EGFR–c-Src-Mediated HDAC3 Phosphorylation Exacerbates Invasion of Breast Cancer Cells

 , , ,
, , ,
Abstract
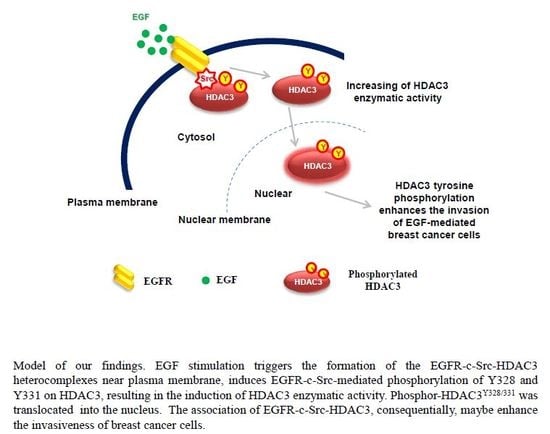
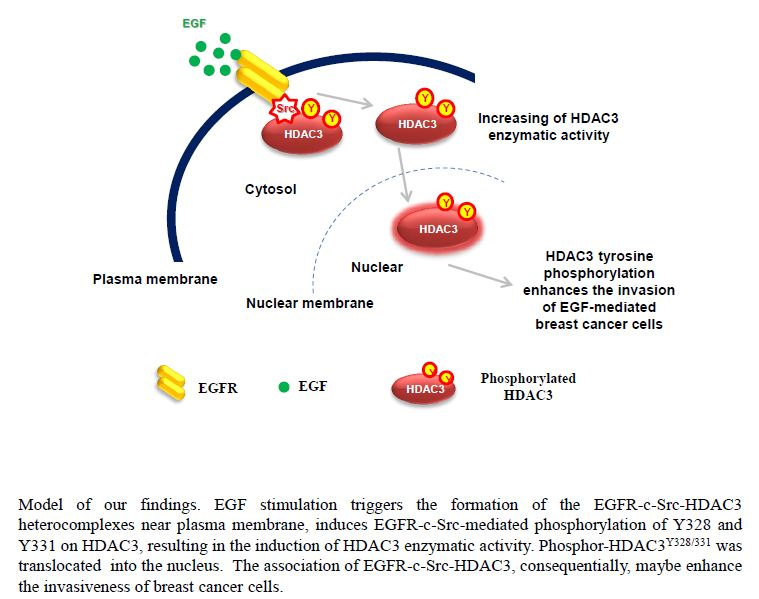
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Transfections
