Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. CAR Constructs and Cloning
2.3. Viral Transfection of T Cells
2.4. T Cell Cytotoxicity
2.5. Binding Assay
2.6. Flow Cytometry
2.7. ELISA
2.8. RT-PCR, qPCR
2.9. Apoptosis Assay
2.10. Proliferation Assay
2.11. Imaging
2.12. Combination Therapy with Drugs and Blocking Antibody
2.13. Animal Study
2.14. Statistical Analysis
3. Results
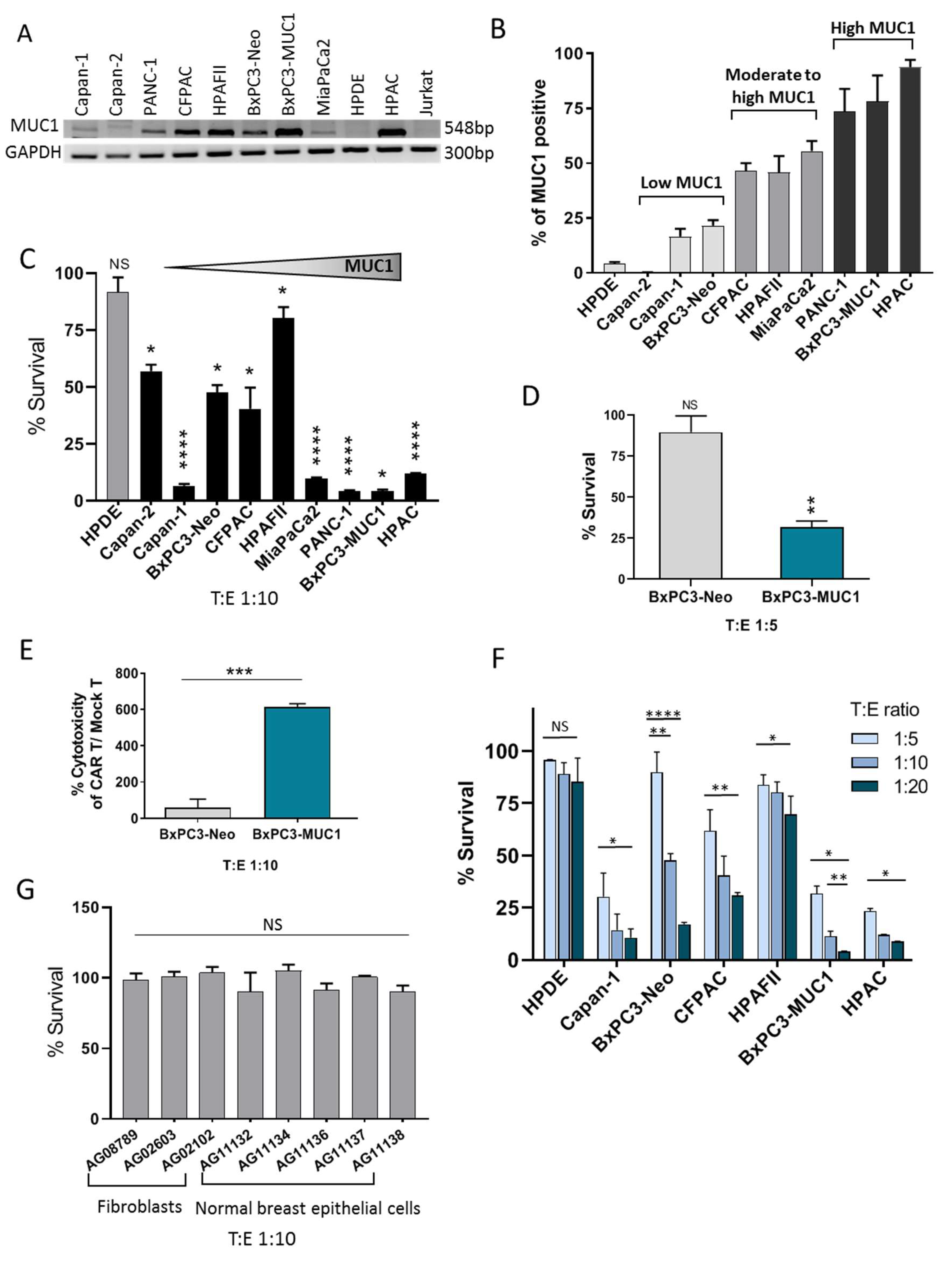
3.1. CAR Architecture, CAR Expression on Engineered T Cells, and Binding of CAR T Cells to Target PDA Cells
3.2. tMUC1-CAR T Cells Show Robust Cytotoxicity against PDA Cells but not Normal Cells
3.3. tMUC1-CAR T Cells Produce IFN-γ and Granzyme B upon Activation and Antigen Recognition
3.4. tMUC1-CAR T Cells Control Pancreatic Tumor Growth In Vivo
3.5. Deciphering the Intrinsic Resistance Mechanism Utilized by PDA Cells to CAR T Cell Therapy: Role of IDO1 and Gal-9
3.6. Battling the Resistance of PDA Cells with Combination Therapy
3.6.1. Targeting Resistance Related Genes with Small Molecule Inhibitors and Blocking Antibody
3.6.2. tMUC1-CAR T Cells Work Synergistically with Common Chemotherapy Drugs to Kill Resistant PDA Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Löhr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.-L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet (London, England) 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Yabar, C.S.; Winter, J.M. Pancreatic Cancer: A Review. Gastroenterol. Clin. N. Am. 2016, 45, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Swayden, M.; Iovanna, J.; Soubeyran, P. Pancreatic cancer chemo-resistance is driven by tumor phenotype rather than tumor genotype. Heliyon 2018, 4, e01055. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Yazdanifar, M.; Zhou, R.; Mukherjee, P. Emerging immunotherapeutics in adenocarcinomas: A focus on CAR-T cells. Curr. Trends Immunol. 2016, 17, 95–115. [Google Scholar]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef]
- Levi, E.; Klimstra, D.S.; Adsay, N.V.; Andea, A.; Basturk, O. MUC1 and MUC2 in pancreatic neoplasia. J. Clin. Pathol. 2004, 57, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Chhieng, D.C.; Benson, E.; Eltoum, I.; Eloubeidi, M.A.; Jhala, N.; Jhala, D.; Siegal, G.P.; Grizzle, W.E.; Manne, U. MUC1 and MUC2 expression in pancreatic ductal carcinoma obtained by fine-needle aspiration. Cancer 2003, 99, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.A.; Masri, A.A.; Adriance, M.C.; Tessier, J.C.; Kotlarczyk, K.L.; Thompson, M.C.; Gendler, S.J. MUC1 overexpression results in mammary gland tumorigenesis and prolonged alveolar differentiation. Oncogene 2004, 23, 5739–5747. [Google Scholar] [CrossRef] [PubMed]
