Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism


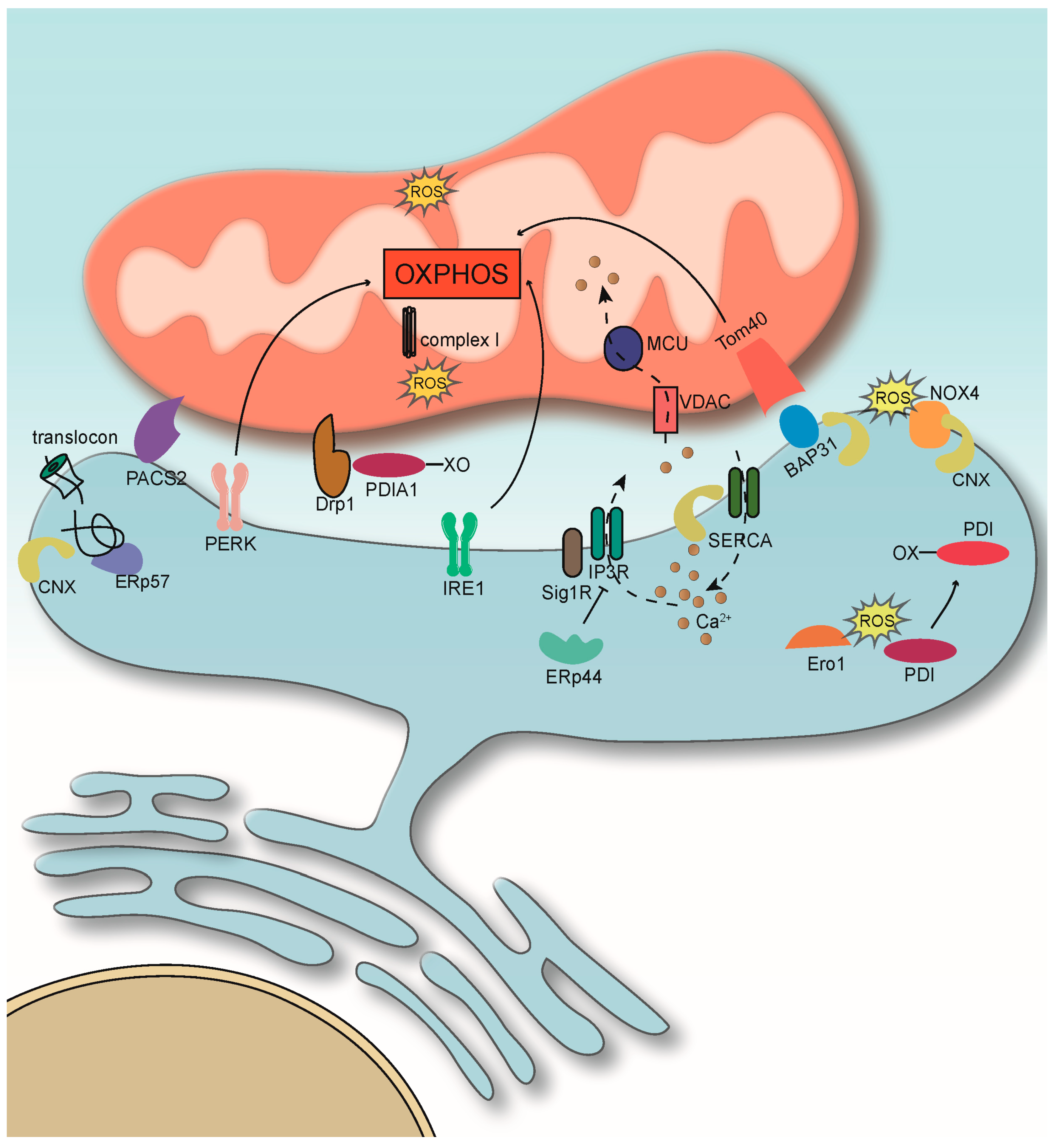{kind=link}
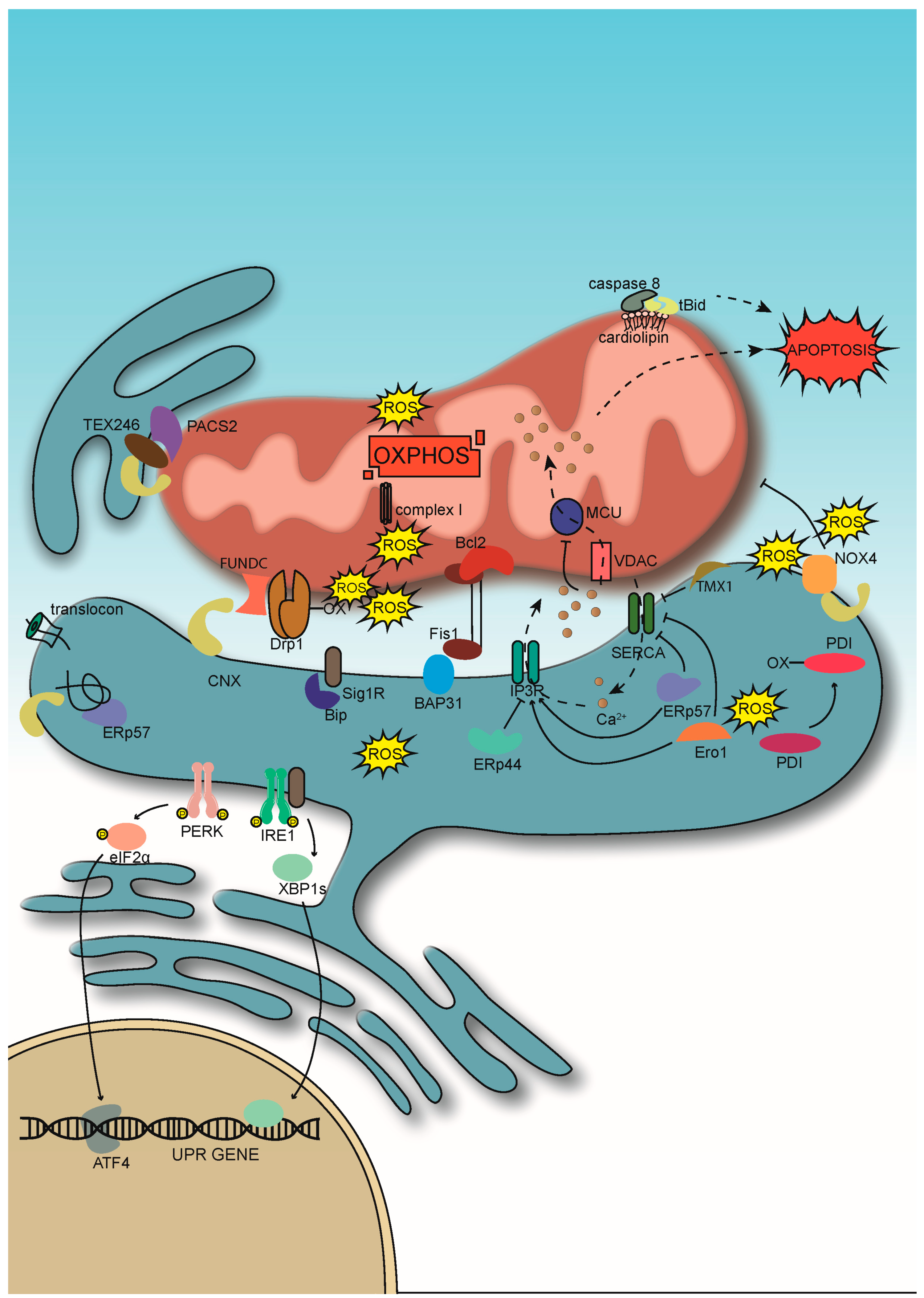{kind=link}
Abstract
1. Introduction
2. Sigma-1 Receptor (Sig1R) Controls a Subset of Contact Site Regulators
3. Calnexin and BAP31 Form a Chaperone-Based Nexus to Control Multiple MERC Functions
4. MERC Redox Control by Protein Disulfide Isomerase (PDI) Family Proteins and the Ero1 Oxidoreductase
5. Connections between UPR Signaling and Metabolism Mediated by PERK and Ire1
6. MERC Tethering Factors: PACS-2 and Mitofusin-2
7. Links to Autophagy
8. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simmen, T.; Lynes, E.M.; Gesson, K.; Thomas, G. Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2010, 1798, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. ER stress and its functional link to mitochondria: Role in cell survival and death. Cold Spring Harb. Perspect. Biol. 2011, 3, a004424. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Gilady, S.Y.; Bui, M.; Lynes, E.M.; Benson, M.D.; Watts, R.; Vance, J.E.; Simmen, T. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 2010, 15, 619–629. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Reimertz, C.; Kogel, D.; Rami, A.; Chittenden, T.; Prehn, J.H. Gene expression during ER stress-induced apoptosis in neurons: Induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 2003, 162, 587–597. [Google Scholar] [CrossRef]
- Schamel, W.W.; Kuppig, S.; Becker, B.; Gimborn, K.; Hauri, H.P.; Reth, M. A high-molecular-weight complex of membrane proteins BAP29/BAP31 is involved in the retention of membrane-bound IgD in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2003, 100, 9861–9866. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.D.; Barlowe, C. Yet1p and Yet3p, the yeast homologs of BAP29 and BAP31, interact with the endoplasmic reticulum translocation apparatus and are required for inositol prototrophy. J. Biol. Chem. 2010, 285, 18252–18261. [Google Scholar] [CrossRef] [PubMed]
- Zuppini, A.; Groenendyk, J.; Cormack, L.A.; Shore, G.; Opas, M.; Bleackley, R.C.; Michalak, M. Calnexin deficiency and endoplasmic reticulum stress-induced apoptosis. Biochemistry 2002, 41, 2850–2858. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5, eaaw1386. [Google Scholar] [CrossRef]
- Lee, G.H.; Lee, H.Y.; Li, B.; Kim, H.R.; Chae, H.J. Bax inhibitor-1-mediated inhibition of mitochondrial Ca2+ intake regulates mitochondrial permeability transition pore opening and cell death. Sci. Rep. 2014, 4, 5194. [Google Scholar] [CrossRef]
- Lee, G.H.; Kim, H.K.; Chae, S.W.; Kim, D.S.; Ha, K.C.; Cuddy, M.; Kress, C.; Reed, J.C.; Kim, H.R.; Chae, H.J. Bax inhibitor-1 regulates endoplasmic reticulum stress-associated reactive oxygen species and heme oxygenase-1 expression. J. Biol. Chem. 2007, 282, 21618–21628. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028 e7. [Google Scholar] [CrossRef]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Bischof, H.; Burgstaller, S.; Siirin, M.; Murphy, A.; Malli, R.; Kaufman, R.J. Mitochondria supply ATP to the ER through a mechanism antagonized by cytosolic Ca(2). BioRxiv 2019. [Google Scholar] [CrossRef]
- Klein, M.C.; Zimmermann, K.; Schorr, S.; Landini, M.; Klemens, P.A.W.; Altensell, J.; Jung, M.; Krause, E.; Nguyen, D.; Helms, V.; et al. AXER is an ATP/ADP exchanger in the membrane of the endoplasmic reticulum. Nat. Commun. 2018, 9, 3489. [Google Scholar] [CrossRef]
