Next Generation Sequencing Identifies Five Novel Mutations in Lebanese Patients with Bardet–Biedl and Usher Syndromes

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Clinical Examinations
2.2. Molecular Analysis and Mutations Detection
2.2.1. DNA Extraction
2.2.2. Targeted Next-Generation Sequencing
2.2.3. Whole-Exome Sequencing
2.2.4. Analysis of Annotated Sequencing Data
2.2.5. In-Silico Evaluation of the Pathogenicity of Candidate Mutations
2.2.6. Polymerase Chain Reaction, Sanger Sequencing, and Co-Segregation Analysis
2.3. Genotype–Phenotype Associations
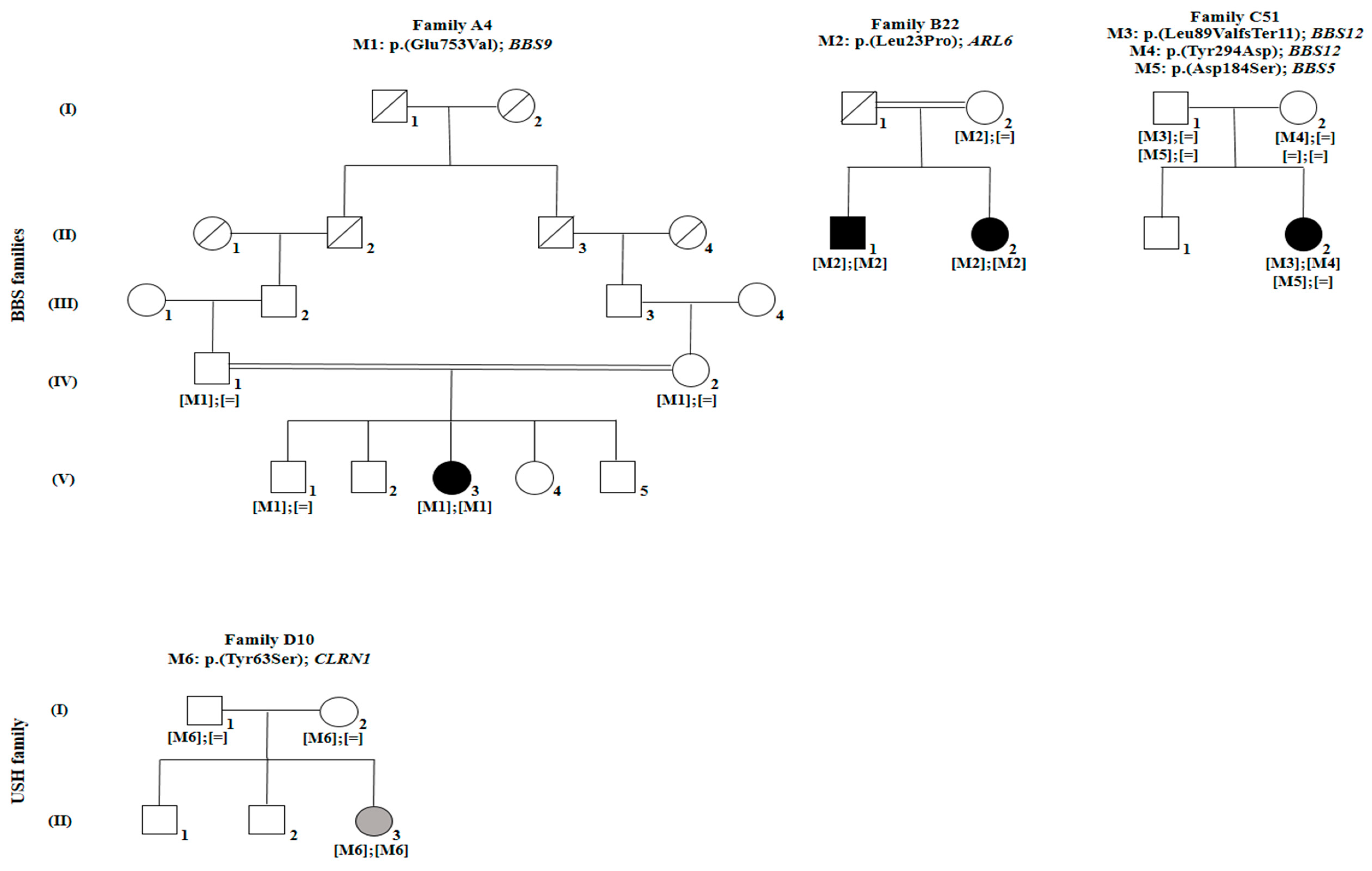
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef] [PubMed]
- Bessant, D.A.; Ali, R.R.; Bhattacharya, S.S. Molecular genetics and prospects for therapy of the inherited retinal dystrophies. Curr. Opin. Genet. Dev. 2001, 11, 307–316. [Google Scholar] [CrossRef]
- Sahel, J.A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef] [PubMed]
- Astuti, G.D.N.; van den Born, L.I.; Khan, M.I.; Hamel, C.P.; Bocquet, B.; Manes, G.; Quinodoz, M.; Ali, M.; Toomes, C.; McKibbin, M.; et al. Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes. Genes 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, P.J. Genetics of inherited retinal disease. J. R. Soc. Med. 2006, 99, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Boughman, J.A.; Vernon, M.; Shaver, K.A. Usher syndrome: Definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 1983, 36, 595–603. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar]
- Mockel, A.; Perdomo, Y.; Stutzmann, F.; Letsch, J.; Marion, V.; Dollfus, H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011, 30, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet-Biedl Syndrome—Now and in the Future. Front. Pediatr. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farag, T.I.; Teebi, A.S. Bardet-Biedl and Laurence-Moon syndromes in a mixed Arab population. Clin. Genet. 1988, 33, 78–82. [Google Scholar] [CrossRef]