- Sahraei, M.; Roy, L.D.; Curry, J.M.; Teresa, T.L.; Nath, S.; Besmer, D.; Kidiyoor, A.; Dalia, R.; Gendler, S.J.; Mukherjee, P. MUC1 regulates PDGFA expression during pancreatic cancer progression. Oncogene 2012, 31, 4935–4945. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Anurathapan, U.; Chan, R.C.; Hindi, H.F.; Mucharla, R.; Bajgain, P.; Hayes, B.C.; Fisher, W.E.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther. 2014, 22, 623–633. [Google Scholar] [CrossRef]
- You, F.; Jiang, L.; Zhang, B.; Lu, Q.; Zhou, Q.; Liao, X.; Wu, H.; Du, K.; Zhu, Y.; Meng, H.; et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci. China Life Sci. 2016, 59, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.D.; Dillon, L.M.; Zhou, R.; Moore, L.J.; Livasy, C.; El-Khoury, J.M.; Puri, R.; Mukherjee, P. A tumor specific antibody to aid breast cancer screening in women with dense breast tissue. Genes Cancer 2017, 8, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.J.; Roy, L.D.; Zhou, R.; Grover, P.; Wu, S.T.; Curry, J.M.; Dillon, L.M.; Puri, P.M.; Yazdanifar, M.; Puri, R.; et al. Antibody-Guided In Vivo Imaging for Early Detection of Mammary Gland Tumors. Transl. Oncol. 2016, 9, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Curry, J.M.; Roy, L.D.; Grover, P.; Haider, J.; Moore, L.J.; Wu, S.T.; Kamesh, A.; Yazdanifar, M.; Ahrens, W.A.; et al. A novel association of neuropilin-1 and MUC1 in pancreatic ductal adenocarcinoma: Role in induction of VEGF signaling and angiogenesis. Oncogene 2016, 35, 5608–5618. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.M.; Thompson, K.J.; Rao, S.G.; Besmer, D.M.; Murphy, A.M.; Grdzelishvili, V.Z.; Ahrens, W.A.; McKillop, I.H.; Sindram, D.; Iannitti, D.A.; et al. The use of a novel MUC1 antibody to identify cancer stem cells and circulating MUC1 in mice and patients with pancreatic cancer. J. Surg. Oncol. 2013, 107, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Das Roy, L.; Zhou, R.; Moore, L.J.; Dillon, L.M.; Puri, R.; Lyerly, K.; Marks, J.R.; Mukherjee, P. Early detection of breast cancer using a unique tumor specific antibody. J. Clin. Oncol. 2015, 33, abstr–e22153. [Google Scholar] [CrossRef]
- Wu, S.-T.; Fowler, A.J.; Garmon, C.B.; Fessler, A.B.; Ogle, J.D.; Grover, K.R.; Allen, B.C.; Williams, C.D.; Zhou, R.; Yazdanifar, M.; et al. Treatment of pancreatic ductal adenocarcinoma with tumor antigen specific-targeted delivery of paclitaxel loaded PLGA nanoparticles. BMC Cancer 2018, 18, 457. [Google Scholar] [CrossRef] [PubMed]