- Zhang, D.; Lu, C.; Whiteman, M.; Chance, B.; Armstrong, J.S. The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J. Biol. Chem. 2008, 283, 3476–3486. [Google Scholar] [CrossRef]
- Rusinol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018, 87, 783–807. [Google Scholar] [CrossRef]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145 e15. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Hayashi, T.; Su, T.P. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 2012, 417, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: Implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alvarez, M.I.; Sebastian, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernandez, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895 e17. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human sigma1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Mavylutov, T.; Chen, X.; Guo, L.; Yang, J. APEX2- tagging of Sigma 1-receptor indicates subcellular protein topology with cytosolic N-terminus and ER luminal C-terminus. Protein Cell 2018, 9, 733–737. [Google Scholar] [CrossRef]
- Gromek, K.A.; Suchy, F.P.; Meddaugh, H.R.; Wrobel, R.L.; LaPointe, L.M.; Chu, U.B.; Primm, J.G.; Ruoho, A.E.; Senes, A.; Fox, B.G. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J. Biol. Chem. 2014, 289, 20333–20344. [Google Scholar] [CrossRef]
- Moebius, F.F.; Reiter, R.J.; Hanner, M.; Glossmann, H. High affinity of sigma 1-binding sites for sterol isomerization inhibitors: Evidence for a pharmacological relationship with the yeast sterol C8-C7 isomerase. Br. J. Pharmacol. 1997, 121, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef]
- Morales-Lazaro, S.L.; Gonzalez-Ramirez, R.; Rosenbaum, T. Molecular Interplay Between the Sigma-1 Receptor, Steroids, and Ion Channels. Front. Pharmacol. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Aydar, E.; Palmer, C.P.; Klyachko, V.A.; Jackson, M.B. The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 2002, 34, 399–410. [Google Scholar] [CrossRef]
- Zhang, H.; Cuevas, J. Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J. Neurophysiol. 2002, 87, 2867–2879. [Google Scholar] [CrossRef] [PubMed]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox. Res. 2019, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Fontanilla, D.; Gopalakrishnan, A.; Chae, Y.K.; Markley, J.L.; Ruoho, A.E. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur. J. Pharmacol. 2012, 682, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Y.; Rothman, R.K.; Su, T.P. Insights into the Sigma-1 receptor chaperone’s cellular functions: A microarray report. Synapse 2012, 66, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Gregianin, E.; Pallafacchina, G.; Zanin, S.; Crippa, V.; Rusmini, P.; Poletti, A.; Fang, M.; Li, Z.; Diano, L.; Petrucci, A.; et al. Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum. Mol. Genet. 2016, 25, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Saul, A.; Tawfik, A.; Williams, C.; Bollinger, K.; Smith, R.; Tachikawa, M.; Zorrilla, E.; Ganapathy, V.; Smith, S.B. Late-onset inner retinal dysfunction in mice lacking sigma receptor 1 (sigmaR1). Investig. Ophthalmol. Vis. Sci. 2011, 52, 7749–7760. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shanmugam, A.; Markand, S.; Zorrilla, E.; Ganapathy, V.; Smith, S.B. Sigma 1 receptor regulates the oxidative stress response in primary retinal Muller glial cells via NRF2 signaling and system xc(-), the Na(+)-independent glutamate-cystine exchanger. Free Radic. Biol. Med. 2015, 86, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, J.; Cui, X.; Mysona, B.A.; Navneet, S.; Saul, A.; Ahuja, M.; Lambert, N.; Gazaryan, I.G.; Thomas, B.; et al. The molecular chaperone sigma 1 receptor mediates rescue of retinal cone photoreceptor cells via modulation of NRF2. Free Radic. Biol. Med. 2019, 134, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Rutkevich, L.A.; Williams, D.B. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Curr. Opin. Cell Biol. 2011, 23, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Lynes, E.M.; Raturi, A.; Shenkman, M.; Ortiz Sandoval, C.; Yap, M.C.; Wu, J.; Janowicz, A.; Myhill, N.; Benson, M.D.; Campbell, R.E.; et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J. Cell Sci. 2013, 126, 3893–3903. [Google Scholar] [CrossRef]
- Lynes, E.M.; Bui, M.; Yap, M.C.; Benson, M.D.; Schneider, B.; Ellgaard, L.; Berthiaume, L.G.; Simmen, T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012, 31, 457–470. [Google Scholar] [CrossRef]
- Cho, I.T.; Adelmant, G.; Lim, Y.; Marto, J.A.; Cho, G.; Golden, J.A. Ascorbate peroxidase proximity labeling coupled with biochemical fractionation identifies promoters of endoplasmic reticulum-mitochondrial contacts. J. Biol. Chem. 2017, 292, 16382–16392. [Google Scholar] [CrossRef]
- Hung, V.; Lam, S.S.; Udeshi, N.D.; Svinkina, T.; Guzman, G.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 2017, 6. [Google Scholar] [CrossRef]
- Lakkaraju, A.K.; Abrami, L.; Lemmin, T.; Blaskovic, S.; Kunz, B.; Kihara, A.; Dal Peraro, M.; van der Goot, F.G. Palmitoylated calnexin is a key component of the ribosome-translocon complex. EMBO J. 2012, 31, 1823–1835. [Google Scholar] [CrossRef]
- Delom, F.; Fessart, D.; Chevet, E. Regulation of calnexin sub-cellular localization modulates endoplasmic reticulum stress-induced apoptosis in MCF-7 cells. Apoptosis 2007, 12, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S.; et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; McFie, P.J.; Vu, H.; Chumala, P.; Katselis, G.S.; Stone, S.J. Identification of calnexin as a diacylglycerol acyltransferase-2 interacting protein. PLoS ONE 2019, 14, e0210396. [Google Scholar] [CrossRef] [PubMed]
- Jennelle, L.; Hunegnaw, R.; Dubrovsky, L.; Pushkarsky, T.; Fitzgerald, M.L.; Sviridov, D.; Popratiloff, A.; Brichacek, B.; Bukrinsky, M. HIV-1 protein Nef inhibits activity of ATP-binding cassette transporter A1 by targeting endoplasmic reticulum chaperone calnexin. J. Biol. Chem. 2014, 289, 28870–28884. [Google Scholar] [CrossRef] [PubMed]
- Prior, K.K.; Wittig, I.; Leisegang, M.S.; Groenendyk, J.; Weissmann, N.; Michalak, M.; Jansen-Durr, P.; Shah, A.M.; Brandes, R.P. The Endoplasmic Reticulum Chaperone Calnexin Is a NADPH Oxidase NOX4 Interacting Protein. J. Biol. Chem. 2016, 291, 7045–7059. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef]