- Farag, T.I.; Teebi, A.S. High incidence of Bardet Biedl syndrome among the Bedouin. Clin. Genet. 1989, 36, 463–464. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Green, J.S.; Fan, Y.; Bhogal, A.K.; Dicks, E.; Fernandez, B.A.; Stefanelli, M.; Murphy, C.; Cramer, B.C.; Dean, J.C.; et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: A 22-year prospective, population-based, cohort study. Am. J. Med. Genet. Part A 2005, 132, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbour, B.; Salameh, P. Consanguinity in Lebanon: Prevalence, distribution and determinants. J. Biosoc. Sci. 2009, 41, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]
- Lee, K.; Garg, S. Navigating the current landscape of clinical genetic testing for inherited retinal dystrophies. Genet. Med. 2015, 17, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Audo, I.; Bujakowska, K.M.; Leveillard, T.; Mohand-Said, S.; Lancelot, M.E.; Germain, A.; Antonio, A.; Michiels, C.; Saraiva, J.P.; Letexier, M.; et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J. Rare Dis. 2012, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Jaffal, L.; Joumaa, W.H.; Assi, A.; Helou, C.; Condroyer, C.; El Dor, M.; Cherfan, G.; Zeitz, C.; Audo, I.; Zibara, K.; et al. Novel Missense Mutations in BEST1 Are Associated with Bestrophinopathies in Lebanese Patients. Genes 2019, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019, 531210. [Google Scholar] [CrossRef] [Green Version]
- The NHLBI Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Program. BRAVO Variant Browser: University of Michigan and NHLBI. 2018. Available online: https://bravo.sph.umich.edu/freeze5/hg38/ (accessed on 1 July 2019).
