Regulation of IKs Potassium Current by Isoproterenol in Adult Cardiomyocytes Requires Type 9 Adenylyl Cyclase

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Generation of IKs-AC9KO Gene-Targeted Mice
2.2. Plasmids and Adenoviruses
2.3. Cell Culture and Transfections
2.4. Antibodies Used for Immunoprecipitation and Western Blotting
2.5. Adenylyl Cyclase Activity and IP-AC Assays
2.6. Adult Cardiomyocyte Isolation
2.7. Single Cell Electrophysiology
2.8. Neonatal Cardiomyocyte Isolation and Adenoviral Infection
2.9. Cardiomyocyte Immunocytochemistry
2.10. Statistical Analysis.
3. Results
3.1. Genetic Ablation of AC9 Results in Preweaning Subviability
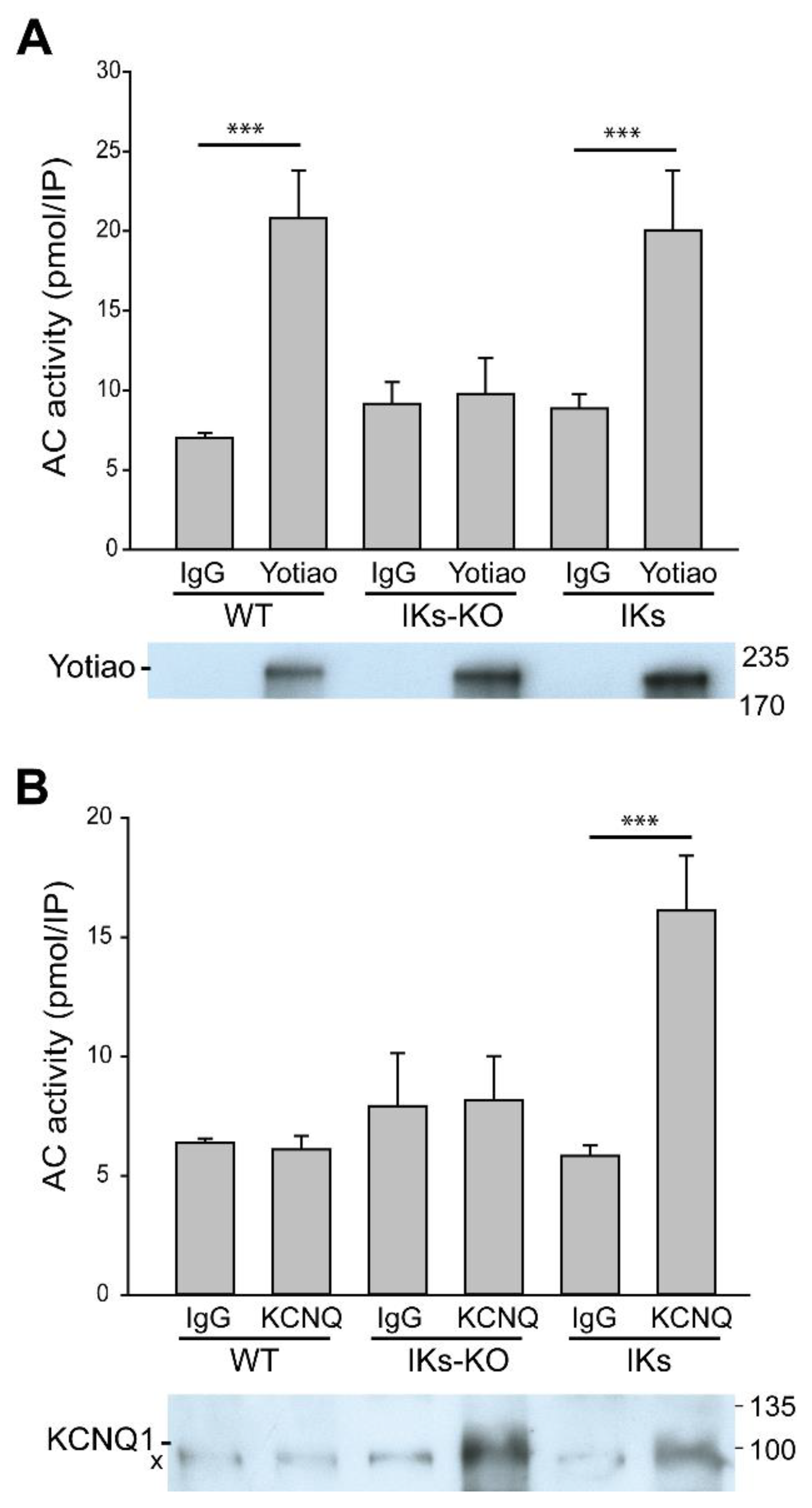
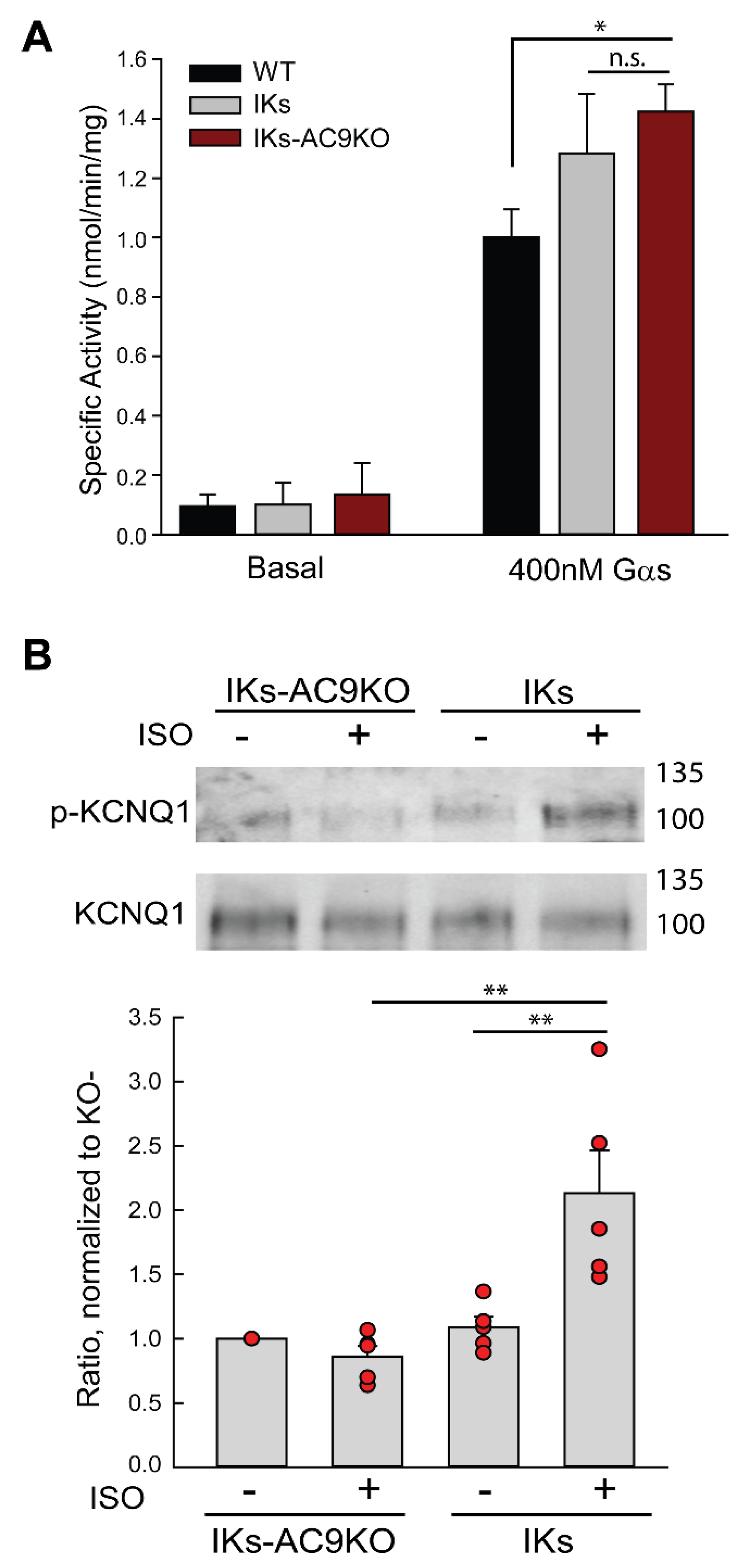
3.2. Deletion of AC9 Results in Loss of Yotiao- and KCNQ1-Associated AC Activity but no Alterations in Total Cardiac AC Activity
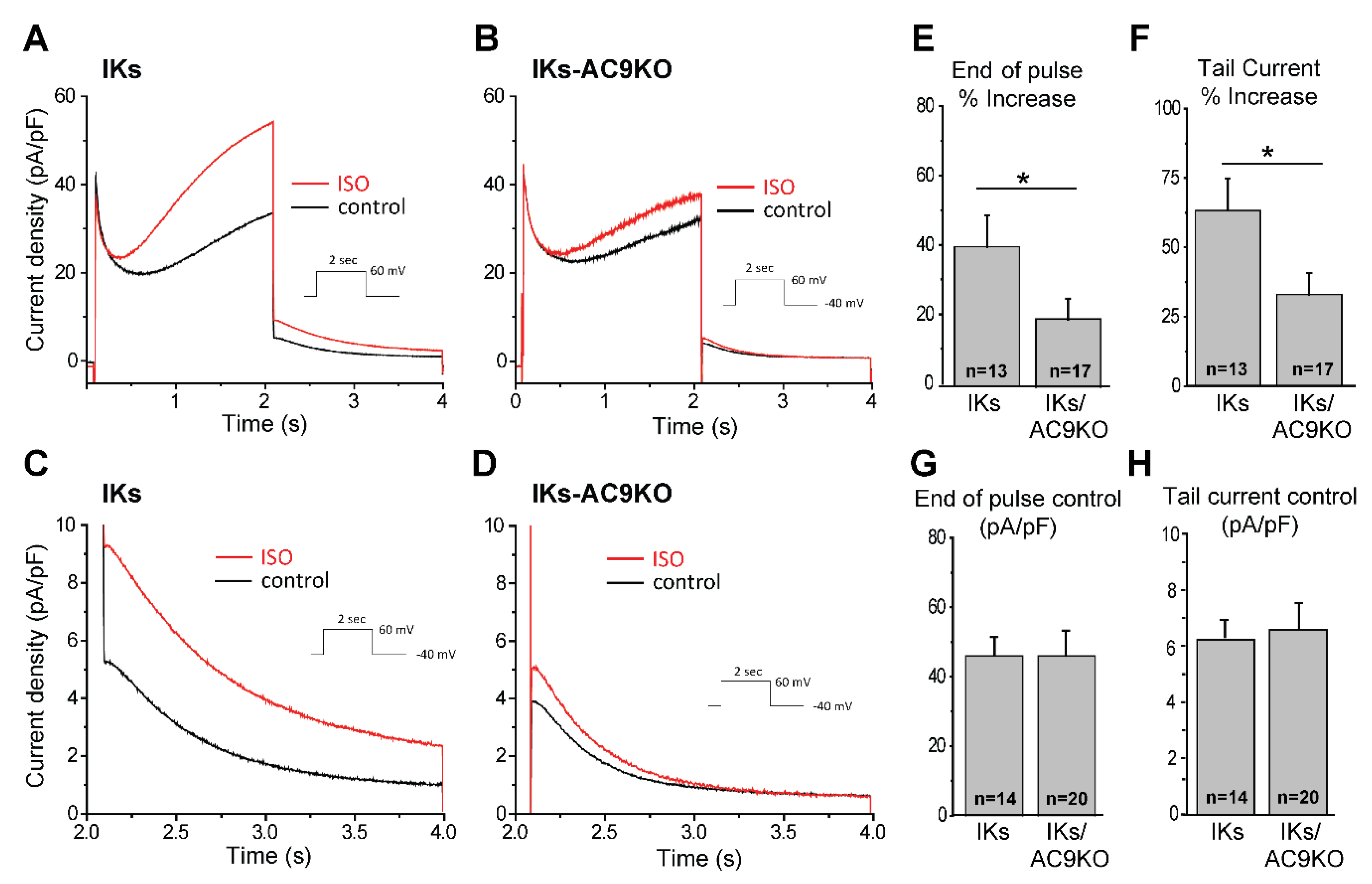
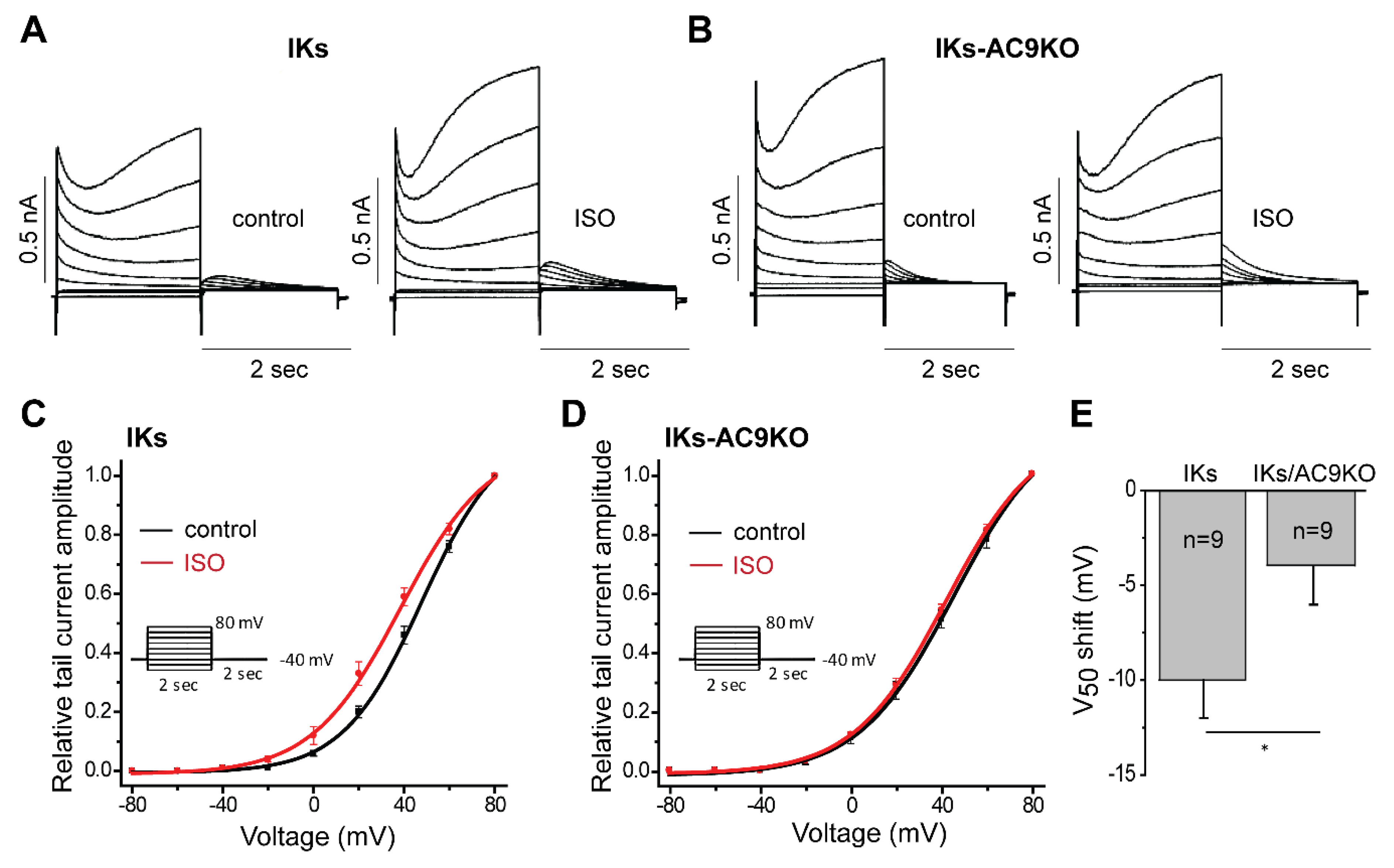
3.3. Loss of AC9 Reduces β-Adrenergic Regulation of IKs Currents in Adult Cardiomyocytes
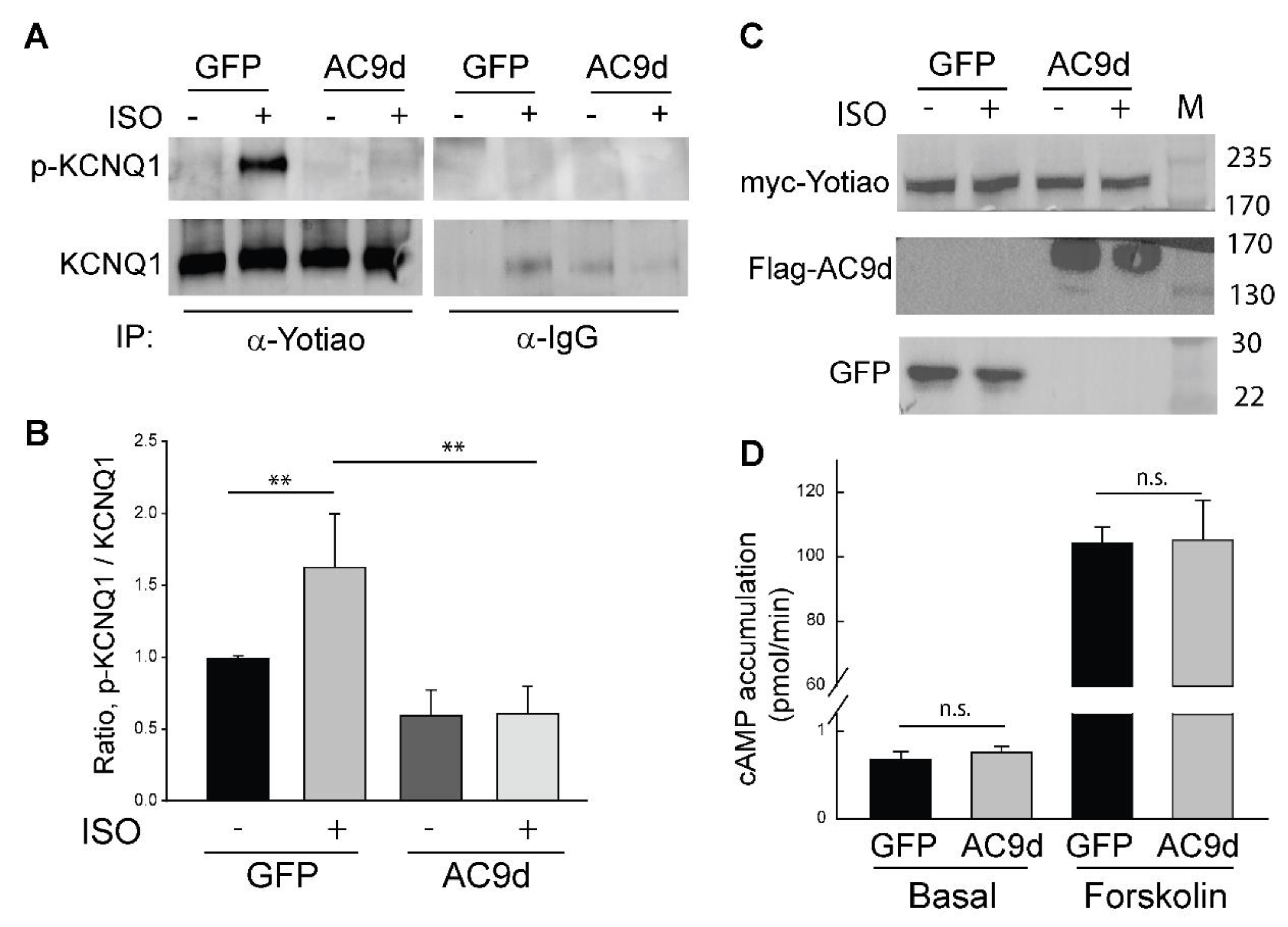
3.4. Expression of Catalytically Inactive AC9 Blocks KCNQ1 Phosphorylation
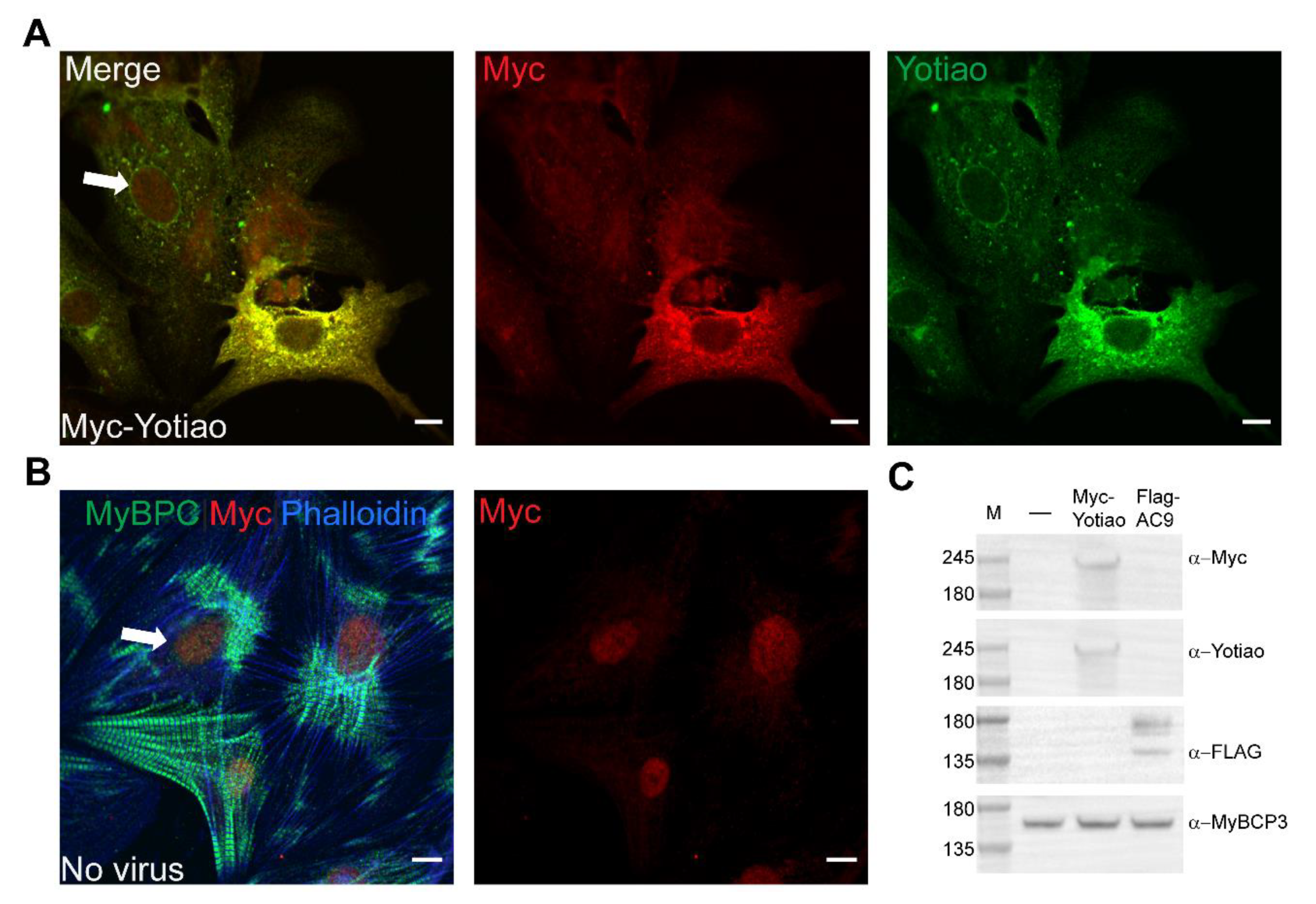
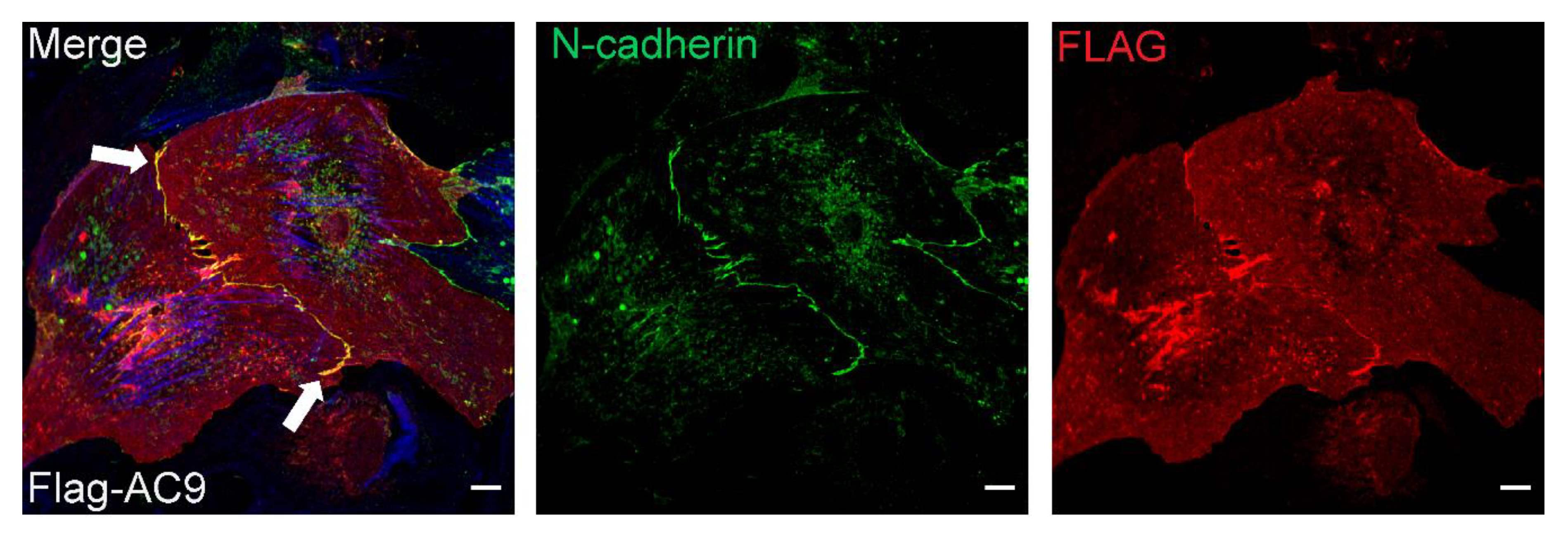
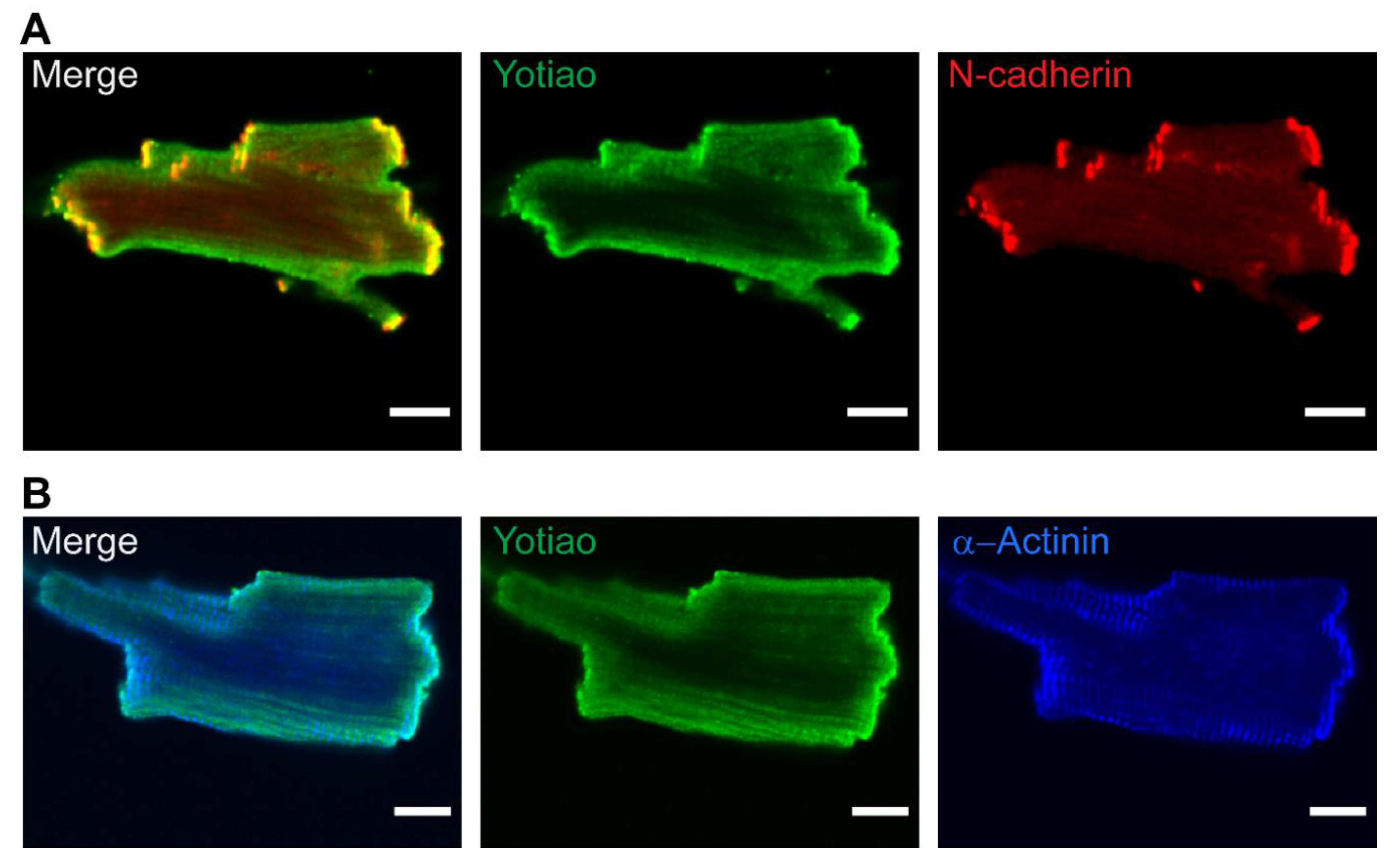
3.5. Subcellular Distribution of AC9 and Yotiao in Cardiomyocytes
4. Discussion
4.1. Association of IKs with AC9
4.2. AC9 Regulation and Physiology
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef]