- Tang, P.; Dang, H.; Huang, J.; Xu, T.; Yuan, P.; Hu, J.; Sheng, J.F. NADPH oxidase NOX4 is a glycolytic regulator through mROS-HIF1alpha axis in thyroid carcinomas. Sci. Rep. 2018, 8, 15897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gibhardt, C.S.; Will, T.; Stanisz, H.; Korbel, C.; Mitkovski, M.; Stejerean, I.; Cappello, S.; Pacheu-Grau, D.; Dudek, J.; et al. Redox signals at the ER-mitochondria interface control melanoma progression. EMBO J. 2019, e100871. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Pawlak, K.J.; Burak, W.E.; Perry, E.E.; Marshall, B.; Whittal, R.M.; Bose, H.S. Mitochondrial metabolic regulation by GRP78. Sci. Adv. 2017, 3, e1602038. [Google Scholar] [CrossRef] [PubMed]
- Claypool, S.M.; Koehler, C.M. The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 2012, 37, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schagger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [PubMed]
- Bustillo-Zabalbeitia, I.; Montessuit, S.; Raemy, E.; Basanez, G.; Terrones, O.; Martinou, J.C. Specific interaction with cardiolipin triggers functional activation of Dynamin-Related Protein 1. PLoS ONE 2014, 9, e102738. [Google Scholar] [CrossRef] [PubMed]
- Stepanyants, N.; Macdonald, P.J.; Francy, C.A.; Mears, J.A.; Qi, X.; Ramachandran, R. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol. Biol. Cell 2015, 26, 3104–3116. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Korytowski, W.; Basova, L.V.; Pilat, A.; Kernstock, R.M.; Girotti, A.W. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: Mechanistic implications for the intrinsic pathway of oxidative apoptosis. J. Biol. Chem. 2011, 286, 26334–26343. [Google Scholar] [CrossRef]
- Jalmar, O.; Francois-Moutal, L.; Garcia-Saez, A.J.; Perry, M.; Granjon, T.; Gonzalvez, F.; Gottlieb, E.; Ayala-Sanmartin, J.; Klosgen, B.; Schwille, P.; et al. Caspase-8 binding to cardiolipin in giant unilamellar vesicles provides a functional docking platform for bid. PLoS ONE 2013, 8, e55250. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef]
- Joseph, S.K.; Young, M.P.; Alzayady, K.; Yule, D.I.; Ali, M.; Booth, D.M.; Hajnoczky, G. Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 2018, 293, 17464–17476. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Camacho, P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. J. Cell Biol. 2004, 164, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Siwik, D.A.; Pimentel, D.R.; Morgan, R.J.; Biolo, A.; Tu, V.H.; Kang, Y.J.; Cohen, R.A.; Colucci, W.S. Cytosolic H2O2 mediates hypertrophy, apoptosis, and decreased SERCA activity in mice with chronic hemodynamic overload. Am. J. Physiol.-Heart Circ. Physiol. 2014, 306, H1453–H1463. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Malinouski, M.; Zhou, Y.; Belousov, V.V.; Hatfield, D.L.; Gladyshev, V.N. Hydrogen peroxide probes directed to different cellular compartments. PLoS ONE 2011, 6, e14564. [Google Scholar] [CrossRef] [PubMed]
- Soares Moretti, A.I.; Martins Laurindo, F.R. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch. Biochem. Biophys. 2017, 617, 106–119. [Google Scholar] [CrossRef]
- Matsusaki, M.; Kanemura, S.; Kinoshita, M.; Lee, Y.H.; Inaba, K.; Okumura, M. The Protein Disulfide Isomerase Family: From proteostasis to pathogenesis. Biochim. Biophys. Acta Gen. Subj. 2019. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta 2008, 1783, 535–548. [Google Scholar] [CrossRef]
