Comparative Transcriptomic Analysis of the Larval and Adult Stages of Taenia pisiformis

Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Illumina Hi-Seq Sequencing
2.3. De novo Assembly, Expression, and Annotation Analysis
2.4. Differentially Expressed Genes Identification between TpA and TpM
2.5. Quantitative Polymerase Chain Reaction (qPCR) Analysis
3. Results
3.1. Sequencing and De Novo Transcriptome Assembly
3.2. Gene Transcription Profile and Annotation
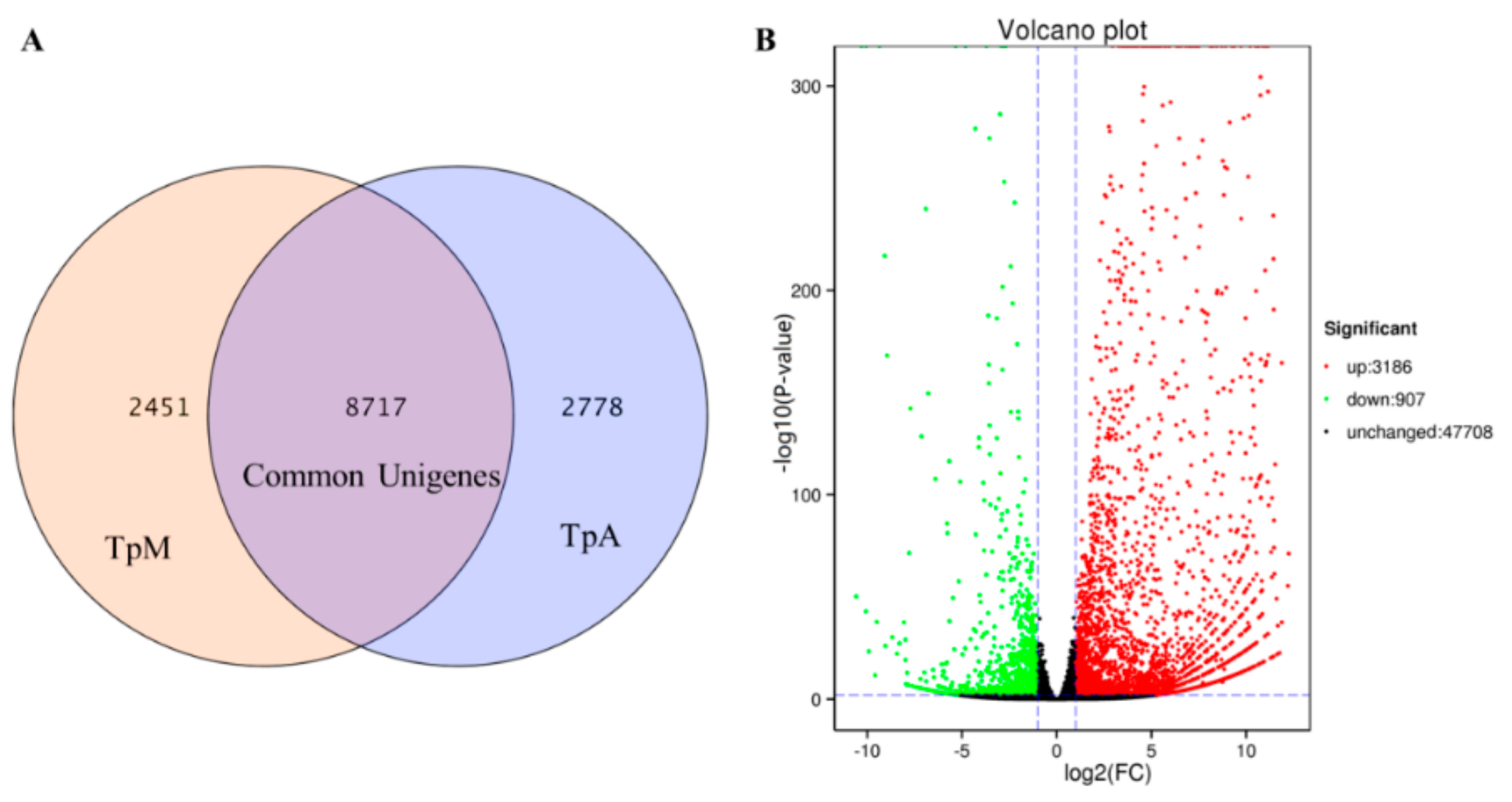
3.3. Identification and Classification of Differentially Expressed Genes
3.4. Key Candidate Differentially Expressed Genes and Pathways Involved in TpA Development
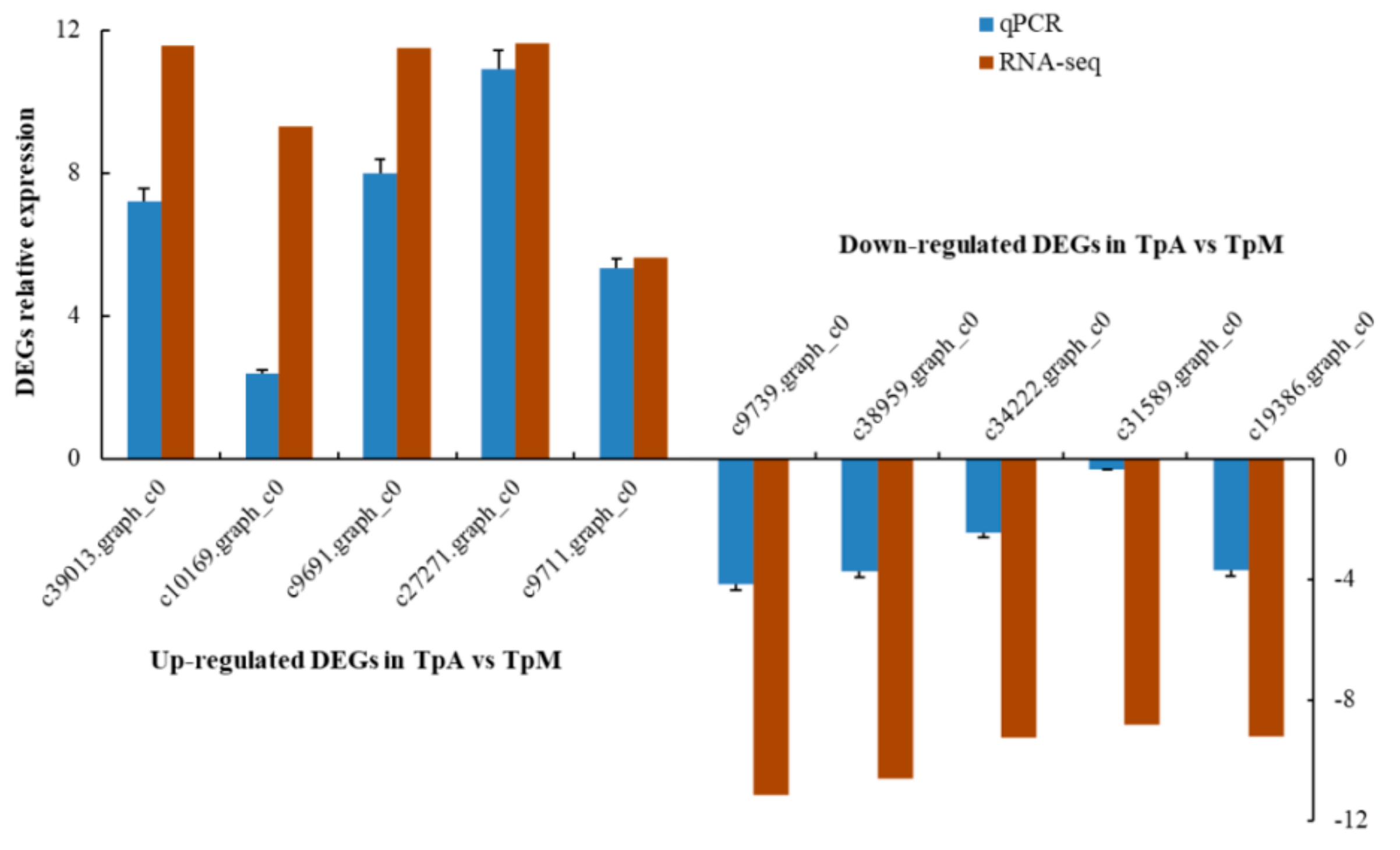
3.5. Quantitative Polymerase Chain Reaction (qPCR) Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loos-Frank, B. An up-date of verster’s (1969) ‘taxonomic revision of the genus Taenia linnaeus’ (cestoda) in table format. Syst. Parasitol. 2000, 45, 155–183. [Google Scholar] [CrossRef] [PubMed]
- Foronda, P.; Valladares, B.; Lorenzo-Morales, J.; Ribas, A.; Feliu, C.; Casanova, J.C. Helminths of the wild rabbit (Oryctolagus cuniculus) in Macaronesia. J. Parasitol. 2003, 89, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Toral-Bastida, E.; Garza-Rodriguez, A.; Jimenez-Gonzalez, D.E.; Garcia-Cortes, R.; Avila-Ramirez, G.; Maravilla, P.; Flisser, A. Development of Taenia pisiformis in golden hamster (Mesocricetus auratus). Parasit. Vectors 2011, 4, 147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Du, A.; Zhang, X.; Wu, Y.; Tong, F.; Wu, G. Research of harmfulness of Cysticercus pisiformis in rabbit. J. Zhejiang Agric. Sci. 2008, 3, 372–373. (In Chinese) [Google Scholar]
- Martinez-Moreno, F.J.; Hernandez, S.; Lopez-Cobos, E.; Becerra, C.; Acosta, I.; Martinez-Moreno, A. Estimation of canine intestinal parasites in Cordoba (Spain) and their risk to public health. Vet. Parasitol. 2007, 143, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Coman, B.J.; Rickard, M.D. The location of Taenia pisiformis, Taenia ovis and Taenia hydatigena in the gut of the dog and its effect on net environmental contamination with ova. Z. Parasitenkd. 1975, 47, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Chabalgoity, J.A.; Harrison, J.A.; Esteves, A.; Demarco de Hormaeche, R.; Ehrlich, R.; Khan, C.M.; Hormaeche, C.E. Expression and immunogenicity of an Echinococcus granulosus fatty acid-binding protein in live attenuated Salmonella vaccine strains. Infect. Immun. 1997, 65, 2402–2412. [Google Scholar]