- Liu, Z.; Du, L.; Li, M. Update on the slow delayed rectifier potassium current (I(Ks)): Role in modulating cardiac function. Curr. Med. Chem. 2012, 19, 1405–1420. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, J.; Motoike, H.K.; Rao, J.; Kass, R.S. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. PNAS 2004, 101, 16374–16378. [Google Scholar] [CrossRef]
- Marx, S.O.; Kurokawa, J.; Reiken, S.; Motoike, H.; D’Armiento, J.; Marks, A.R.; Kass, R.S. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 2002, 295, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, C.S.; Park, K.H.; El Harchi, A.; Camonis, J.; Kass, R.S.; Escande, D.; Merot, J.; Loussouarn, G.; Le Bouffant, F.; Baro, I. IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiovasc. Res. 2008, 79, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.; Eldstrom, J.; Fedida, D. Single channel kinetic analysis of the cAMP effect on IKs mutants, S209F and S27D/S92D. Channels 2018, 12, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Terrenoire, C.; Houslay, M.D.; Baillie, G.S.; Kass, R.S. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J. Biol. Chem. 2009, 284, 9140–9146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Boulet, I.R.; Raes, A.L.; Ottschytsch, N.; Snyders, D.J. Functional effects of a KCNQ1 mutation associated with the long QT syndrome. Cardiovasc. Res. 2006, 70, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Kass, R.S. Recent progress in congenital long QT syndrome. Curr. Opin. Cardiol. 2010, 25, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. PNAS 2007, 104, 20990–20995. [Google Scholar] [CrossRef]