- Caramelo, J.J.; Parodi, A.J. A sweet code for glycoprotein folding. FEBS Lett. 2015, 589, 3379–3387. [Google Scholar] [CrossRef]
- Gutierrez, T.; Simmen, T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Simmen, T. ER-luminal thiol/selenol-mediated regulation of Ca2+ signalling. Biochem. Soc. Trans. 2016, 44, 452–459. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shi, W.; Guo, Y.; Chai, Z. ERp57 modulates mitochondrial calcium uptake through the MCU. FEBS Lett. 2014, 588, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Benham, A.M. Endoplasmic Reticulum redox pathways: In sickness and in health. FEBS J. 2019, 286, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Graven, K.K.; Molvar, C.; Roncarati, J.S.; Klahn, B.D.; Lowrey, S.; Farber, H.W. Identification of protein disulfide isomerase as an endothelial hypoxic stress protein. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2002, 282, L996–L1003. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, Y.M.; Youn, S.W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Kim, J.E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Lu, H.; Li, C. Proapoptotic activities of protein disulfide isomerase (PDI) and PDIA3 protein, a role of the Bcl-2 protein Bak. J. Biol. Chem. 2015, 290, 8949–8963. [Google Scholar] [CrossRef]
- Nakato, R.; Ohkubo, Y.; Konishi, A.; Shibata, M.; Kaneko, Y.; Iwawaki, T.; Nakamura, T.; Lipton, S.A.; Uehara, T. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep. 2015, 5, 14812. [Google Scholar] [CrossRef]
- Hoffstrom, B.G.; Kaplan, A.; Letso, R.; Schmid, R.S.; Turmel, G.J.; Lo, D.C.; Stockwell, B.R. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat. Chem. Biol. 2010, 6, 900–906. [Google Scholar] [CrossRef]
- Manuel, A.M.; Walla, M.D.; Faccenda, A.; Martin, S.L.; Tanis, R.M.; Piroli, G.G.; Adam, J.; Kantor, B.; Mutus, B.; Townsend, D.M.; et al. Succination of Protein Disulfide Isomerase Links Mitochondrial Stress and Endoplasmic Reticulum Stress in the Adipocyte During Diabetes. Antioxid. Redox Signal. 2017, 27, 1281–1296. [Google Scholar] [CrossRef]
- Kimura, T.; Horibe, T.; Sakamoto, C.; Shitara, Y.; Fujiwara, F.; Komiya, T.; Yamamoto, A.; Hayano, T.; Takahashi, N.; Kikuchi, M. Evidence for mitochondrial localization of P5, a member of the protein disulphide isomerase family. J. Biochem. 2008, 144, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Tonohora, Y.; Goto, T.; Yamada, Y.; Miki, T.; Makino, H.; Miwa, M.; Komiya, T. Mitochondrial P5, a member of protein disulphide isomerase family, suppresses oxidative stress-induced cell death. J. Biochem. 2012, 152, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Ortiz-Sandoval, C.; Simmen, T. Redox dependence of endoplasmic reticulum (ER) Ca(2)(+) signaling. Hist. Histopat. 2014, 29, 543–552. [Google Scholar]
- Roth, D.; Lynes, E.; Riemer, J.; Hansen, H.G.; Althaus, N.; Simmen, T.; Ellgaard, L. A di-arginine motif contributes to the ER localization of the type I transmembrane ER oxidoreductase TMX4. Biochem. J. 2010, 425, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Gutierrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.H.; Lucchinetti, E.; et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Frand, A.R.; Kaiser, C.A. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell 1999, 4, 469–477. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Riemer, J.; Christensen, B.; Sorensen, E.S.; Ellgaard, L. A novel disulphide switch mechanism in Ero1alpha balances ER oxidation in human cells. EMBO J. 2008, 27, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Ramming, T.; Hansen, H.G.; Nagata, K.; Ellgaard, L.; Appenzeller-Herzog, C. GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radic. Biol. Med. 2014, 70, 106–116. [Google Scholar] [CrossRef]