- Dréau, D.; Moore, L.J.; Alvarez-Berrios, M.P.; Tarannum, M.; Mukherjee, P.; Vivero-Escoto, J.L. Mucin-1-antibody-conjugated mesoporous silica nanoparticles for selective breast cancer detection in a mucin-1 transgenic murine mouse model. J. Biomed. Nanotechnol. 2016, 12, 2172–2184. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, I.; Cattarino, S.; Giantulli, S.; Nazzari, C.; Collalti, G.; Sciarra, A. A perspective of immunotherapy for prostate cancer. Cancers 2016, 8, 64. [Google Scholar] [CrossRef]
- Delitto, D.; Wallet, S.M.; Hughes, S.J. Targeting tumor tolerance: A new hope for pancreatic cancer therapy? Pharmacol. Therapeut. 2016, 166, 9–29. [Google Scholar] [CrossRef]
- Whilding, L.M.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Petrovic, R.M.G.; Kao, Y.V.; Saxena, S.A.; Romain, A.; Costa-Guerra, J.A.; Violette, S.; et al. Targeting of aberrant alphavbeta6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol. Ther. 2017, 25, 259–273. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sutton, V.R.; Sedelies, K.A.; Ciccone, A.; Jenkins, M.; Turner, S.J.; Bird, P.I.; Trapani, J.A. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J. Cell Biol. 2006, 173, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.K.; Lockhart, D.C.; von Bernstorff, W.; Spanjaard, R.A.; Joo, H.G.; Eberlein, T.J.; Goedegebuure, P.S. Soluble MUC1 secreted by human epithelial cancer cells mediates immune suppression by blocking T-cell activation. Int. J. Cancer 1999, 82, 721–726. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, S.; Mori, H.; Kobara, H.; Rafiq, K.; Niki, T.; Hirashima, M.; Masaki, T. Galectin-9 in cancer therapy. Recent Pat. Endocr. Metab. Immune Drug Discov. 2013, 7, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commum. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Front. Immunol. 2015, 6, 34. [Google Scholar] [CrossRef]
- Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolusic, E.; Frederick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc. Natl. Acad. Sci. USA 2012, 109, 2497–2502. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Thaker, A.I.; Rao, M.S.; Bishnupuri, K.S.; Kerr, T.A.; Foster, L.; Marinshaw, J.M.; Newberry, R.D.; Stenson, W.F.; Ciorba, M.A. IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology 2013, 145, 416–425. [Google Scholar] [CrossRef]
- Nagahara, K.; Arikawa, T.; Oomizu, S.; Kontani, K.; Nobumoto, A.; Tateno, H.; Watanabe, K.; Niki, T.; Katoh, S.; Miyake, M.; et al. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J. Immunol. 2008, 181, 7660. [Google Scholar] [CrossRef]