- Felsmann, M.; Michalski, M.; Felsmann, M.; Sokol, R.; Szarek, J.; Strzyzewska-Worotynska, E. Invasive forms of canine endoparasites as a potential threat to public health—A review and own studies. Ann. Agric. Environ. Med. 2017, 24, 245–249. [Google Scholar] [CrossRef]
- Kohansal, M.H.; Nourian, A.; Haniloo, A.; Fazaeli, A. Molecular detection of Taenia spp. In dogs’ feces in Zanjan province, northwest of Iran. Vet. World 2017, 10, 445–449. [Google Scholar] [CrossRef][Green Version]
- Smyth, J.D.; Miller, H.J.; Howkins, A.B. Further analysis of the factors controlling strobilization, differentiation, and maturation of Echinococcus granulosus in vitro. Exp. Parasitol. 1967, 21, 31–41. [Google Scholar] [CrossRef]
- Liu, G.H.; Xu, M.J.; Chang, Q.C.; Gao, J.F.; Wang, C.R.; Zhu, X.Q. De novo transcriptomic analysis of the female and male adults of the blood fluke Schistosoma turkestanicum. Parasit. Vectors 2016, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.Q.; Li, Y.W.; Wang, H.Q.; Wang, J.L.; Ni, L.Y.; Yang, M.; Lao, G.F.; Luo, X.C.; Li, A.X.; Dan, X.M. Comparative transcriptional profile of the fish parasite Cryptocaryon irritans. Parasit. Vectors 2016, 9, 630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Cong, W.; Elsheikha, H.M.; Liu, G.H.; Ma, J.G.; Huang, W.Y.; Zhao, Q.; Zhu, X.Q. De novo transcriptome sequencing and analysis of the juvenile and adult stages of Fasciola gigantica. Infect. Genet. Evol. 2017, 51, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Basika, T.; Paludo, G.P.; Araujo, F.M.; Salim, A.C.; Pais, F.; Maldonado, L.; Macchiaroli, N.; Camargo de Lima, J.; Rosenzvit, M.; Oliveira, G.C.; et al. Transcriptomic profile of two developmental stages of the cestode parasite Mesocestoides corti. Mol. Biochem. Parasitol. 2019, 229, 35–46. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Zhang, S.; Cai, X.; Luo, X.; Wang, S.; Guo, A.; Hou, J.; Wu, R. Molecular cloning and characterization of leucine aminopeptidase gene from Taenia pisiformis. Exp. Parasitol. 2018, 186, 1–9. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yu, J.; Xu, J.; Wang, W.; Ji, L.L.; Yang, C.Z.; Yu, H. Comparative transcriptomes analysis of Taenia pisiformis at different development stages. bioRxiv 2018, 490276. [Google Scholar] [CrossRef]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, X.; Cui, Y.; Zheng, X.; Yang, H. Comparative transcriptome analysis reveals hormone signaling genes involved in the launch of culm-shape differentiation in Dendrocalamus sinicus. Genes 