- Ramming, T.; Okumura, M.; Kanemura, S.; Baday, S.; Birk, J.; Moes, S.; Spiess, M.; Jeno, P.; Berneche, S.; Inaba, K.; et al. A PDI-catalyzed thiol-disulfide switch regulates the production of hydrogen peroxide by human Ero1. Free Radic. Biol. Med. 2015, 83, 361–372. [Google Scholar] [CrossRef]
- Yoboue, E.D.; Rimessi, A.; Anelli, T.; Pinton, P.; Sitia, R. Regulation of Calcium Fluxes by GPX8, a Type-II Transmembrane Peroxidase Enriched at the Mitochondria-Associated Endoplasmic Reticulum Membrane. Antioxid. Redox Signal. 2017, 27, 583–595. [Google Scholar] [CrossRef]
- Marino, M.; Stoilova, T.; Giorgi, C.; Bachi, A.; Cattaneo, A.; Auricchio, A.; Pinton, P.; Zito, E. SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Hum. Mol. Genet. 2015, 24, 1843–1855. [Google Scholar] [CrossRef]
- Ushioda, R.; Miyamoto, A.; Inoue, M.; Watanabe, S.; Okumura, M.; Maegawa, K.I.; Uegaki, K.; Fujii, S.; Fukuda, Y.; Umitsu, M.; et al. Redox-assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc. Natl. Acad. Sci. USA 2016, 113, E6055–E6063. [Google Scholar] [CrossRef]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha Regulates Ca2+ Fluxes at the Endoplasmic Reticulum-Mitochondria Interface (MAM). Antioxid. Redox Signal. 2011. [Google Scholar] [CrossRef]
- Higo, T.; Hattori, M.; Nakamura, T.; Natsume, T.; Michikawa, T.; Mikoshiba, K. Subtype-Specific and ER Lumenal Environment-Dependent Regulation of Inositol 1,4,5-Trisphosphate Receptor Type 1 by ERp44. Cell 2005, 120, 85–98. [Google Scholar] [CrossRef]
- Princiotta, M.F.; Finzi, D.; Qian, S.B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef]
- Culic, O.; Gruwel, M.L.; Schrader, J. Energy turnover of vascular endothelial cells. Am. J. Physiol. 1997, 273, C205–C213. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; Garcia-Poyatos, C.; Martin-Garcia, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol. Cell 2019, 74, 877–890 e6. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Van Vliet, A.R.; Giordano, F.; Gerlo, S.; Segura, I.; Van Eygen, S.; Molenberghs, G.; Rocha, S.; Houcine, A.; Derua, R.; Verfaillie, T.; et al. The ER Stress Sensor PERK Coordinates ER-Plasma Membrane Contact Site Formation through Interaction with Filamin-A and F-Actin Remodeling. Mol. Cell 2017, 65, 885–899 e6. [Google Scholar] [CrossRef]
- Van der Harg, J.M.; van Heest, J.C.; Bangel, F.N.; Patiwael, S.; van Weering, J.R.; Scheper, W. The UPR reduces glucose metabolism via IRE1 signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 655–665. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jana, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernandez, E.; Sassano, M.L.; et al. Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. MITOL prevents ER stress-induced apoptosis by IRE1alpha ubiquitylation at ER-mitochondria contact sites. EMBO J. 2019, 38, e100999. [Google Scholar] [CrossRef]
