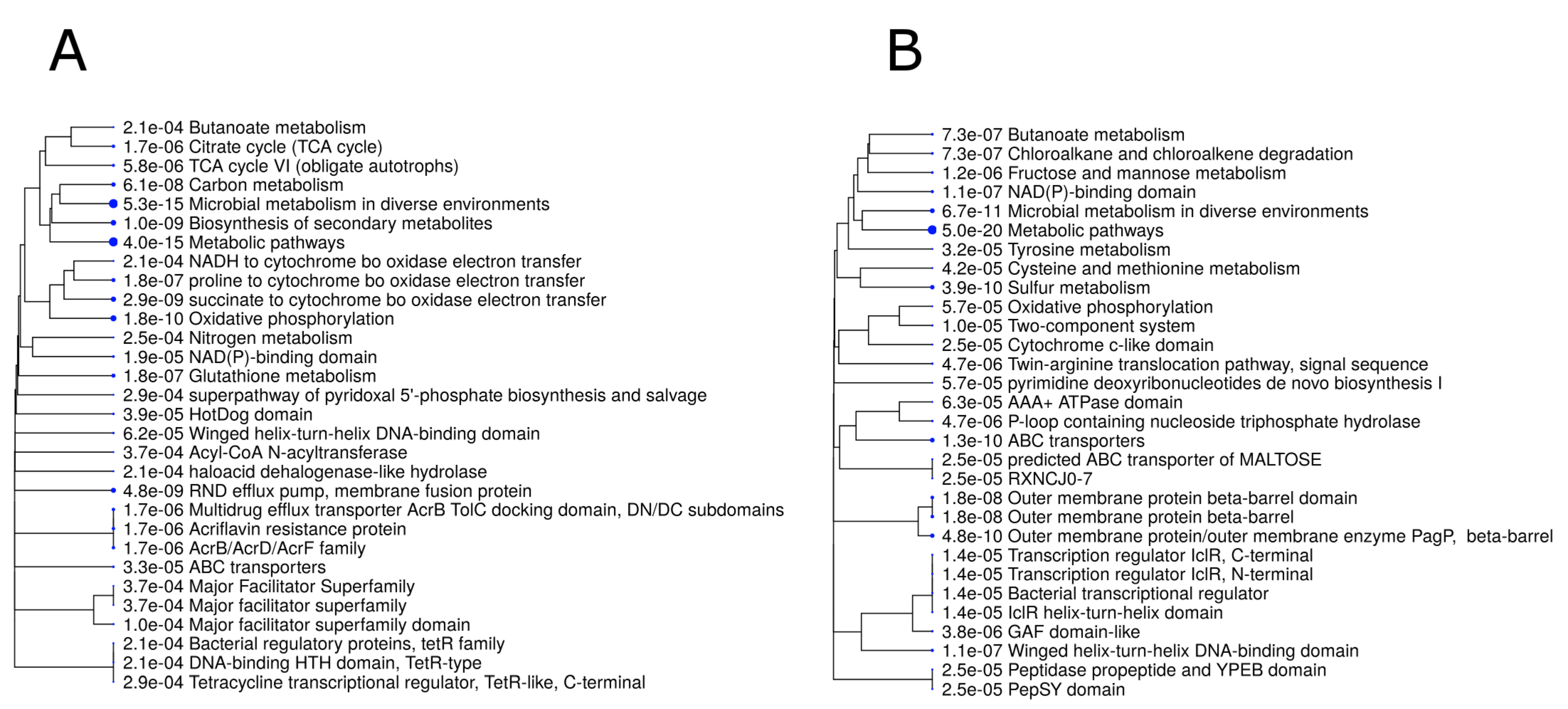
3.1. Gene Expression in B. melitensis 16M Grown under Either Acidic or Neutral pH Conditions
3.2. The Effects of Acid Stress on Gene Expression in B. melitensis 16M, as Compared with Its Previously Reported Transcriptional Profile in an In Cellulo Hela Cell Model
3.3. The Effects of Acid Stress on Gene Expression in B. melitensis 16M, as Compared with Its Previously Reported Transcriptional Profile in an In Vivo Infection Model
3.4. Key Genes in B. melitensis 16M That Are DE Specifically in Response to Acidic Stress
3.5. RT-qPCR Validation of the RNA-Seq Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lund, P.; Tramonti, A.; de Biase, D. Coping with low pH: Molecular strategies in neutralophilic bacteria. FEMS Microbiol. Rev. 2014, 38, 1091–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, D.L.; Tucker, N.; Conway, T. Gene expression profiling of the pH response in Escherichia coli. J. Bacteriol. 2002, 184, 6551–6558. [Google Scholar] [CrossRef] [Green Version]
- Guan, N.; Liu, L. Microbial response to acid stress: Mechanisms and applications. Appl. Microbiol. Biotechnol. 2020, 104, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivijs, B.; Aertsen, A.; Michiels, C.W. Identification of genes required for growth of Escherichia coli MG1655 at moderately low pH. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Marcus, E.A.; Matrubutham, U.; Gleeson, M.A.; Scott, D.R.; Sachs, G. Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 2003, 71, 5921–5939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.W. Escherichia coli acid resistance: Tales of an amateur acidophile. Nat. Rev. Microbiol. 2004, 2, 898–907. [Google Scholar] [CrossRef]
- Sun, Y.; Fukamachi, T.; Saito, H.; Kobayashi, H. Respiration and the F1Fo-ATPase enhance survival under acidic conditions in Escherichia coli. PLoS ONE 2012, 7, e52577. [Google Scholar] [CrossRef] [Green Version]
- Bearson, S.; Bearson, B.; Foster, J.W. Acid stress responses in enterobacteria. FEMS Microbiol. Lett. 2006, 147, 173–180. [Google Scholar] [CrossRef]
- Ryan, S.; Begley, M.; Gahan, C.G.M.; Hill, C. Molecular characterization of the arginine deiminase system in Listeria monocytogenes: Regulation and role in acid tolerance. Environ. Microbiol. 2009, 11, 432–445. [Google Scholar] [CrossRef]
- Freddi, L.; Damiano, M.A.; Chaloin, L.; Pennacchietti, E.; Al Dahouk, S.; Köhler, S.; de Biase, D.; Occhialini, A. The glutaminase-dependent system confers extreme acid resistance to new species and atypical strains of Brucella. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Pennacchietti, E.; D’alonzo, C.; Freddi, L.; Occhialini, A.; de Biase, D. The glutaminase-dependent acid resistance system: Qualitative and quantitative assays and analysis of its distribution in enteric bacteria. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.R.; Marcus, E.A.; Wen, Y.; Singh, S.; Feng, J.; Sachs, G. Cytoplasmic histidine kinase (HP0244)-regulated assembly of urease with UreI, a channel for urea and its metabolites, CO2, NH3, and NH4+, is necessary for acid survival of Helicobacter pylori. J. Bacteriol. 