2017, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Cao, B.; Long, Y.; Tukayo, M.; Feng, C.; Fang, W.; Luo, D. Comparative transcriptomic analysis of two important life stages of Angiostrongylus cantonensis: Fifth-stage larvae and female adults. Genet. Mol. Biol. 2017, 40, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.Y.; Fu, Y.; Wu, X.H.; Xie, Y.; Nie, H.M.; Chen, L.; Nong, X.; Gu, X.B.; Wang, S.X.; Peng, X.R.; et al. Annotation of the transcriptome from Taenia pisiformis and its comparative analysis with three Taeniidae species. PLoS ONE 2012, 7, e32283. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yuan, G.; Zheng, Y.; Luo, X.; Zhang, S.; Ding, J.; Jing, Z.; Lu, C. Effective production and purification of the glycosylated TSOL18 antigen, which is protective against pig cysticercosis. Infect. Immun. 2008, 76, 767–770. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dezaki, E.S.; Yaghoobi, M.M.; Taheri, E.; Almani, P.G.; Tohidi, F.; Gottstein, B.; Harandi, M.F. Differential expression of Hox and Notch genes in larval and adult stages of Echinococcus granulosus. Korean J. Parasitol. 2016, 54, 653–658. [Google Scholar] [CrossRef]
- Wu, X.; Fu, Y.; Yang, D.; Zhang, R.; Zheng, W.; Nie, H.; Xie, Y.; Yan, N.; Hao, G.; Gu, X.; et al. Detailed transcriptome description of the neglected cestode Taenia multiceps. PLoS ONE 2012, 7, e45830. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, W.; Zhang, L.; Zhang, Z.; Li, J.; Lu, G.; Zhu, Y.; Wang, Y.; Huang, Y.; Liu, J.; et al. The genome of the hydatid tapeworm Echinococcus granulosus. Nat. Genet. 2013, 45, 1168–1175. [Google Scholar] [CrossRef]
- Wang, S.; Wang, S.; Luo, Y.; Xiao, L.; Luo, X.; Gao, S.; Dou, Y.; Zhang, H.; Guo, A.; Meng, Q.; et al. Comparative genomics reveals adaptive evolution of Asian tapeworm in switching to a new intermediate host. Nat. Commun. 2016, 7, 12845. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.J.; Zarowiecki, M.; Holroyd, N.; Garciarrubio, A.; Sanchez-Flores, A.; Brooks, K.L.; Tracey, A.; Bobes, R.J.; Fragoso, G.; Sciutto, E.; et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature 2013, 496, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Halton, D.W. Nutritional adaptations to parasitism within the platyhelminthes. Int. J. Parasitol. 1997, 27, 693–704. [Google Scholar] [CrossRef]
- Huberts, D.H.; van der Klei, I.J. Moonlighting proteins: An intriguing mode of multitasking. Biochim. Biophys. Acta 2010, 1803, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef]