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- El Shamieh, S.; Boulanger-Scemama, E.; Lancelot, M.E.; Antonio, A.; Demontant, V.; Condroyer, C.; Letexier, M.; Saraiva, J.P.; Mohand-Said, S.; Sahel, J.A.; et al. Targeted next generation sequencing identifies novel mutations in RP1 as a relatively common cause of autosomal recessive rod-cone dystrophy. BioMed Res. Int. 2015, 2015, 485624. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitz, C.; Kloeckener-Gruissem, B.; Forster, U.; Kohl, S.; Magyar, I.; Wissinger, B.; Matyas, G.; Borruat, F.X.; Schorderet, D.F.; Zrenner, E.; et al. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 2006, 79, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokkema, I.F.; den Dunnen, J.T.; Taschner, P.E. LOVD: Easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 2005, 26, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Berg, J.S.; Milko, L.; Crooks, K.; Lu, M.; Bizon, C.; Owen, P.; Wilhelmsen, K.C.; Weck, K.E.; Evans, J.P.; et al. High Diagnostic Yield of Whole Exome Sequencing in Participants With Retinal Dystrophies in a Clinical Ophthalmology Setting. Am. J. Ophthalmol. 2015, 160, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Li, J.B.; Gerdes, J.M.; Haycraft, C.J.; Fan, Y.; Teslovich, T.M.; May-Simera, H.; Li, H.; Blacque, O.E.; Li, L.; Leitch, C.C.; et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004, 117, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Abu Safieh, L.; Aldahmesh, M.A.; Shamseldin, H.; Hashem, M.; Shaheen, R.; Alkuraya, H.; Al Hazzaa, S.A.; Al-Rajhi, A.; Alkuraya, F.S. Clinical and molecular characterisation of Bardet-Biedl syndrome in consanguineous populations: The power of homozygosity mapping. J. Med. Genet. 2010, 47, 236–241. [Google Scholar] [CrossRef]
- Chandrasekar, S.P.; Namboothiri, S.; Sen, P.; Sarangapani, S. Screening for mutation hotspots in Bardet-Biedl syndrome patients from India. Indian J. Med. Res. 2018, 147, 177–182. [Google Scholar] [CrossRef]
- Beales, P.L.; Warner, A.M.; Hitman, G.A.; Thakker, R.; Flinter, F.A. Bardet-Biedl syndrome: A molecular and phenotypic study of 18 families. J. Med. Genet. 1997, 34, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heon, E.; Kim, G.; Qin, S.; Garrison, J.E.; Tavares, E.; Vincent, A.; Nuangchamnong, N.; Scott, C.A.; Slusarski, D.C.; Sheffield, V.C. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum. Mol. Genet. 2016, 25, 2283–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, E.; Stoetzel, C.; Scheidecker, S.; Geoffroy, V.; Prasad, M.K.; Redin, C.; Missotte, I.; Lacombe, D.; Mandel, J.L.; Muller, J.; et al. Identification of a novel mutation confirms the implication of IFT172 (BBS20) in Bardet-Biedl syndrome. J. Hum. Genet. 2016, 61, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, D.; Seo, S.; Stone, E.M.; Sheffield, V.C. Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome. J. Biol. Chem. 2012, 287, 20625–20635. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef] [Green Version]