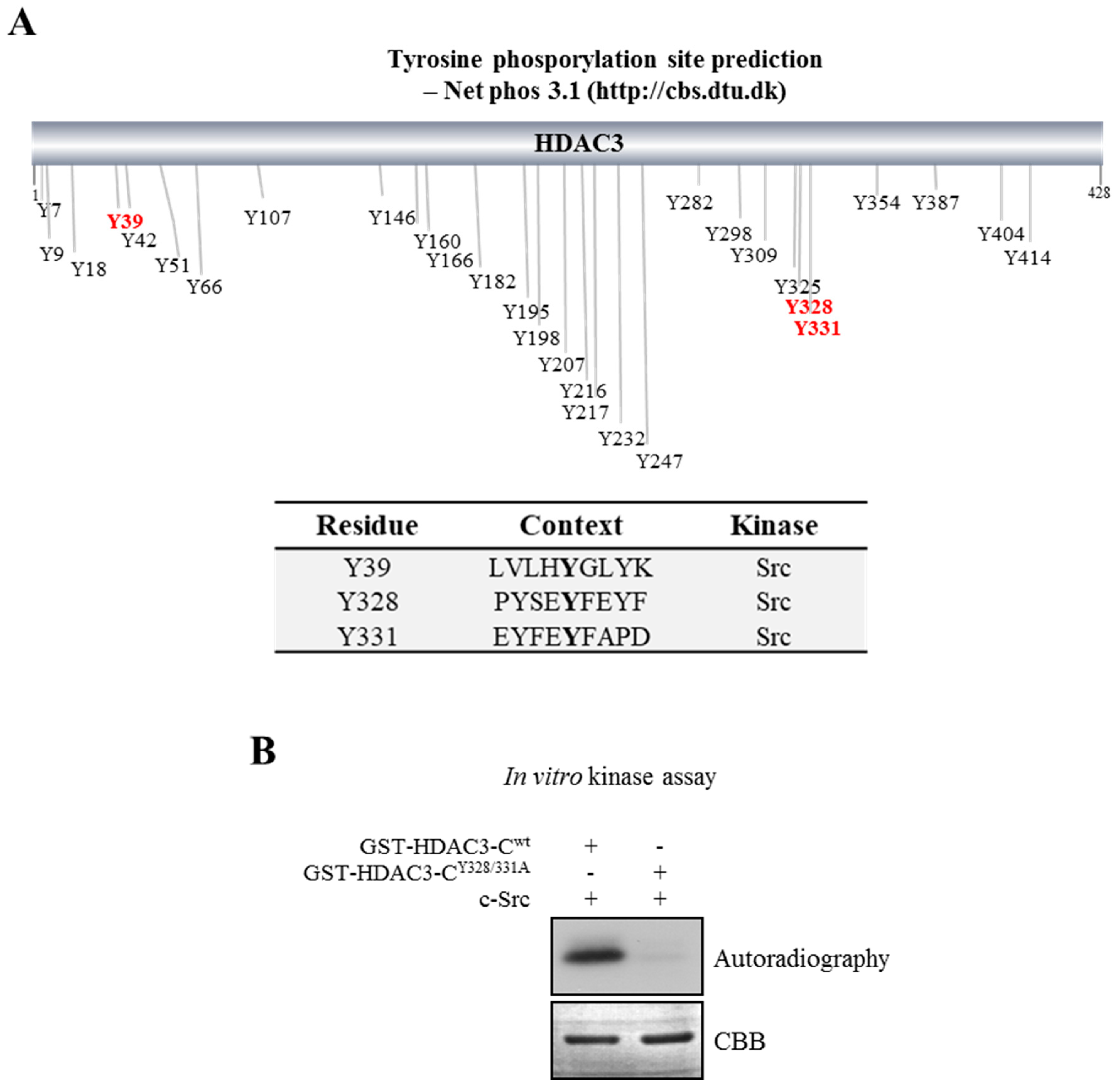
2.3. Prediction of Tyrosine Phosphorylation of HDAC3 and in vitro Kinase Assay
2.4. Western Blot Analysis and Antibodies
2.5. Immunoprecipitation (IP) Assay
2.6. siRNA and Transfection
2.7. Total Internal Reflection Fluorescence Microscopy (TIRF-M)