- Lorenzatto, K.R.; Monteiro, K.M.; Paredes, R.; Paludo, G.P.; da Fonseca, M.M.; Galanti, N.; Zaha, A.; Ferreira, H.B. Fructose-bisphosphate aldolase and enolase from Echinococcus granulosus: Genes, expression patterns and protein interactions of two potential moonlighting proteins. Gene 2012, 506, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Guo, A.; Zhu, X.; You, Y.; Hou, J.; Wang, Q.; Luo, X.; Cai, X. Identification and functional characterization of alpha-enolase from Taenia pisiformis metacestode. Acta Trop. 2015, 144, 31–40. [Google Scholar] [CrossRef]
- Matsui, M.; Fowler, J.H.; Walling, L.L. Leucine aminopeptidases: Diversity in structure and function. Biol. Chem. 2006, 387, 1535–1544. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of peptidases. Biochem. J. 1993, 290 Pt 1, 205–218. [Google Scholar] [CrossRef]
- Muhamad, N.; Simcock, D.C.; Pedley, K.C.; Simpson, H.V.; Brown, S. The kinetic properties of the glutamate dehydrogenase of Teladorsagia circumcincta and their significance for the lifestyle of the parasite. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2011, 159, 71–77. [Google Scholar] [CrossRef]
- Sako, Y.; Yamasaki, H.; Nakaya, K.; Nakao, M.; Ito, A. Cloning and characterization of cathepsin L-like peptidases of Echinococcus multilocularis metacestodes. Mol. Biochem. Parasitol. 2007, 154, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Li, A.H.; Moon, S.U.; Park, Y.K.; Na, B.K.; Hwang, M.G.; Oh, C.M.; Cho, S.H.; Kong, Y.; Kim, T.S.; Chung, P.R. Identification and characterization of a cathepsin L-like cysteine protease from Taenia solium metacestode. Vet. Parasitol. 2006, 141, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, S.; Luo, X.; Hou, J.; Zhu, X.; Cai, X. Cloning and characterization of a cathepsin L-like cysteine protease from Taenia pisiformis. Vet. Parasitol. 2013, 194, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Kim, J.G.; Han, X.; Bae, Y.A.; Park, W.J.; Kang, I.; Wang, H.; Kong, Y. Biochemical characterization of Echinococcus multilocularis antigen B3 reveals insight into adaptation and maintenance of parasitic homeostasis at the host-parasite interface. J. Proteome Res. 2017, 16, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Willms, K. Morphology and biochemistry of the pork tapeworm, Taenia solium. Curr. Top. Med. Chem. 2008, 8, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Aceves-Ramos, A.; de la Torre, P.; Hinojosa, L.; Ponce, A.; Garcia-Villegas, R.; Laclette, J.P.; Bobes, R.J.; Romano, M.C. Cloning, characterization and functional expression of Taenia solium 17 beta-hydroxysteroid dehydrogenase. Gen. Comp. Endocrinol. 2014, 203, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Croce, J.C.; McClay, D.R. Evolution of the Wnt pathways. Methods Mol. Biol. 2008, 469, 3–18. [Google Scholar]