- Dvir, M.; Strulovich, R.; Sachyani, D.; Ben-Tal Cohen, I.; Haitin, Y.; Dessauer, C.; Pongs, O.; Kass, R.; Hirsch, J.A.; Attali, B. Long QT mutations at the interface between KCNQ1 helix C and KCNE1 disrupt I(KS) regulation by PKA and PIP(2). J. Cell Sci. 2014, 127, 3943–3955. [Google Scholar] [CrossRef] [PubMed]
- Sadana, R.; Dessauer, C.W. Physiological Roles for G Protein-Regulated Adenylyl Cyclase Isoforms: Insights from Knockout and Overexpression Studies. NeuroSignals 2009, 17, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W.; Watts, V.J.; Ostrom, R.S.; Conti, M.; Dove, S.; Seifert, R. International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases. Pharmacol. Rev. 2017, 69, 93–139. [Google Scholar] [CrossRef] [PubMed]
- Bauman, A.L.; Soughayer, J.; Nguyen, B.T.; Willoughby, D.; Carnegie, G.K.; Wong, W.; Hoshi, N.; Langeberg, L.K.; Cooper, D.M.; Dessauer, C.W.; et al. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol. Cell 2006, 23, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Piggott, L.A.; Bauman, A.L.; Scott, J.D.; Dessauer, C.W. The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. PANS 2008, 105, 13835–13840. [Google Scholar] [CrossRef] [PubMed]