2.8. Cell Fractionation
2.9. Immunofluorescence Analysis
2.10. Assay for HDAC3 Activity
2.11. Matrigel Invasion Assays
2.12. Lentiviral shRNAs
2.13. Statistical Analysis
3. Results
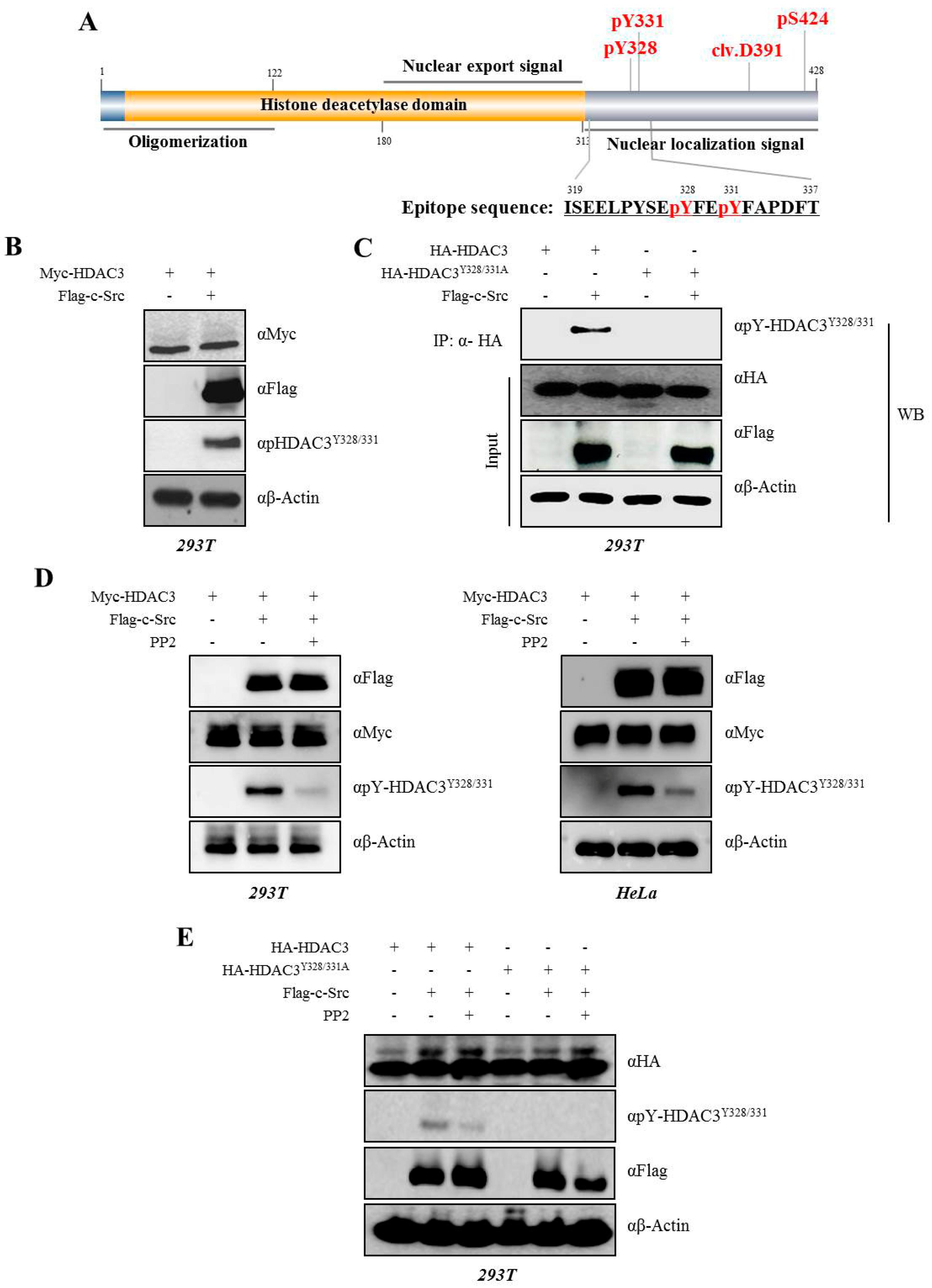
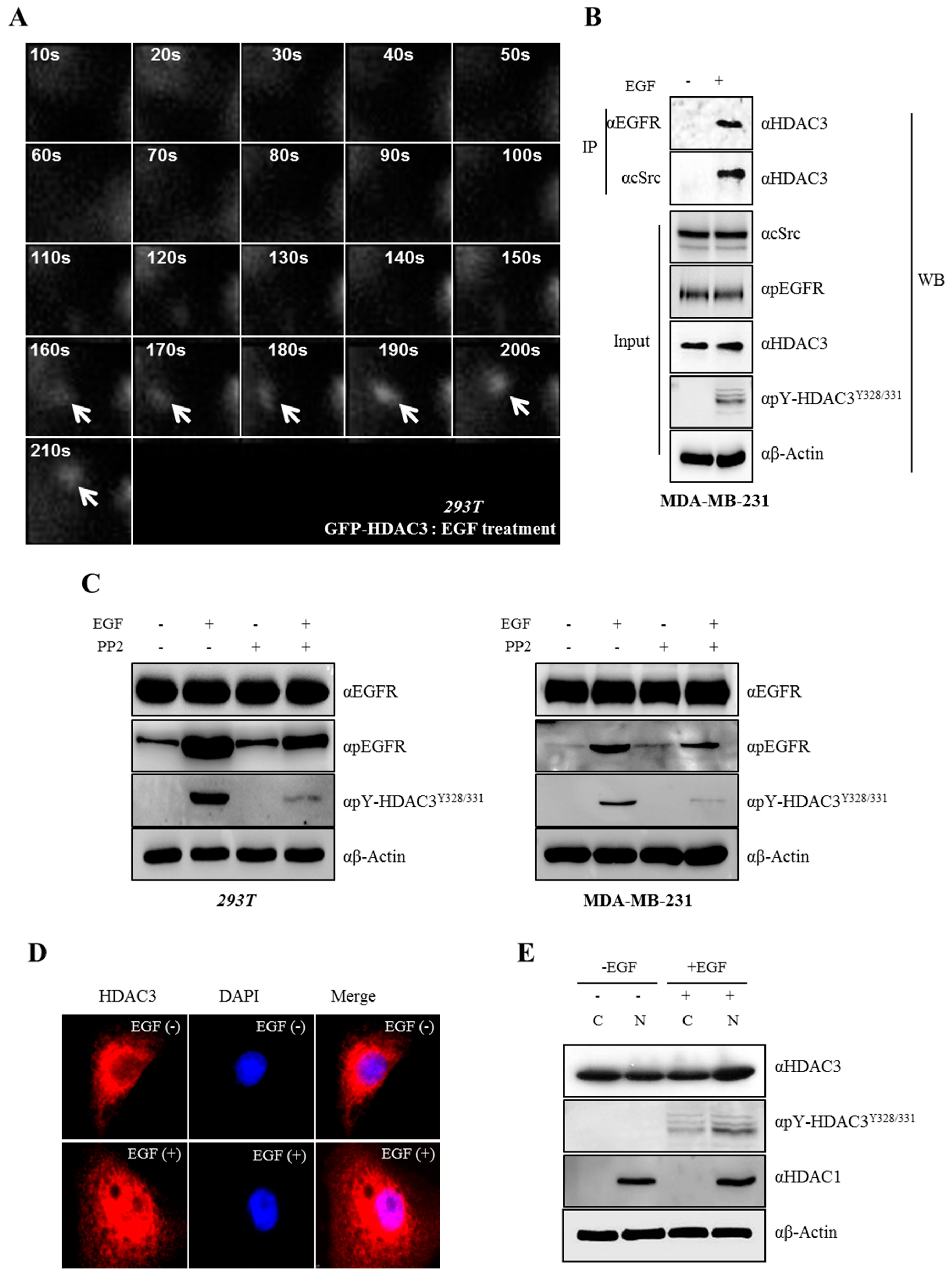