- Wu, M.; Herman, M.A. A novel noncanonical Wnt pathway is involved in the regulation of the asymmetric B cell division in C. elegans. Dev. Biol. 2006, 293, 316–329. [Google Scholar] [CrossRef]
- Koziol, U.; Jarero, F.; Olson, P.D.; Brehm, K. Comparative analysis of Wnt expression identifies a highly conserved developmental transition in flatworms. BMC Biol. 2016, 14, 10. [Google Scholar] [CrossRef]
- Dezaki, E.S.; Yaghoubi, M.M.; Spiliotis, M.; Boubaker, G.; Taheri, E.; Almani, P.G.; Tohidi, F.; Harandi, M.F.; Gottstein, B. Comparison of ex vivo harvested and in vitro cultured materials from Echinococcus granulosus by measuring expression levels of five genes putatively involved in the development and maturation of adult worms. Parasitol. Res. 2016, 115, 4405–4416. [Google Scholar] [CrossRef]
- Riddiford, N.; Olson, P.D. Wnt gene loss in flatworms. Dev. Genes Evol. 2011, 221, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Gurley, K.A.; Elliott, S.A.; Simakov, O.; Schmidt, H.A.; Holstein, T.W.; Sanchez Alvarado, A. Expression of secreted Wnt pathway components reveals unexpected complexity of the planarian amputation response. Dev. Biol. 2010, 347, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Luo, X.; Wang, S.; Yin, C.; Zhang, S.; Zhu, X.; Dou, Y.; Cai, X. Sequence analysis and molecular characterization of Wnt4 gene in metacestodes of Taenia solium. Korean J. Parasitol. 2014, 52, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Li, H.F.; Wang, X.B.; Jin, Y.P.; Xia, Y.X.; Feng, X.G.; Yang, J.M.; Qi, X.Y.; Yuan, C.X.; Lin, J.J. Wnt4, the first member of the Wnt family identified in Schistosoma japonicum, regulates worm development by the canonical pathway. Parasitol. Res. 2010, 107, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.E.; Zhou, B.; Solomon, N.; Sunohara, M.; Li, C.; Nguyen, C.; Liu, Y.; Pan, J.H.; Minoo, P.; Crandall, E.D.; et al. p300/β-catenin interactions regulate adult progenitor cell differentiation downstream of Wnt5a/protein kinase c (PKC). J. Biol. Chem. 2016, 291, 6569–6582. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R. Alternative wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a007914. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Vo, N.; Goodman, R.H. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef]