- Kapiloff, M.S.; Piggott, L.A.; Sadana, R.; Li, J.; Heredia, L.A.; Henson, E.; Efendiev, R.; Dessauer, C.W. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem. 2009, 284, 23540–23546. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.; Halls, M.L.; Everett, K.L.; Ciruela, A.; Skroblin, P.; Klussmann, E.; Cooper, D.M. A key phosphorylation site in AC8 mediates regulation of Ca(2+)-dependent cAMP dynamics by an AC8-AKAP79-PKA signalling complex. J. Cell Sci. 2012, 125, 5850–5859. [Google Scholar] [CrossRef]
- Shen, J.X.; Cooper, D.M. AKAP79, PKC, PKA and PDE4 participate in a Gq-linked muscarinic receptor and adenylate cyclase 2 cAMP signalling complex. Biochem. J. 2013, 455, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Guinzberg, R.; Diaz-Cruz, A.; Acosta-Trujillo, C.; Vilchis-Landeros, M.M.; Vazquez-Meza, H.; Lozano-Flores, C.; Chiquete-Felix, N.; Varela-Echavarria, A.; Uribe-Carvajal, S.; Riveros-Rosas, H.; et al. Newly synthesized cAMP is integrated at a membrane protein complex signalosome to ensure receptor response specificity. FEBS J. 2017, 284, 258–276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-kinase anchoring protein Yotiao facilitates complex formation between type 9 adenylyl cyclase and the IKs potassium channel in heart. J. Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Baldwin, T.A.; Wang, Y.; Subramaniam, J.; Carbajal, A.G.; Brand, C.S.; Cunha, S.R.; Dessauer, C.W. Loss of type 9 adenylyl cyclase triggers reduced phosphorylation of Hsp20 and diastolic dysfunction. Sci. Rep. 2017, 7, 5522. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, J.; Chen, L.; Kass, R.S. Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. PNAS 2003, 100, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dessauer, C.W. Identifying Complexes of Adenylyl Cyclase with A-kinase Anchoring Proteins. In Cyclic Nucleotide Signaling; Cheng, X., Ed.; CRC Press: Boca Raton, FL, USA, 2015; pp. 147–164. [Google Scholar]
- Dessauer, C.W. Kinetic analysis of the action of P-site analogs. Methods in Enzymology 2002, 345, 112–126. [Google Scholar] [PubMed]
- Sharma, S.; Guthrie, P.H.; Chan, S.S.; Haq, S.; Taegtmeyer, H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc. Res. 2007, 76, 71–80. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.D.; Rodrigo, M.C.; Simpson, P.C. Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 2007, 357, 271–296. [Google Scholar] [PubMed]
- Drici, M.D.; Arrighi, I.; Chouabe, C.; Mann, J.R.; Lazdunski, M.; Romey, G.; Barhanin, J. Involvement of IsK-associated K+ channel in heart rate control of repolarization in a murine engineered model of Jervell and Lange-Nielsen syndrome. Circ. Res. 1998, 83, 95–102. [Google Scholar] [CrossRef]
- Honore, E.; Attali, B.; Romey, G.; Heurteaux, C.; Ricard, P.; Lesage, F.; Lazdunski, M.; Barhanin, J. Cloning, expression, pharmacology and regulation of a delayed rectifier K+ channel in mouse heart. EMBO J. 1991, 10, 2805–2811. [Google Scholar] [CrossRef]