2010, 192, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuk, H.G.; Marshall, D.L. Adaptation of Escherichia coli O157:H7 to pH alters membrane lipid composition, verotoxin secretion, and resistance to simulated gastric fluid acid. Appl. Environ. Microbiol. 2004, 70, 3500–3505. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.; Splitter, G.A. Molecular host-pathogen interaction in brucellosis: Current understanding and future approaches to vaccine development for mice and humans. Clin. Microbiol. Rev. 2003, 16, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bargen, K.; Gorvel, J.P.; Salcedo, S.P. Internal affairs: Investigating the Brucella intracellular lifestyle. FEMS Microbiol. Rev. 2012, 36, 533–562. [Google Scholar] [CrossRef] [Green Version]
- Poester, F.P.; Samartino, L.E.; Santos, R.L. Pathogenesis and pathobiology of brucellosis in livestock. Rev. Sci. Tech. 2013, 32, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Sangari, F.J.; Seoane, A.; Rodríguez, M.C.; Agüero, J.; García Lobo, J.M. Characterization of the urease operon of Brucella abortus and assessment of its role in virulence of the bacterium. Infect. Immun. 2007, 75, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Celli, J.; de Chastellier, C.; Franchini, D.M.; Pizarro-Cerda, J.; Moreno, E.; Gorvel, J.P. Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 2003, 198, 545–556. [Google Scholar] [CrossRef]
- Salcedo, S.P.; Chevrier, N.; Lacerda, T.L.S.; Ben Amara, A.; Gerart, S.; Gorvel, V.A.; de Chastellier, C.; Blasco, J.M.; Mege, J.L.; Gorvel, J.P.; et al. Pathogenic Brucellae replicate in human trophoblasts. J. Infect. Dis. 2013, 207, 1075–1083. [Google Scholar] [CrossRef]
- Delrue, R.M.; Lestrate, P.; Tibor, A.; Letesson, J.J.; de Bolle, X. Brucella pathogenesis, genes identified from random large-scale screens. FEMS Microbiol. Lett. 2004, 231, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Celli, J. Surviving inside a macrophage: The many ways of Brucella. Res. Microbiol. 2006, 157, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.; Ng, T.W.; Wehrly, T.D.; Knodler, L.A.; Celli, J. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic 2008, 9, 678–694. [Google Scholar] [CrossRef] [PubMed]
- Boschiroli, M.L.; Ouahrani-Bettache, S.; Foulongne, V.; Michaux-Charachon, S.; Bourg, G.; Allardet-Servent, A.; Cazevieille, C.; Liautard, J.P.; Ramuz, M.; O’Callaghan, D.; et al. The Brucella suis virB operon is induced intracellularly in macrophages. Proc. Natl. Acad. Sci. USA 2002, 99, 1544–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porte, F.; Liautard, J.P.; Köhler, S. Early acidification of phagosomes containing Brucella suis is essential for intracellular survival in murine macrophages. Infect. Immun. 1999, 67, 4041–4047. [Google Scholar] [CrossRef] [Green Version]