- Rana, A.K.; Misra-Bhattacharya, S. Current drug targets for helminthic diseases. Parasitol. Res. 2013, 112, 1819–1831. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef]
- EI Kouni, M.H. Pyrimidine metabolism in Schistosomes: A comparison with other parasites and the search for potential chemotherapeutic targets. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2017, 213, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.K.; Datta, R.; Sen, B. Antiparasitic chemotherapy: Tinkering with the purine salvage pathway. Adv. Exp. Med. Biol. 2008, 625, 116–132. [Google Scholar] [PubMed]
- Manneck, T.; Keiser, J.; Muller, J. Mefloquine interferes with glycolysis in schistosomula of Schistosoma mansoni via inhibition of enolase. Parasitology 2012, 139, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Contreras, D.; Skelly, P.J.; Landa, A.; Shoemaker, C.B.; Laclette, J.P. Molecular and functional characterization and tissue localization of 2 glucose transporter homologues (TGTP1 and TGTP2) from the tapeworm Taenia solium. Parasitology 1998, 117 Pt 6, 579–588. [Google Scholar] [CrossRef]
- Roy, B.; Giri, B.R. α-viniferin and resveratrol induced alteration in the activities of some energy metabolism related enzymes in the cestode parasite Raillietina echinobothrida. Acta Trop. 2016, 154, 102–106. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | TpM | TpA | TpM + TpA |
|---|---|---|---|
| clean reads | 53,815,898 | 45,351,168 | 99,167,066 |
| Total bases (Gb) | 16.0 | 13.48 | 29.48 |
| (G + C) % | 49.58 | 49.66 | 49.62 |
| Q20 of clean reads (%) | 96.79 | 96.65 | 96.72 |
| Q30 of clean reads (%) | 92.26 | 92.04 | 92.15 |
| Numbers of transcripts | / | / | 354,182 |
| Mean length (transcript) | / | / | 3209 |
| N50 length (transcript) | / | / | 4893 |
| Total unigene | / | / | 68,588 |
| Mean length (unigene) | / | / | 789 |
| N50 length (unigene) | / | / | 1485 |
| Annotated Databases | Annotated Number of Unigenes | 300 ≤ Length < 1000nt | Length ≥ 1000 nt |
|---|---|---|---|
| COG | 8613 | 3118 | 2362 |
| GO | 6149 | 2100 | 2493 |
| KEGG | 6076 | 1828 | 3195 |
| KOG | 10,920 | 3292 | 4802 |
| Pfam | 13,342 | 4476 | 5334 |
| Swissprot | 7794 | 2327 | 4031 |
| eggNOG | 17,413 | 5800 | 5430 |
| Nr | 14,607 | 4562 | 7003 |
| All | 20,720 | 6846 | 7097 |
| KEGG pathway | Pathway ID | DEGs No. | p-value |
|---|---|---|---|
| ECM-receptor interaction | ko04512 | 10 | 8.61 × 10−7 |
| Vasopressin-regulated water reabsorption | ko04962 | 8 | 2.15 × 10−6 |