3.1. Phosphorylation of HDAC3 at Tyrosine
3.2. c-Src Phosphorylates Y328 and Y331 on HDAC3
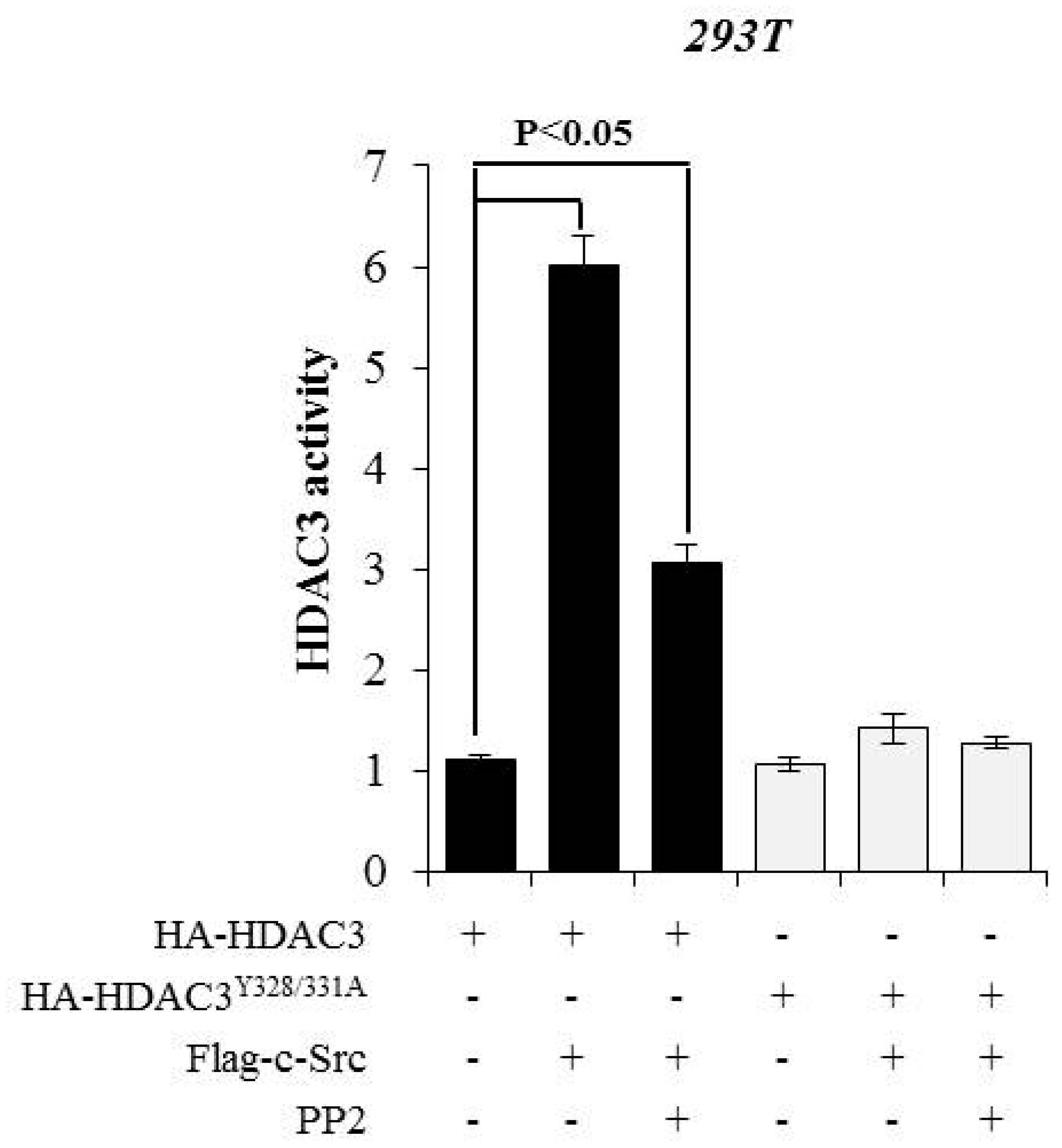
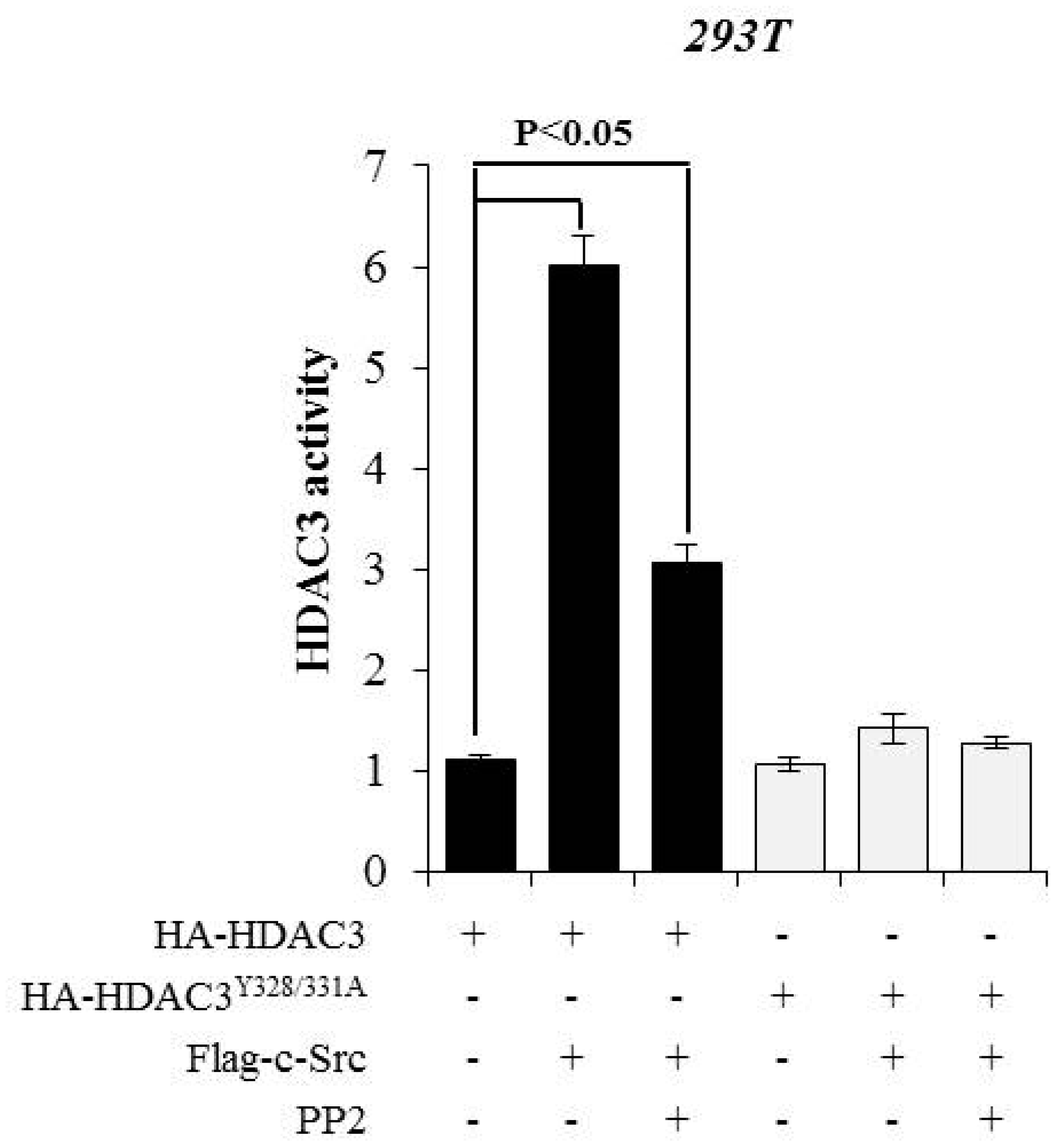
3.3. c-Src-mediated Phosphorylation of HDAC3 at Y328 and Y331 Alters HDAC3 Enzymatic Activity