- Golden-Mason, L.; McMahan, R.H.; Strong, M.; Reisdorph, R.; Mahaffey, S.; Palmer, B.E.; Cheng, L.; Kulesza, C.; Hirashima, M.; Niki, T.; et al. Galectin-9 functionally impairs natural killer cells in humans and mice. J. Virol. 2013, 87, 4835. [Google Scholar] [CrossRef]
- Goncalves Silva, I.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Siligardi, G.; Ceccone, G.; et al. The Tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine 2017, 22, 44–57. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.-W.; Dutta, A.; Chang, L.-Y.; Mahalingam, J.; Lin, Y.-C.; Chiang, J.-M.; Hsu, C.-Y.; Huang, C.-T.; Su, W.-T.; Chu, Y.-Y.; et al. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci. Rep. 2015, 5, 15659. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- Galore-Haskel, G.; Nemlich, Y.; Greenberg, E.; Ashkenazi, S.; Hakim, M.; Itzhaki, O.; Shoshani, N.; Shapira-Fromer, R.; Ben-Ami, E.; Ofek, E.; et al. A novel immune resistance mechanism of melanoma cells controlled by the ADAR1 enzyme. Oncotarget 2015, 6, 28999–29015. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.Y.; Hurst, E.A.; Argyle, D.J. Cyclooxygenase-2: A role in cancer stem cell survival and repopulation of cancer cells during therapy. Stem Cells Int. 2016, 2016, 2048731. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. J. Cell Biol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Koike, N.; Pilon-Thomas, S.; Mule, J.J. Nonmyeloablative chemotherapy followed by T-cell adoptive transfer and dendritic cell-based vaccination results in rejection of established melanoma. J. Immunother. 2008, 31, 402–412. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yazdanifar, M.; Zhou, R.; Grover, P.; Williams, C.; Bose, M.; Moore, L.J.; Wu, S.-t.; Maher, J.; Dreau, D.; Mukherjee, P. Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 1070. https://doi.org/10.3390/cells8091070
Yazdanifar M, Zhou R, Grover P, Williams C, Bose M, Moore LJ, Wu S-t, Maher J, Dreau D, Mukherjee P. Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells. 2019; 8(9):1070. https://doi.org/10.3390/cells8091070
Chicago/Turabian StyleYazdanifar, Mahboubeh, Ru Zhou, Priyanka Grover, Chandra Williams, Mukulika Bose, Laura J. Moore, Shu-ta Wu, John Maher, Didier Dreau, and Pinku Mukherjee. 2019. "Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma" Cells 8, no. 9: 1070. https://doi.org/10.3390/cells8091070
APA StyleYazdanifar, M., Zhou, R., Grover, P., Williams, C., Bose, M., Moore, L. J., Wu, S.-t., Maher, J., Dreau, D., & Mukherjee, P. (2019). Overcoming Immunological Resistance Enhances the Efficacy of a Novel Anti-tMUC1-CAR T Cell Treatment against Pancreatic Ductal Adenocarcinoma. Cells, 8(9), 1070. https://doi.org/10.3390/cells8091070