| Neuroactive ligand-receptor interaction | ko04080 | 12 | 9.20 × 10−5 |
| Phagosome | ko04145 | 26 | 6.38 × 10−4 |
| MAPK signaling pathway | ko04010 | 12 | 9.15 × 10−4 |
| Sphingolipid metabolism | ko00600 | 8 | 9.92 × 10−4 |
| Fructose and mannose metabolism | ko00051 | 14 | 1.08 × 10−3 |
| Estrogen signaling pathway | ko04915 | 8 | 2.68 × 10−3 |
| Fanconi anemia pathway | ko03460 | 9 | 3.32 × 10−3 |
| Nicotinate and nicotinamide metabolism | ko00760 | 7 | 3.63 × 10−3 |
| Lysosome | ko04142 | 19 | 4.17 × 10−3 |
| Morphine addiction | ko05032 | 3 | 4.62 × 10−3 |
| Folate biosynthesis | ko00790 | 5 | 5.99 × 10−3 |
| Wnt signaling pathway | ko04310 | 19 | 5.99 × 10−3 |
| Platelet activation | ko04611 | 4 | 6.61 × 10−3 |
| Glutathione metabolism | ko00480 | 13 | 8.78 × 10−3 |
| Fatty acid biosynthesis | ko00061 | 6 | 9.19 × 10−3 |
| Gap junction | ko04540 | 9 | 1.00 × 10−2 |
| Galactose metabolism | ko00052 | 10 | 1.00 × 10−2 |
| Melanogenesis | ko04916 | 8 | 1.11 × 10−2 |
| Transcriptome gene ID | log2FC | FDR | Gene annotation |
|---|---|---|---|
| Carbohydrate transport and metabolism | |||
| c18758.graph_c0 | 11.3187 | 1.19 × 10−72 | Fructose 1,6 bisphosphate aldolase |
| c10141.graph_c0 | 11.0122 | 5.32 × 10−164 | Hexokinase |
| c39583.graph_c0 | 9.7607 | 1.55 × 10−32 | Glycosyltransferase |
| c39382.graph_c0 | 8.1210 | 4.30 × 10−169 | Pyruvate kinase |
| c39314.graph_c0 | 6.8155 | 1.71 × 10−245 | Enolase |
| c9826.graph_c0 | 6.4117 | 0 | Glyceraldehyde-3-phosphate dehydrogenase |
| c29987.graph_c0 | 4.8445 | 7.00 × 10−138 | Phosphoglycerate mutase |
| c18823.graph_c0 | 2.5367 | 1.87 × 10−247 | alpha glucosidase |
| Amino acid transport and metabolism | |||
| c39047.graph_c0 | 9.1324 | 6.51 × 10−283 | Cathepsin L cysteine proteinase |
| c31605.graph_c0 | 8.1481 | 0 | Cytosolic carboxypeptidase protein 5 |
| c37268.graph_c0 | 8.0330 | 1.63 × 10−87 | Neutral amino acid transporter A |
| c38431.graph_c0 | 7.5075 | 1.58 × 10−40 | Enteropeptidase |
| c38964.graph_c0 | 7.4278 | 0 | Glutamate dehydrogenase |
| c39380.graph_c0 | 6.7885 | 1.18 × 10−216 | Cytosolic non specific dipeptidase |
| c9617.graph_c0 | 5.9653 | 0 | Excitatory amino acid transporter |
| c9711.graph_c0 | 5.7216 | 0 | Leucyl aminopeptidase |
| c18022.graph_c0 | 5.2819 | 0 | Arginase-2 |
| c35080.graph_c0 | 4.1657 | 3.16 × 10−195 | Aromatic amino acid decarboxylase |
| c31258.graph_c0 | 3.9168 | 1.57 × 10−195 | Cytosolic carboxypeptidase 1 |
| c32085.graph_c0 | 3.2382 | 3.90 × 10−113 | Cationic amino acid transporter |