- Comerci, D.J.; Martínez-Lorenzo, M.J.; Sieira, R.; Gorvel, J.P.; Ugalde, R.A. Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cell. Microbiol. 2001, 3, 159–168. [Google Scholar] [CrossRef]
- Ke, Y.; Wang, Y.; Li, W.; Chen, Z. Type IV secretion system of Brucella spp. and its effectors. Front. Cell. Infect. Microbiol. 2015, 5, 72. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Dong, H.; Li, J.; Ou, Q.; Lv, Y.; Wang, X.; Xiang, Z.; He, Y.; Wu, Q. RNA-seq reveals the critical role of OtpR in regulating Brucella melitensis metabolism and virulence under acidic stress. Sci. Rep. 2015, 5, 10864. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, X.; Yan, F.; He, Y.; Wei, J.; Zhang, Y.; Liu, L.; Sun, Y. Comparative transcriptome analysis of Brucella melitensis in an acidic environment: Identification of the two-component response regulator involved in the acid resistance and virulence of Brucella. Microb. Pathog. 2016, 91, 92–98. [Google Scholar] [CrossRef]
- Salmon-Divon, M.; Zahavi, T.; Kornspan, D. Transcriptomic analysis of the Brucella melitensis rev.1 vaccine strain in an acidic environment: Insights into virulence attenuation. Front. Microbiol. 2019, 10, 250. [Google Scholar] [CrossRef]
- Rossetti, C.A.; Galindo, C.L.; Garner, H.R.; Adams, L.G. Transcriptional profile of the intracellular pathogen Brucella melitensis following HeLa cells infection. Microb. Pathog. 2011, 51, 338–344. [Google Scholar] [CrossRef] [Green Version]
- Boggiatto, P.M.; Fitzsimmons, D.; Bayles, D.O.; Alt, D.; Vrentas, C.E.; Olsen, S.C. Coincidence cloning recovery of Brucella melitensis RNA from goat tissues: Advancing the in vivo analysis of pathogen gene expression in brucellosis. BMC Mol. Biol. 2018, 19, s12867. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive-PubMed. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants-PubMed. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. EggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2007, 36, D250–D254. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.C.; Frawley, E.R.; Tapscott, T.; Vázquez-Torres, A. Bacterial stress responses during host infection. Cell Host Microbe 2016, 20, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellaire, B.H.; Roop, R.M.; Cardelli, J.A. Opsonized virulent Brucella abortus replicates within nonacidic, endoplasmic reticulum-negative, LAMP-1-positive phagosomes in human monocytes. Infect. Immun. 2005, 73, 3702–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roop, R.M.; Gaines, J.M.; Anderson, E.S.; Caswell, C.C.; Martin, D.W. Survival of the fittest: How Brucella strains adapt to their intracellular niche in the host. Med. Microbiol. Immunol. 2009, 198, 221–238. [Google Scholar] [CrossRef] [Green Version]
- Maloney, P.C.; Kashket, E.R.; Wilson, T.H. A protonmotive force drives ATP synthesis in bacteria. Proc. Natl. Acad. Sci. USA 1974, 71, 3896–3900. [Google Scholar] [CrossRef] [Green Version]
- Mobley, H.L.; Island, M.D.; Hausinger, R.P. Molecular biology of microbial ureases. Microbiol. Mol. Biol. Rev. 1995, 59, 451–480. [Google Scholar] [CrossRef]