- Liew, G.M.; Ye, F.; Nager, A.R.; Murphy, J.P.; Lee, J.S.; Aguiar, M.; Breslow, D.K.; Gygi, S.P.; Nachury, M.V. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 2014, 31, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef]
- Seo, S.; Baye, L.M.; Schulz, N.P.; Beck, J.S.; Zhang, Q.; Slusarski, D.C.; Sheffield, V.C. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. USA 2010, 107, 1488–1493. [Google Scholar] [CrossRef] [Green Version]
- Marion, V.; Stoetzel, C.; Schlicht, D.; Messaddeq, N.; Koch, M.; Flori, E.; Danse, J.M.; Mandel, J.L.; Dollfus, H. Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 1820–1825. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Kulozik, A.E. A perfect message: RNA surveillance and nonsense-mediated decay. Cell 1999, 96, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Abu-Safieh, L.; Al-Anazi, S.; Al-Abdi, L.; Hashem, M.; Alkuraya, H.; Alamr, M.; Sirelkhatim, M.O.; Al-Hassnan, Z.; Alkuraya, B.; Mohamed, J.Y.; et al. In search of triallelism in Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2012, 20, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Badano, J.L.; Leitch, C.C.; Ansley, S.J.; May-Simera, H.; Lawson, S.; Lewis, R.A.; Beales, P.L.; Dietz, H.C.; Fisher, S.; Katsanis, N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 2006, 439, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Beales, P.L.; Katsanis, N.; Lewis, R.A.; Ansley, S.J.; Elcioglu, N.; Raza, J.; Woods, M.O.; Green, J.S.; Parfrey, P.S.; Davidson, W.S.; et al. Genetic and mutational analyses of a large multiethnic Bardet-Biedl cohort reveal a minor involvement of BBS6 and delineate the critical intervals of other loci. Am. J. Hum. Genet. 2001, 68, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Katsanis, N.; Ansley, S.J.; Badano, J.L.; Eichers, E.R.; Lewis, R.A.; Hoskins, B.E.; Scambler, P.J.; Davidson, W.S.; Beales, P.L.; Lupski, J.R. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001, 293, 2256–2259. [Google Scholar] [CrossRef] [Green Version]
- Katsanis, N.; Eichers, E.R.; Ansley, S.J.; Lewis, R.A.; Kayserili, H.; Hoskins, B.E.; Scambler, P.J.; Beales, P.L.; Lupski, J.R. BBS4 is a minor contributor to Bardet-Biedl syndrome and may also participate in triallelic inheritance. Am. J. Hum. Genet. 2002, 71, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Lindstrand, A.; Frangakis, S.; Carvalho, C.M.; Richardson, E.B.; McFadden, K.A.; Willer, J.R.; Pehlivan, D.; Liu, P.; Pediaditakis, I.L.; Sabo, A.; et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am. J. Hum. Genet. 2016, 99, 318–336. [Google Scholar] [CrossRef] [Green Version]