| c9897.graph_c0 | 2.9532 | 0 | Aspartate aminotransferase |
| c24988.graph_c0 | 2.5384 | 3.29 × 10−23 | L-asparaginase |
| Lipid transport and metabolism | |||
| c36506.graph_c1 | 8.5344 | 1.20 × 10−9 | 2-acylglycerol O-acyltransferase 2-A |
| c36947.graph_c0 | 6.9948 | 1.91 × 10−85 | Phospholipase D1 |
| c33881.graph_c0 | 4.4314 | 1.95 × 10−67 | Protein bicaudal C1 |
| c38848.graph_c0 | 3.4956 | 1.95 × 10−23 | Cytosolic fatty acid binding protein |
| c9852.graph_c0 | 3.3705 | 1.64 × 10−204 | Elongation of very long chain fatty acids |
| c32099.graph_c0 | 3.2596 | 8.83 × 10−152 | Clavesin-2 |
| c19373.graph_c0 | 2.6551 | 3.73 × 10−100 | SEC14-like protein |
| c35827.graph_c0 | 2.4747 | 1.79 × 10−35 | Choline O acetyltransferase |
| c39590.graph_c0 | 2.2559 | 1.65 × 10−26 | Acetylcholinesterase |
| c37723.graph_c0 | 2.2167 | 8.66 × 10−98 | Phosphatidate phosphatase |
| Inorganic ion transport and metabolism | |||
| c10169.graph_c0 | 8.9320 | 1.21 × 10−33 | Major intrinsic protein |
| c32341.graph_c0 | 7.0015 | 2.85 × 10−63 | Sodium/hydrogen exchanger family |
| c18882.graph_c0 | 5.3420 | 0 | Alkaline phosphatase |
| c24171.graph_c0 | 4.9420 | 9.49 × 10−50 | ZIP Zinc transporter |
| c35064.graph_c0 | 3.8998 | 2.39 × 10−33 | Sodium/hydrogen exchanger family |
| c37766.graph_c0 | 3.5535 | 1.23 × 10−7 | KCNQ voltage-gated potassium channel |
| Nucleotide transport and metabolism | |||
| c23643.graph_c0 | 7.8158 | 2.29 × 10−117 | Adenylate cyclase 9 |
| c10172.graph_c0 | 5.0739 | 2.84 × 10−71 | Thymidine kinase |
| c10055.graph_c0 | 4.9673 | 2.52 × 10−7 | Purine nucleoside phosphorylase |
| c31659.graph_c0 | 4.1400 | 0 | Thioredoxin domain containing protein 3 |
| c35152.graph_c0 | 4.0682 | 0 | tRNA-specific adenosine deaminase |
| c31658.graph_c0 | 2.9613 | 2.23 × 10−147 | Equilibrative nucleoside transporter 3 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S. Comparative Transcriptomic Analysis of the Larval and Adult Stages of Taenia pisiformis. Genes 2019, 10, 507. https://doi.org/10.3390/genes10070507
Zhang S. Comparative Transcriptomic Analysis of the Larval and Adult Stages of Taenia pisiformis. Genes. 2019; 10(7):507. https://doi.org/10.3390/genes10070507
Chicago/Turabian StyleZhang, Shaohua. 2019. "Comparative Transcriptomic Analysis of the Larval and Adult Stages of Taenia pisiformis" Genes 10, no. 7: 507. https://doi.org/10.3390/genes10070507
APA StyleZhang, S. (2019). Comparative Transcriptomic Analysis of the Larval and Adult Stages of Taenia pisiformis. Genes, 10(7), 507. https://doi.org/10.3390/genes10070507