- Stingl, K.; Altendorf, K.; Bakker, E.P. Acid survival of Helicobacter pylori: How does urease activity trigger cytoplasmic pH homeostasis? Trends Microbiol. 2002, 10, 70–74. [Google Scholar] [CrossRef]
- Chen, Y.Y.M.; Weaver, C.A.; Burne, R.A. Dual functions of Streptococcus salivarius urease. J. Bacteriol. 2000, 182, 4667–4669. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Bhinderwala, F.; Lehman, M.K.; Thomas, V.C.; Chaudhari, S.S.; Yamada, K.J.; Foster, K.W.; Powers, R.; Kielian, T.; Fey, P.D.; et al. Urease is an essential component of the acid response network of Staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef]
- Corbel, M.J.; Hendry, D.M. Urease activity of Brucella species. Res. Vet. Sci. 1985, 38, 252–253. [Google Scholar] [CrossRef]
- Sun, Y.; Fukamachi, T.; Saito, H.; Kobayashi, H. Adenosine deamination increases the survival under acidic conditions in Escherichia coli. J. Appl. Microbiol. 2012, 112, 775–781. [Google Scholar] [CrossRef]
- Piddock, L.J.V. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 2006, 19, 382–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqua, M.; Grossi, M.; Scinicariello, S.; Aussel, L.; Barras, F.; Colonna, B.; Prosseda, G. The MFS efflux pump EmrKY contributes to the survival of Shigella within macrophages. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, A.M.; Webber, M.A.; Cooles, S.; Randall, L.P.; La Ragione, R.M.; Woodward, M.J.; Piddock, L.J.V. The AcrAB-TolC efflux system of Salmonella enterica serovar typhimurium plays a role in pathogenesis. Cell. Microbiol. 2006, 8, 847–856. [Google Scholar] [CrossRef]
- Quillin, S.J.; Schwartz, K.T.; Leber, J.H. The novel Listeria monocytogenes bile sensor BrtA controls expression of the cholic acid efflux pump MdrT. Mol. Microbiol. 2011, 81, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Bina, X.R.; Provenzano, D.; Nguyen, N.; Bina, J.E. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect. Immun. 2008, 76, 3595–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang-Kan, X.; Blair, J.M.A.; Chirullo, B.; Betts, J.; la Ragione, R.M.; Ivens, A.; Ricci, V.; Opperman, T.J.; Piddock, L.J.V. Lack of AcrB efflux function confers loss of virulence on Salmonella enterica serovar typhimurium. MBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, J.; Xu, H.; Li, D. Role of a major facilitator superfamily transporter in adaptation capacity of Penicillium funiculosum under extreme acidic stress. Fungal Genet. Biol. 2014, 69, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Maleki, F.; Khosravi, A.; Nasser, A.; Taghinejad, H.; Azizian, M. Bacterial heat shock protein activity. J. Clin. Diagn. Res. 2016, 10, BE01-3. [Google Scholar] [CrossRef]
- Uversky, V.; Goto, Y. Acid denaturation and anion-induced folding of globular proteins: Multitude of equilibrium partially folded intermediates. Curr. Protein Pept. Sci. 2009, 10, 447–455. [Google Scholar] [CrossRef]