- Petit, C. Usher syndrome: From genetics to pathogenesis. Annu. Rev. Genom. Hum. Genet. 2001, 2, 271–297. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.R.; Zhou, G.; Huang, D.; Davis, J.R.; Moller, C.; Jacobson, S.G.; Kimberling, W.J.; Sumegi, J. Usher syndrome type III: Revised genomic structure of the USH3 gene and identification of novel mutations. Am. J. Hum. Genet. 2002, 71, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, T.; Hamalainen, R.; Yuan, B.; Johnson, C.; Tegelberg, S.; Gasparini, P.; Zelante, L.; Pirvola, U.; Pakarinen, L.; Lehesjoki, A.E.; et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am. J. Hum. Genet. 2001, 69, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Zallocchi, M.; Meehan, D.T.; Delimont, D.; Askew, C.; Garige, S.; Gratton, M.A.; Rothermund-Franklin, C.A.; Cosgrove, D. Localization and expression of clarin-1, the Clrn1 gene product, in auditory hair cells and photoreceptors. Hear. Res. 2009, 255, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akoury, E.; El Zir, E.; Mansour, A.; Megarbane, A.; Majewski, J.; Slim, R. A novel 5-bp deletion in Clarin 1 in a family with Usher syndrome. Ophthalmic Genet. 2011, 32, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Laurier, V.; Stoetzel, C.; Muller, J.; Thibault, C.; Corbani, S.; Jalkh, N.; Salem, N.; Chouery, E.; Poch, O.; Licaire, S.; et al. Pitfalls of homozygosity mapping: An extended consanguineous Bardet-Biedl syndrome family with two mutant genes (BBS2, BBS10), three mutations, but no triallelism. Eur. J. Hum. Genet. 2006, 14, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Alfadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Fahiminiya, S.; El Zir, E.; Mansour, A.; Megarbane, A.; Majewski, J.; Slim, R. Molecular genetics of the Usher syndrome in Lebanon: Identification of 11 novel protein truncating mutations by whole exome sequencing. PLoS ONE 2014, 9, e107326. [Google Scholar] [CrossRef]
- Saouda, M.; Mansour, A.; Bou Moglabey, Y.; El Zir, E.; Mustapha, M.; Chaib, H.; Nehme, A.; Megarbane, A.; Loiselet, J.; Petit, C.; et al. The Usher syndrome in the Lebanese population and further refinement of the USH2A candidate region. Hum. Genet. 1998, 103, 193–198. [Google Scholar] [CrossRef]
- Stoetzel, C.; Laurier, V.; Davis, E.E.; Muller, J.; Rix, S.; Badano, J.L.; Leitch, C.C.; Salem, N.; Chouery, E.; Corbani, S.; et al. BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 2006, 38, 521–524. [Google Scholar] [CrossRef]