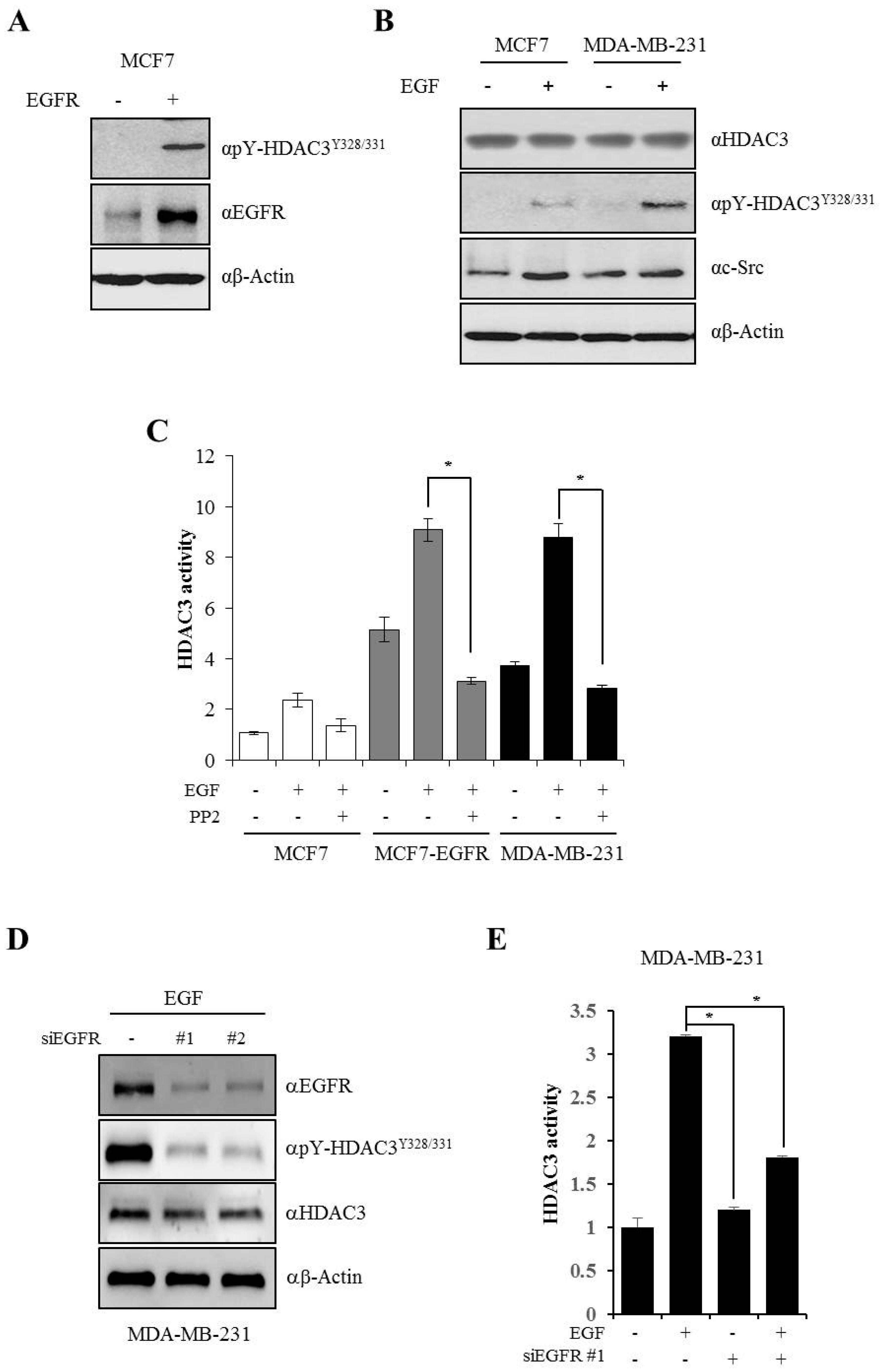
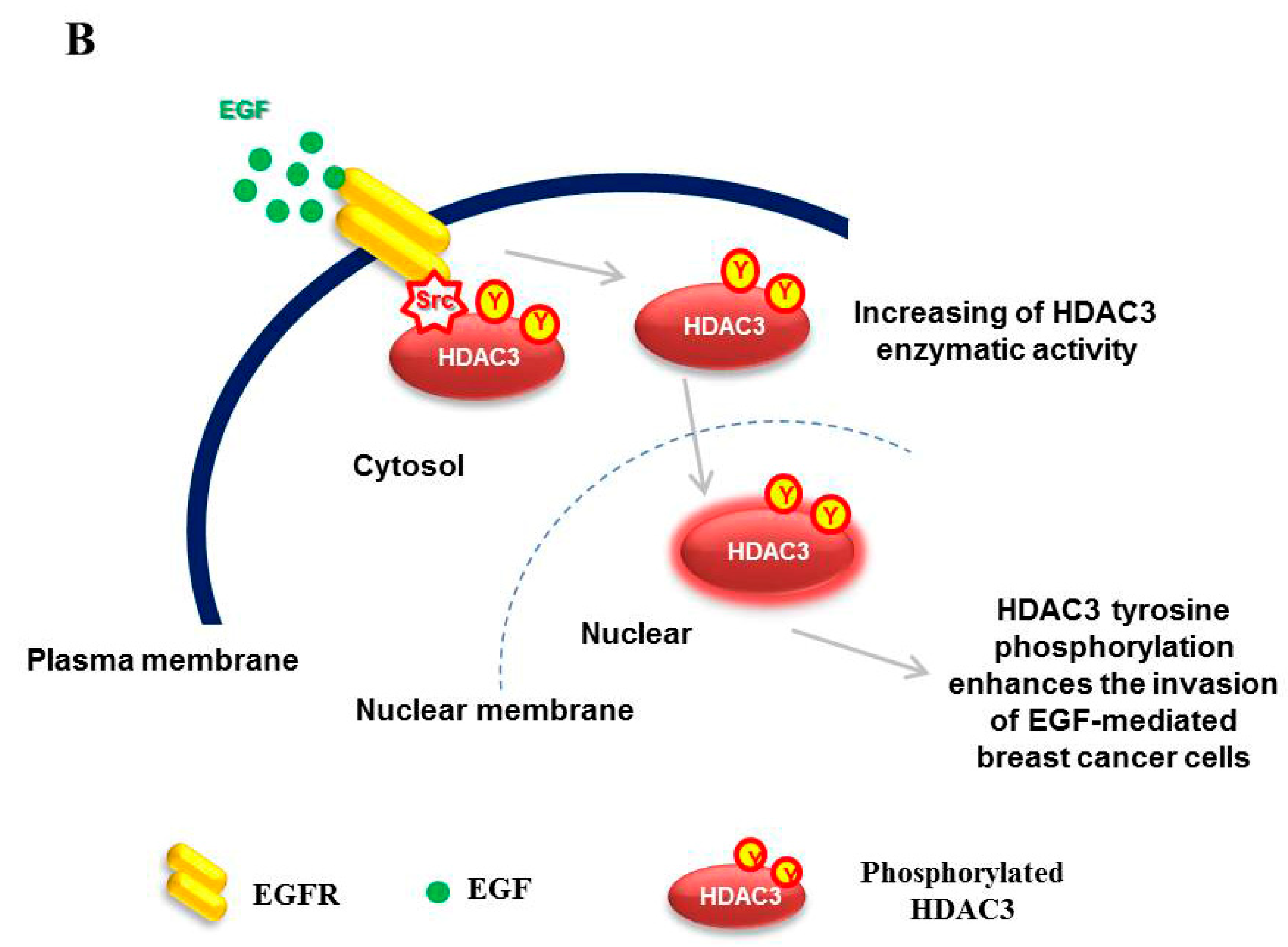
3.4. EGFR is Essential for c-Src-mediated HDAC3 Phosphorylation
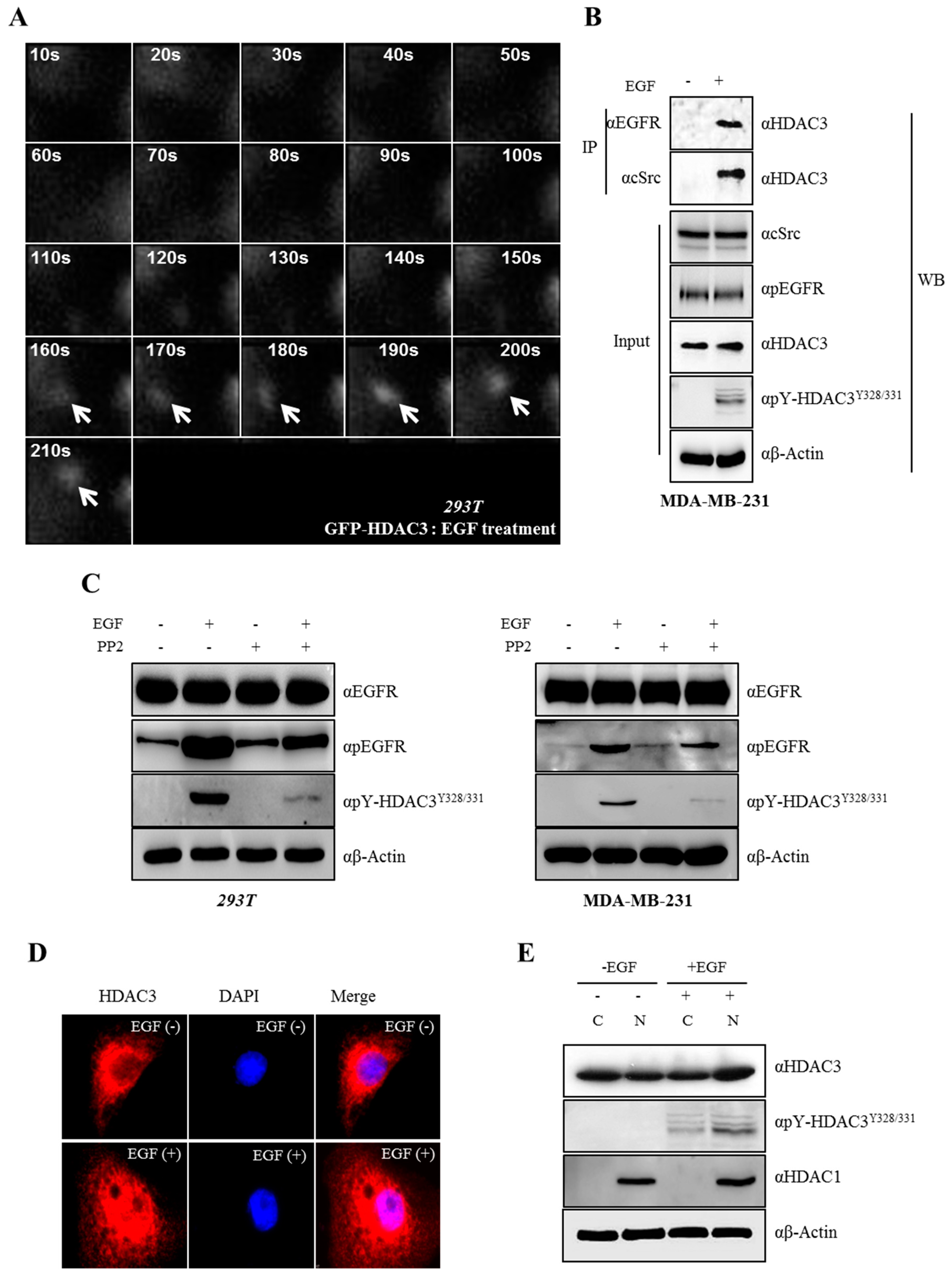
3.5. EGF-Induced Phosphorylation of HDAC3 Affects Its Localization
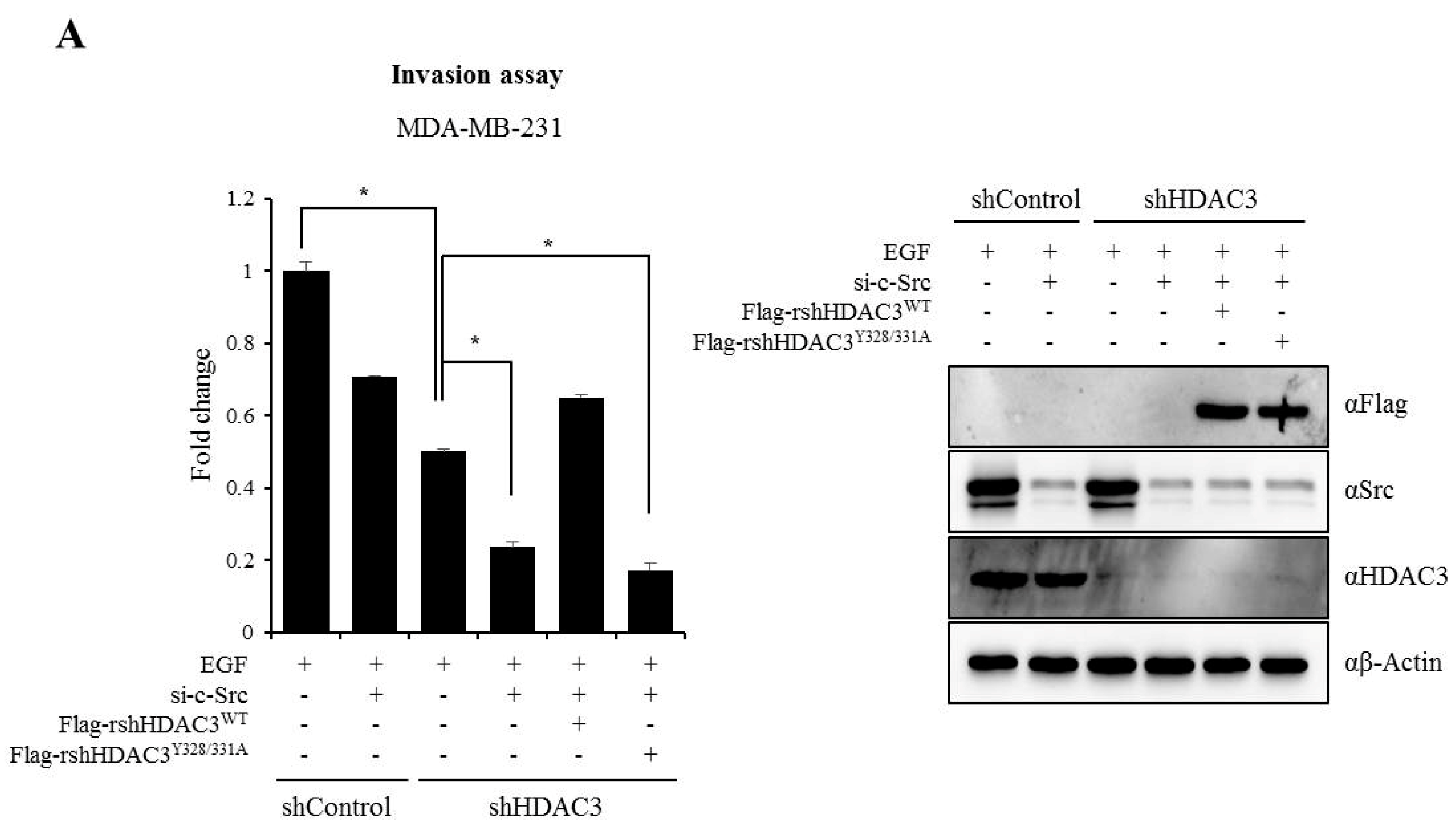