- Thompson, E.; Eldstrom, J.; Westhoff, M.; McAfee, D.; Balse, E.; Fedida, D. cAMP-dependent regulation of I(Ks) single-channel kinetics. J. Gen. Physiol. 2017, 149, 781–798. [Google Scholar] [CrossRef] [PubMed]
- Dilly, K.W.; Kurokawa, J.; Terrenoire, C.; Reiken, S.; Lederer, W.J.; Marks, A.R.; Kass, R.S. Overexpression of beta2-adrenergic receptors cAMP-dependent protein kinase phosphorylates and modulates slow delayed rectifier potassium channels expressed in murine heart: Evidence for receptor/channel co-localization. J. Biol. Chem. 2004, 279, 40778–40787. [Google Scholar] [CrossRef] [PubMed]
- Terrenoire, C.; Clancy, C.E.; Cormier, J.W.; Sampson, K.J.; Kass, R.S. Autonomic control of cardiac action potentials - Role of potassium channel kinetics in response to sympathetic stimulation. Circ. Res. 2005, 96, E25–E34. [Google Scholar] [CrossRef] [PubMed]
- Policarova, M.; Novotny, T.; Bebarova, M. Impaired Adrenergic/Protein Kinase A Response of Slow Delayed Rectifier Potassium Channels as a Long QT Syndrome Motif: Importance and Unknowns. Can. J. Cardiol. 2019, 35, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Efendiev, R.; Bavencoffe, A.; Hu, H.; Zhu, M.X.; Dessauer, C.W. Scaffolding by A-kinase anchoring protein enhances functional coupling between adenylyl cyclase and TRPV1 channel. J. Biol. Chem. 2013, 288, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, T.A.; Li, Y.; Brand, C.S.; Watts, V.J.; Dessauer, C.W. Insights into the Regulatory Properties of Human Adenylyl Cyclase Type 9. Mol. Pharmacol. 