- Son, S.M.; Byun, J.; Roh, S.E.; Kim, S.J.; Mook-Jung, I. Reduced IRE1alpha mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis. 2014, 5, e1188. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernandez-Cardenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef]
- Arruda, A.P.; Pers, B.M.; Parlakgul, G.; Guney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Moulis, M.; Grousset, E.; Faccini, J.; Richetin, K.; Thomas, G.; Vindis, C. The Multifunctional Sorting Protein PACS-2 Controls Mitophagosome Formation in Human Vascular Smooth Muscle Cells through Mitochondria-ER Contact Sites. Cells 2019, 8, 638. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef]
- Atkins, K.M.; Thomas, L.L.; Barroso-Gonzalez, J.; Thomas, L.; Auclair, S.; Yin, J.; Kang, H.; Chung, J.H.; Dikeakos, J.D.; Thomas, G. The multifunctional sorting protein PACS-2 regulates SIRT1-mediated deacetylation of p53 to modulate p21-dependent cell-cycle arrest. Cell Rep. 2014, 8, 1545–1557. [Google Scholar] [CrossRef]
- Aslan, J.E.; You, H.; Williamson, D.M.; Endig, J.; Youker, R.T.; Thomas, L.; Shu, H.; Du, Y.; Milewski, R.L.; Brush, M.H.; et al. Akt and 14-3-3 control a PACS-2 homeostatic switch that integrates membrane traffic with TRAIL-induced apoptosis. Mol. Cell 2009, 34, 497–509. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef]
- Wang, P.T.; Garcin, P.O.; Fu, M.; Masoudi, M.; St-Pierre, P.; Pante, N.; Nabi, I.R. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J. Cell Sci. 2015, 128, 2759–2765. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Xin, Y.; Wu, W.; Qu, J.; Wang, X.; Lei, S.; Yuan, L.; Liu, X. Inhibition of Mitofusin-2 Promotes Cardiac Fibroblast Activation via the PERK/ATF4 Pathway and Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2019, 2019, 3649808. [Google Scholar] [CrossRef]
- Munoz, J.P.; Ivanova, S.; Sanchez-Wandelmer, J.; Martinez-Cristobal, P.; Noguera, E.; Sancho, A.; Diaz-Ramos, A.; Hernandez-Alvarez, M.I.; Sebastian, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Papanicolaou, K.N.; Walsh, K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 20321–20332. [Google Scholar] [CrossRef]
- Yao, C.H.; Wang, R.; Wang, Y.; Kung, C.P.; Weber, J.D.; Patti, G.J. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 2019, 8. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef]
- Bernales, S.; Schuck, S.; Walter, P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy 2007, 3, 285–287. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Wilkinson, S. Emerging Principles of Selective ER Autophagy. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Osawa, T.; Kotani, T.; Kawaoka, T.; Hirata, E.; Suzuki, K.; Nakatogawa, H.; Ohsumi, Y.; Noda, N.N. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 2019, 26, 281–288. [Google Scholar] [CrossRef]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Cui, Y.; Parashar, S.; Zahoor, M.; Needham, P.G.; Mari, M.; Zhu, M.; Chen, S.; Ho, H.C.; Reggiori, F.; Farhan, H.; et al. A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science 2019, 365, 53–60. [Google Scholar] [CrossRef]
- Bockler, S.; Westermann, B. Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev. Cell 2014, 28, 450–458. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef]