- Ohnishi, H.; Mizunoe, Y.; Takade, A.; Tanaka, Y.; Miyamoto, H.; Harada, M.; Yoshida, S.I. Legionella dumoffii DjlA, a member of the DnaJ family, is required for intracellular growth. Infect. Immun. 2004, 72, 3592–3603. [Google Scholar] [CrossRef] [Green Version]
- Gajiwala, K.S.; Burley, S.K. HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. J. Mol. Biol. 2000, 295, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Jiao, W.; Hu, J.; Zhang, J.; Liu, C.; Fu, X.; Shen, D.; Xia, B.; Chang, Z. Periplasmic protein HdeA exhibits chaperone-like activity exclusively within stomach pH range by transforming into disordered conformation. J. Biol. Chem. 2005, 280, 27029–27034. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Proc. Free Rad. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef]
- Vuilleumier, S. Bacterial glutathione S-transferases: What are they good for? J. Bacteriol. 1997, 179, 1431–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allocati, N.; Favaloro, B.; Masulli, M.; Alexeyev, M.F.; di Ilio, C. Proteus mirabilis glutathione S-transferase B1-1 is involved in protective mechanisms against oxidative and chemical stresses. Biochem. J. 2003, 373, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, L.M.; Yohannes, E.; Bondurant, S.S.; Radmacher, M.; Slonczewski, J.L. pH regulates genes for flagellar motility, catabolism, and oxidative stress in Escherichia coli K-12. J. Bacteriol. 2005, 187, 304–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabayek, S.; Spellerberg, B. Acid stress response mechanisms of group B streptococci. Front. Cell. Infect. Microbiol. 2017, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Santi, I.; Grifantini, R.; Jiang, S.M.; Brettoni, C.; Grandi, G.; Wessels, M.R.; Soriani, M. CsrRS regulates group B Streptococcus virulence gene expression in response to environmental pH: A new perspective on vaccine development. J. Bacteriol. 2009, 191, 5387–5397. [Google Scholar] [CrossRef] [Green Version]
- Loose, M.; Mitchison, T.J. The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat. Cell Biol. 2014, 16, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Sherratt, D.J.; Arciszewska, L.K.; Crozat, E.; Graham, J.E.; Grainge, I. The Escherichia coli DNA translocase FtsK. Biochem. Soc. Trans. 2010, 38, 395–398. [Google Scholar] [CrossRef]
- Huisman, O.; D’Ari, R.; Gottesman, S. Cell-division control in Escherichia coli: Specific induction of the SOS function SfiA protein is sufficient to block septation. Proc. Natl. Acad. Sci. USA 1984, 81, 4490–4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, Y.Y.; Fang, W.H.; Lee, C.C.; Chen, Y.P.; Yuan, H.S. Structural insights into DNA repair by RNase T-an exonuclease processing 3′ end of structured DNA in repair pathways. PLoS Biol. 2014, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovett, S.T. The DNA Exonucleases of Escherichia coli. EcoSal Plus 2011, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.P.; Miller, C.N.; Child, R.; Cundiff, J.A.; Celli, J. Postreplication roles of the Brucella VirB type IV secretion system uncovered via conditional expression of the VirB11 ATPase. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, S.C.; Palmer, M.V. Advancement of knowledge of Brucella over the past 50 years. Vet. Pathol. 2014, 51, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Mora, D.; Arioli, S. Microbial urease in health and disease. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Zhang, J.; Wang, M.; Du, G.; Chen, J. Lactobacillus casei combats acid stress by maintaining cell membrane functionality. J. Ind. Microbiol. Biotechnol. 2012, 39, 1031–1039. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Kandasamy, S.; Chattha, K.S.; Rajashekara, G.; Saif, L.J. Comparison of probiotic lactobacilli and bifidobacteria effects, immune responses and rotavirus vaccines and infection in different host species. Vet. Immunol. Immunopathol. 2016, 172, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Qin, Q.; Guo, H.; Liu, S.; Ge, S.; Zhang, H.; Cui, J.; Ren, F. Effect of pre-stressing on the acid-stress response in Bifidobacterium revealed using proteomic and physiological approaches. PLoS ONE 2015, 10, e0117702. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene | Fold Change |
|---|---|---|
| BMEI1655 | urease accessory protein ureD 1 | 14.38 |
| BME_RS08240 | urease subunit gamma | 11.29 |
| BMEI1653 | urease subunit beta | 11.22 |
| BMEI1652 | urease subunit alpha 1 | 7.86 |
| BMEI1650 | urease accessory protein UreF 2 | 5.37 |
| BMEI0642 | urea transporter | 4.49 |
| BMEI0556 | MFS transporter | 4.33 |
| BMEII0027 | type IV secretion system protein VirB3 | 2.92 |
| BMEII0025 | type IV secretion system protein VirB1 | 2.70 |
| BMEI0181 | MFS transporter | 2.57 |
| BMEII0280 | MFS transporter | 2.53 |
| BMEII0028 | type IV secretion system protein VirB4 | 2.2 |
| BMEI0564 | molecular chaperone DjlA | 2.07 |
| Gene ID | Gene | Fold Change (Acidic vs. Neutral pH) | p-Value (t-Test) |
|---|---|---|---|
| BMEI1900 | cytochrome o ubiquinol oxidase subunit I | 4.93 | <0.001 |
| BMEII1119 | MFS transporter | 12.7 | 0.02 |
| BMEII0025 | type IV secretion system protein VirB1 | 81.8 | 0.01 |
| BME_RS13825 | DNA translocase FtsK | −17.7 | <0.001 |
| BMEI2002 | molecular chaperone DnaK | −12 | 0.037 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornspan, D.; Zahavi, T.; Salmon-Divon, M. The Acidic Stress Response of the Intracellular Pathogen Brucella melitensis: New Insights from a Comparative, Genome-Wide Transcriptome Analysis. Genes 2020, 11, 1016. https://doi.org/10.3390/genes11091016
Kornspan D, Zahavi T, Salmon-Divon M. The Acidic Stress Response of the Intracellular Pathogen Brucella melitensis: New Insights from a Comparative, Genome-Wide Transcriptome Analysis. Genes. 2020; 11(9):1016. https://doi.org/10.3390/genes11091016
Chicago/Turabian StyleKornspan, David, Tamar Zahavi, and Mali Salmon-Divon. 2020. "The Acidic Stress Response of the Intracellular Pathogen Brucella melitensis: New Insights from a Comparative, Genome-Wide Transcriptome Analysis" Genes 11, no. 9: 1016. https://doi.org/10.3390/genes11091016