2019, 95, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.C.; Kass, R.S. Cholinergic inhibition of slow delayed-rectifier K+ current in guinea pig sino-atrial node is not mediated by muscarinic receptors. Mol. Pharmacol. 1995, 47, 1248–1254. [Google Scholar]
- Leren, I.S.; Hasselberg, N.E.; Saberniak, J.; Håland, T.F.; Kongsgård, E.; Smiseth, O.A.; Edvardsen, T.; Haugaa, K.H. Cardiac Mechanical Alterations and Genotype Specific Differences in Subjects With Long QT Syndrome. JACC: Cardiovasc. Imaging 2015, 8, 501–510. [Google Scholar] [CrossRef]
- Zhang, M.; Patriarchi, T.; Stein, I.S.; Qian, H.; Matt, L.; Nguyen, M.; Xiang, Y.K.; Hell, J.W. Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic beta-adrenergic signaling. J. Biol. Chem. 2013, 288, 17918–17931. [Google Scholar] [CrossRef]
- Bavencoffe, A.; Li, Y.; Wu, Z.; Yang, Q.; Herrera, J.; Kennedy, E.J.; Walters, E.T.; Dessauer, C.W. Persistent Electrical Activity in Primary Nociceptors after Spinal Cord Injury Is Maintained by Scaffolded Adenylyl Cyclase and Protein Kinase A and Is Associated with Altered Adenylyl Cyclase Regulation. J. Neurosci. 2016, 36, 1660–1668. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company/Product Number | Dilution |
|---|---|---|
| rabbit anti-Yotiao | Previously described [18] | 1:75 |
| mouse anti-DYKDDDDK tag (9A3) | Cell Signaling Technologies (8146) | 1:750 |
| mouse anti-Alpha-actinin (sarcomeric) | Sigma (A7811) | 1:1000 |
| rabbit anti-Myosin binding protein C 3 | Santa Cruz Biotechnology (sc 67353) | 1:200 |