- Stoetzel, C.; Laurier, V.; Faivre, L.; Megarbane, A.; Perrin-Schmitt, F.; Verloes, A.; Bonneau, D.; Mandel, J.L.; Cossee, M.; Dollfus, H. BBS8 is rarely mutated in a cohort of 128 Bardet-Biedl syndrome families. J. Hum. Genet. 2006, 51, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Verpy, E.; Leibovici, M.; Zwaenepoel, I.; Liu, X.Z.; Gal, A.; Salem, N.; Mansour, A.; Blanchard, S.; Kobayashi, I.; Keats, B.J.; et al. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat. Genet. 2000, 26, 51–55. [Google Scholar] [CrossRef]
- Shanks, M.E.; Downes, S.M.; Copley, R.R.; Lise, S.; Broxholme, J.; Hudspith, K.A.; Kwasniewska, A.; Davies, W.I.; Hankins, M.W.; Packham, E.R.; et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur. J. Hum. Genet. 2013, 21, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Family | FA4 | FB22 | FB22 | FC51 | FD10 |
| Individual | FA4: V.3 | FB22: II.1 | FB22: II.2 | FC51: II.2 | FD10: III.3 |
| Gender | Female | Male | Female | Female | Female |
| Disease | BBS | BBS | BBS | BBS | USH |
| Age | 24 | 34 | 28 | 16 | 40 |
| Age at Onset of RCD | 6 | 3 | 3 | 11 | 15 |
| Age at Diagnosis | 12 | 5 | 3 | 13 | 28 |
| Visual Acuity (O.D/O.S) | 20/30–20/200 | Only HM—CF | 20/400–20/400 | 20/100–20/125 | 20/30–20/40 |
| ERG | Severely reduced photopic and scotopic ERG | Severely reduced photopic and scotopic ERG | Severely reduced photopic and scotopic ERG | Severely reduced photopic and scotopic ERG | Severely reduced photopic and scotopic ERG |
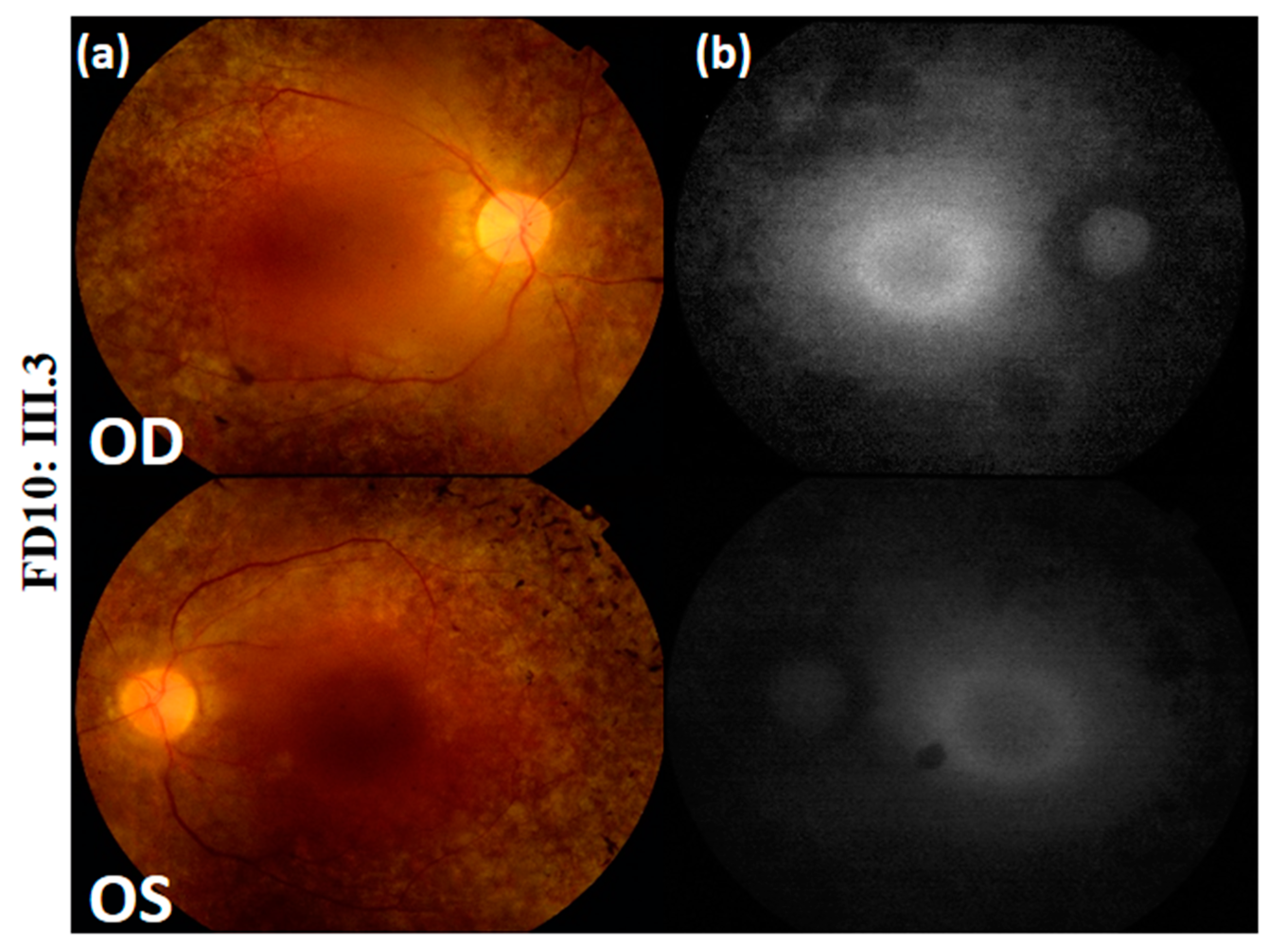
| Fundus Photography | Peripheral pigmentary changes associated with macular involvement | Bilateral widespread pigmentary changes outside the vascular arcades | Bilateral widespread atrophic changes outside the vascular arcades | Atrophy and reduced autofluorescence at the macula | Peripheral retinal pigment and central hyper-fluorescence |
| Optical Coherence Tomography | Bilateral retinal layer thinning at the macula | Reduction of retinal thickness | Bilateral retinal thinning | Bilateral thinning of the neuroretinal layers at the macula | NA |
| Other Symptoms | Table 2 | Table 2 | Table 2 | Table 2 | Hearing loss |
| FA4: V.3 (24 YEARS) | FB22: II.1 (34 YEARS) | FB22: II.2 (28 YEARS) | FC51: II.2 (13 YEARS) | |
|---|---|---|---|---|
| MAJOR FEATURES | ||||
| Rod-cone dystrophy | + | + | + | + |
| Truncal obesity | + | + | + | + |
| Polydactyly | − | − | + | − |
| Genital anomalies | − | − | − | − |
| Renal anomalies | − | − | − | − |
| Learning difficulties | + | + | + | + |
| MINOR FEATURES | − | + | ||
| Speech disorder/delay | − | + | − | − |
| Development delay | + | + | + | − |
| Dental anomalies/hypodontia | + | + | − | + |
| Strabismus/cataracts/astigmatism | − | − | + | − |
| Diabetes mellitus | − | + | + | − |
| Brachydactyly | − | + | + | − |
| Syndactyly | − | + | + | + |
| Clinodactyly | − | + | + | − |
| Imbalance/coordination problems | + | − | + | − |
| Anosmia/hyposmia | − | − | − | − |
