m6A-Dependent RNA Dynamics in T Cell Differentiation

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Description
2.2. Analysis of High-Throughput Sequencing Data
2.3. Expression Quantification and Filtering
2.4. Mathematical Modeling of Synthesis, Processing, and Degradation Rates
2.5. Analysis of Data Distributions
2.6. Functional Enrichment Analysis
2.7. Clustering
2.8. Source Code
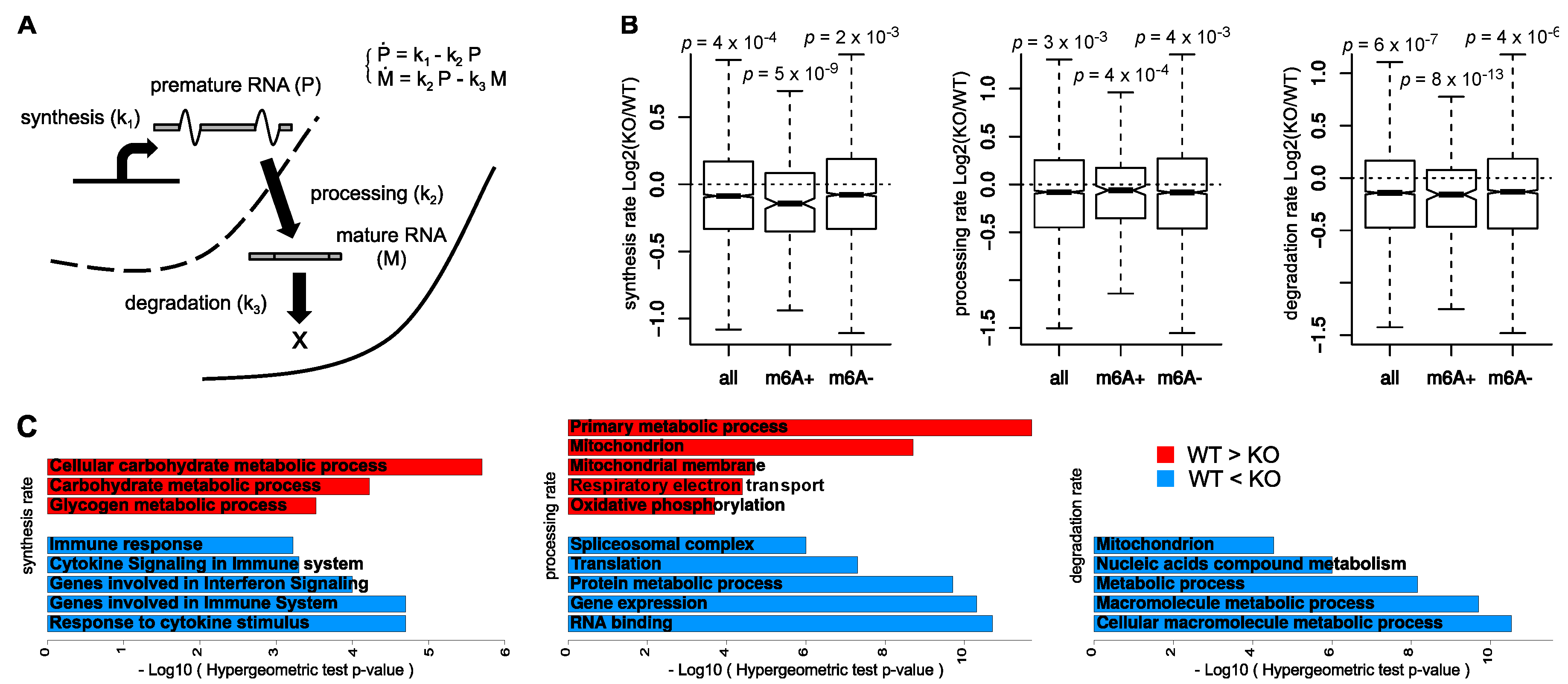
3. Results
3.1. Global Consequences of m6A Depletion on T Cells RNA Dynamics
3.2. Impact of m6A Depletion on T Cells Treated with IL-7
3.3. Clusters Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA modifications in gene expression regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Roignant, J.Y.; Soller, M. m6A in mRNA: An ancient mechanism for fine-tuning gene expression. Trends Genet. 2017, 33, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. Methyltransferases modulate RNA stability in embryonic stem cells. Nat. Cell Biol. 2014, 16, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, Y.; Sun, B.; Wang, L.; Yang, Y.; Ma, D.; Lv, J.; Heng, J.; Ding, Y.; Xue, Y.; et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature 2017, 549, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Hao, Y.J.; Zhang, Y.; Li, M.M.; Wang, M.; Han, W.; Wu, Y.; Lv, Y.; Hao, J.; Wang, L.; et al. A RNA methylation is regulated by MicroRNAs and promotes reprogramming to pluripotency. Stem Cell 2015, 16, 289–301. [Google Scholar]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 1–12. [Google Scholar] [CrossRef]
- Dixit, D.; Xie, Q.; Rich, J.N.; Zhao, J.C. Messenger RNA methylation regulates glioblastoma tumorigenesis. Cancer Cell 2017, 31, 474–475. [Google Scholar] [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vågbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E., Jr.; Darnell, R.B. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef]
- Slobodin, B.; Han, R.; Calderone, V.; Vrielink, J.A.F.O.; Loayza-Puch, F.; Elkon, R.; Agami, R. Transcription Impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell 2017, 169, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Haussmann, I.U.; Bodi, Z.; Sanchez-Moran, E.; Mongan, N.P.; Archer, N.; Fray, R.G.; Soller, M. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 2016, 540, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Lence, T.; Akhtar, J.; Bayer, M.; Schmid, K.; Spindler, L.; Ho, C.H.; Kreim, N.; Andrade-Navarro, M.A.; Poeck, B.; Helm, M.; et al. m6A modulates neuronal functions and sex determination in Drosophila. Nature 2016, 540, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bartosovic, M.; Molares, H.C.; Gregorova, P.; Hrossova, D.; Kudla, G.; Vanacova, S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3′-end processing. Nucleic Acids Res. 2017, 45, 11356–11370. [Google Scholar] [CrossRef] [PubMed]