| mouse anti-N-cadherin (13A9) DyLight550 | Novus Biological (48309R) | 1:500 |
| rabbit anti-N-cadherin | Abcam (Ab18203) | 1:250 |
| mouse anti-myc tag | Invitrogen (MA1-21316) | 1:150 |
| Alexa 647 Phalloidin | Invitrogen (A22287) | 1:1000 |
| Alexa 488 anti-rabbit | Invitrogen (A21206) | 1:500 |
| Alexa 568 anti-mouse | Invitrogen (A10037) | 1:500 |
| Alexa 647 anti-mouse | Invitrogen (A31571) | 1:500 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Hof, T.; Baldwin, T.A.; Chen, L.; Kass, R.S.; Dessauer, C.W. Regulation of IKs Potassium Current by Isoproterenol in Adult Cardiomyocytes Requires Type 9 Adenylyl Cyclase. Cells 2019, 8, 981. https://doi.org/10.3390/cells8090981
Li Y, Hof T, Baldwin TA, Chen L, Kass RS, Dessauer CW. Regulation of IKs Potassium Current by Isoproterenol in Adult Cardiomyocytes Requires Type 9 Adenylyl Cyclase. Cells. 2019; 8(9):981. https://doi.org/10.3390/cells8090981
Chicago/Turabian StyleLi, Yong, Thomas Hof, Tanya A. Baldwin, Lei Chen, Robert S. Kass, and Carmen W. Dessauer. 2019. "Regulation of IKs Potassium Current by Isoproterenol in Adult Cardiomyocytes Requires Type 9 Adenylyl Cyclase" Cells 8, no. 9: 981. https://doi.org/10.3390/cells8090981
APA StyleLi, Y., Hof, T., Baldwin, T. A., Chen, L., Kass, R. S., & Dessauer, C. W. (2019). Regulation of IKs Potassium Current by Isoproterenol in Adult Cardiomyocytes Requires Type 9 Adenylyl Cyclase. Cells, 8(9), 981. https://doi.org/10.3390/cells8090981