| Congenital heart defects | − | − | − | − |
| Hepatic fibrosis/Liver disease | + | − |
| Family | Disease | Gene Reference Sequence | Exon | rs ID | Nucleotide Exchange | Amino Acid Change | Frequencies | PolyPhen-2 | SIFT | Mutation Taster | Novel/Reported |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (Score) | (Score) | (Score) | |||||||||
| FA4 | BBS | BBS9 | 20 | rs61764068 | c.2258A > T | p. (Glu753Val) | 0.0007685 (ExAC) | Probably damaging (0.999) | D | Disease causing (0.998) | Reported by [36] |
| NM_001348041.4 | 0.0007475 (gnomAD) 0.0006769 (TOPMed) | (<0.05) | |||||||||
| Never Hom | |||||||||||
| FB22 | BBS | ARL6 | 3 | rs1359075294 | c.68T > C | p. (Leu23Pro) | 0 (ExAC) | Probably damaging (0.999) | D | Disease causing (0.999) | Novel |
| NM_032146.5 | 0 (gnomAD) | (<0.05) | |||||||||
| 0.0000079 (TOPMed) | |||||||||||
| Never Hom | |||||||||||
| FC51 | BBS | BBS12 | 3 | rs1397714772 | c.265_266delTT | p. (Leu89Valfs*11) | 0 (ExAC) | − | − | Disease causing (1) | Novel |
| NM_001178007.1 | 0.000012 (gnomAD) | ||||||||||
| 0.0000079 (TOPMed) | |||||||||||
| Never Hom | |||||||||||
| BBS12 | 3 | No rs | c.880T > G | p. (Tyr294Asp) | 0 (ExAC) | Benign (0.022) | T | Polymorphism | Novel | ||
| NM_001178007.1 | 0 (gnomAD) | (>0.05) | −0.999 | ||||||||
| 0 (TOPMed) | |||||||||||
| Never Hom | |||||||||||
| BBS5 | 2 | rs137853921 | c.551A > G | p. (Asp184Ser) | 0.004381 (ExAC)/1 Hom | Probably damaging (1) | D | Disease causing (0.999) | Reported by [37] | ||
| NM_152384.3 | 0.00416 (gnomAD)/2 Hom | (<0.05) | |||||||||
| 0.0038 (TOPMed)/2 Hom | |||||||||||
| FD10 | USH | CLRN1 | 1 | No rs | c.188A > C | p. (Tyr63Ser) | 0 (ExAC) | Probably damaging (1) | D | Disease causing (0.999) | Novel |
| NM_001195794.1 | 0 (gnomAD) | (<0.05) | |||||||||
| 0 (TOPMed) | |||||||||||
| Never Hom |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaffal, L.; Joumaa, W.H.; Assi, A.; Helou, C.; Cherfan, G.; Zibara, K.; Audo, I.; Zeitz, C.; El Shamieh, S. Next Generation Sequencing Identifies Five Novel Mutations in Lebanese Patients with Bardet–Biedl and Usher Syndromes. Genes 2019, 10, 1047. https://doi.org/10.3390/genes10121047
Jaffal L, Joumaa WH, Assi A, Helou C, Cherfan G, Zibara K, Audo I, Zeitz C, El Shamieh S. Next Generation Sequencing Identifies Five Novel Mutations in Lebanese Patients with Bardet–Biedl and Usher Syndromes. Genes. 2019; 10(12):1047. https://doi.org/10.3390/genes10121047
Chicago/Turabian StyleJaffal, Lama, Wissam H Joumaa, Alexandre Assi, Charles Helou, George Cherfan, Kazem Zibara, Isabelle Audo, Christina Zeitz, and Said El Shamieh. 2019. "Next Generation Sequencing Identifies Five Novel Mutations in Lebanese Patients with Bardet–Biedl and Usher Syndromes" Genes 10, no. 12: 1047. https://doi.org/10.3390/genes10121047