- Tang, B.; Li, Q.; Zhao, X.H.; Wang, H.G.; Li, N.; Fang, Y.; Wang, K.; Jia, Y.P.; Zhu, P.; Gu, J.; et al. Shiga toxins induce autophagic cell death in intestinal epithelial cells via the endoplasmic reticulum stress pathway. Autophagy 2015, 11, 344–354. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef]
- Luhr, M.; Torgersen, M.L.; Szalai, P.; Hashim, A.; Brech, A.; Staerk, J.; Engedal, N. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J. Biol. Chem. 2019, 294, 8197–8217. [Google Scholar] [CrossRef]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232 e11. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Noack, J.; Bergmann, T.J.; Cebollero, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Solda, T.; et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Fregno, I.; Molinari, M. Endoplasmic reticulum turnover: ER-phagy and other flavors in selective and non-selective ER clearance. F1000Research 2018, 7, 454. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, R.M.; Grumati, P.; Garcia-Pardo, J.; Kalayil, S.; Covarrubias-Pinto, A.; Chen, W.; Kudryashev, M.; Dikic, I.; Hummer, G. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat. Commun. 2019, 10, 2370. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Forrester, A.; De Leonibus, C.; Grumati, P.; Fasana, E.; Piemontese, M.; Staiano, L.; Fregno, I.; Raimondi, A.; Marazza, A.; Bruno, G.; et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 2017, 6. [Google Scholar] [CrossRef]
- Lynes, E.M.; Simmen, T. Urban planning of the endoplasmic reticulum (ER): How diverse mechanisms segregate the many functions of the ER. Biochim. Biophys. Acta 2011, 1813, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Orso, G.; Pendin, D.; Liu, S.; Tosetto, J.; Moss, T.J.; Faust, J.E.; Micaroni, M.; Egorova, A.; Martinuzzi, A.; McNew, J.A.; et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 2009, 460, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Shibata, Y.; Zhu, P.P.; Voss, C.; Rismanchi, N.; Prinz, W.A.; Rapoport, T.A.; Blackstone, C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 2009, 138, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xiao, Y.; Chai, P.; Zheng, P.; Teng, J.; Chen, J. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol. 2019, 29, 846–855 e6. [Google Scholar] [CrossRef] [PubMed]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 2019, 74, 909–921 e6. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Ordureau, A.; Paulo, J.A.; Shoemaker, C.J.; Denic, V.; Harper, J.W. TEX264 Is an Endoplasmic Reticulum-Resident ATG8-Interacting Protein Critical for ER Remodeling during Nutrient Stress. Mol. Cell 2019, 74, 891–908 e10. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [PubMed]
- Andhavarapu, S.; Mubariz, F.; Arvas, M.; Bever, C., Jr.; Makar, T.K. Interplay between ER stress and autophagy: A possible mechanism in multiple sclerosis pathology. Exp. Mol. Pathol. 2019, 108, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. https://doi.org/10.3390/cells8091071
Fan Y, Simmen T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells. 2019; 8(9):1071. https://doi.org/10.3390/cells8091071
Chicago/Turabian StyleFan, Yuxiang, and Thomas Simmen. 2019. "Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism" Cells 8, no. 9: 1071. https://doi.org/10.3390/cells8091071
APA StyleFan, Y., & Simmen, T. (2019). Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells, 8(9), 1071. https://doi.org/10.3390/cells8091071