- Louloupi, A.; Ntini, E.; Conrad, T.; Ørom, U.A.V. Transient N-6-Methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell Rep. 2018, 23, 3429–3437. [Google Scholar] [CrossRef] [PubMed]
- Kasowitz, S.D.; Ma, J.; Anderson, S.J.; Leu, N.A.; Xu, Y.; Gregory, B.D.; Schultz, R.M.; Wang, P.J. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 2018, 14, e1007412–e1007428. [Google Scholar] [CrossRef]
- Darnell, R.B.; Ke, S.; Darnell, J.E. Pre-mRNA processing includes N6 methylation of adenosine residues that are retained in mRNA exons and the fallacy of “RNA epigenetics”. RNA 2018, 24, 262–267. [Google Scholar] [CrossRef]
- Li, H.B.; Tong, J.; Zhu, S.; Batista, P.J.; Duffy, E.E.; Zhao, J.; Bailis, W.; Cao, G.; Kroehling, L.; Chen, Y.; et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 2017, 548, 338–342. [Google Scholar] [CrossRef]
- De Pretis, S.; Kress, T.; Morelli, M.J.; Melloni, G.E.M.; Riva, L.; Amati, B.; Pelizzola, M. INSPEcT: A computational tool to infer mRNA synthesis, processing and degradation dynamics from RNA—and 4sU–seq time course experiments. Bioinformatics 2015, 31, 2829–2835. [Google Scholar] [CrossRef]
- Bianchi, V.; Céol, A.; Ogier, A.G.E.; de Pretis, S.; Galeota, E.; Kishore, K.; Bora, P.; Croci, O.; Campaner, S.; Amati, B.; et al. Integrated systems for NGS data management and analysis: open issues and available solutions. Front Genet. 2016, 7, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup the sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Mclean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Meth. 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.X.; Van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Meth. 2016, 13, 692–698. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furlan, M.; Galeota, E.; De Pretis, S.; Caselle, M.; Pelizzola, M. m6A-Dependent RNA Dynamics in T Cell Differentiation. Genes 2019, 10, 28. https://doi.org/10.3390/genes10010028
Furlan M, Galeota E, De Pretis S, Caselle M, Pelizzola M. m6A-Dependent RNA Dynamics in T Cell Differentiation. Genes. 2019; 10(1):28. https://doi.org/10.3390/genes10010028
Chicago/Turabian StyleFurlan, Mattia, Eugenia Galeota, Stefano De Pretis, Michele Caselle, and Mattia Pelizzola. 2019. "m6A-Dependent RNA Dynamics in T Cell Differentiation" Genes 10, no. 1: 28. https://doi.org/10.3390/genes10010028
APA StyleFurlan, M., Galeota, E., De Pretis, S., Caselle, M., & Pelizzola, M. (2019). m6A-Dependent RNA Dynamics in T Cell Differentiation. Genes, 10(1), 28. https://doi.org/10.3390/genes10010028