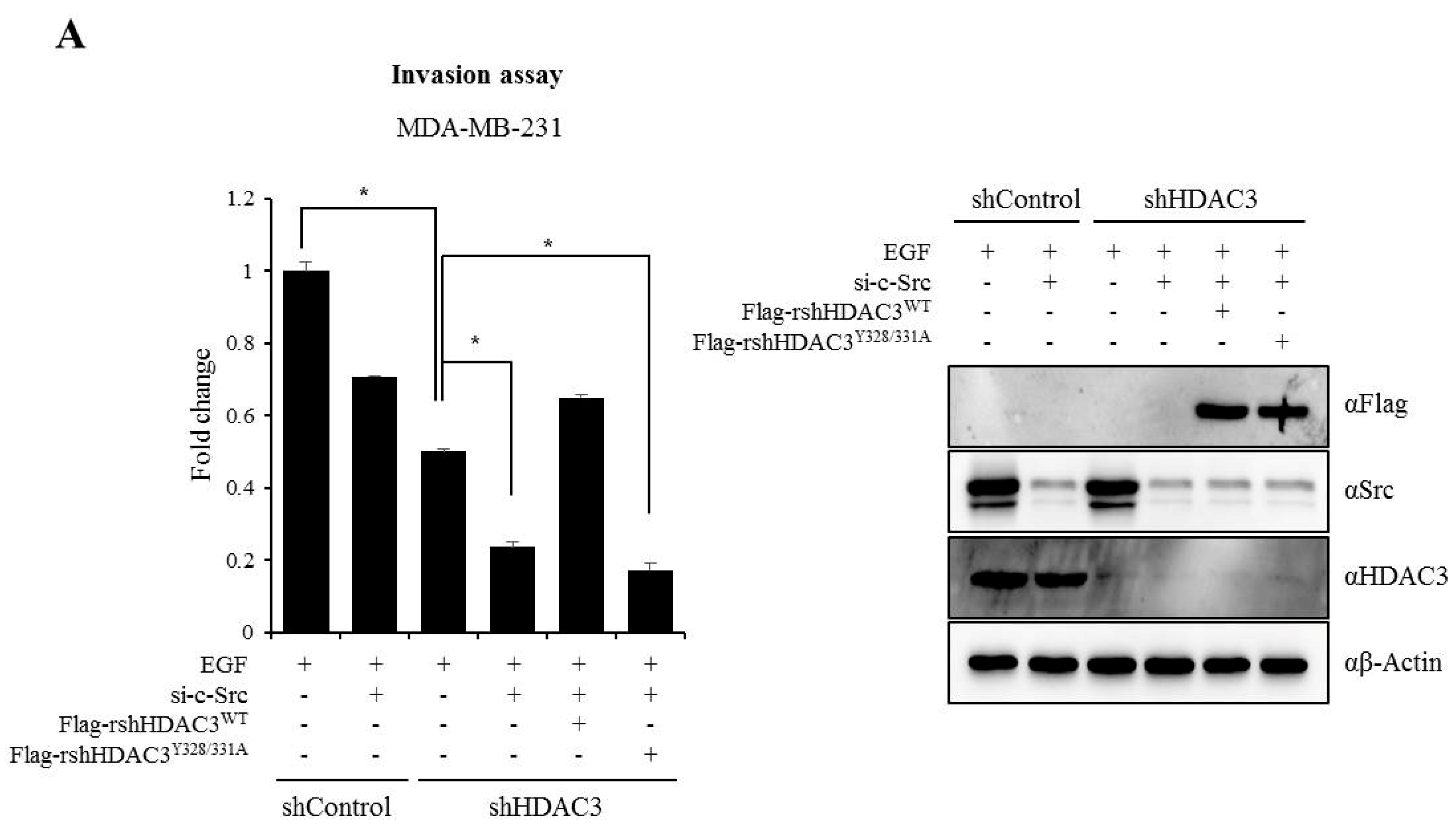
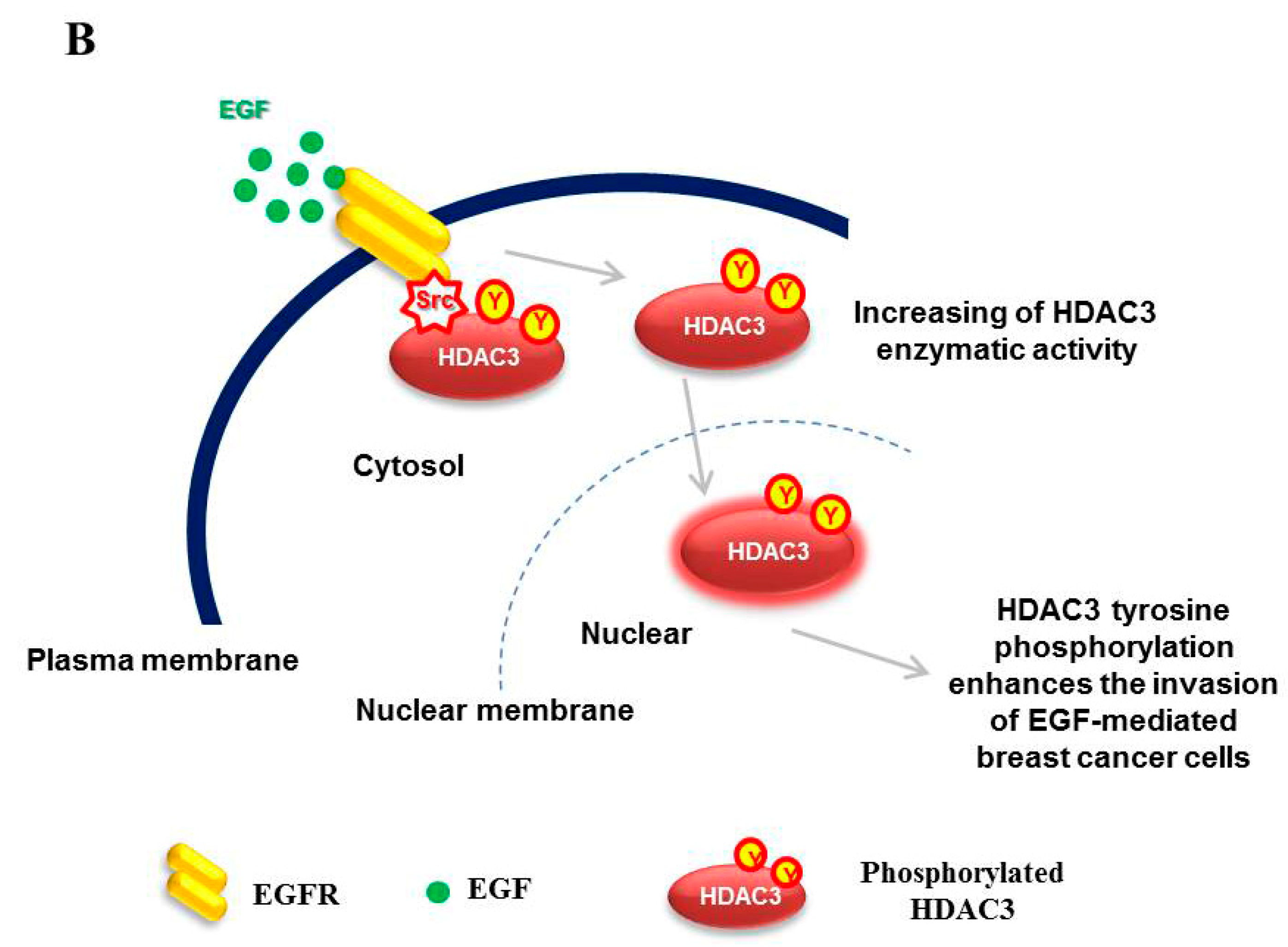
3.6. EGFR–c-Scr-mediated HDAC3 Phosphorylation Is Crucial for the Invasion of Breast Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libson, S.; Lippman, M. A review of clinical aspects of breast cancer. Int. Rev. Psychiatry 2014, 26, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Meric, F.; Lee, W.P.; Sahin, A.; Zhang, H.; Kung, H.J.; Hung, M.C. Expression profile of tyrosine kinases in breast cancer. Clin. Cancer Res. 2002, 8, 361–367. [Google Scholar] [PubMed]
- Hynes, N.E. Tyrosine kinase signaling in breast cancer. Breast Cancer Res. 2000, 2, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Biscardi, J.S.; Ishizawar, R.C.; Silva, C.M.; Parsons, S.J. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000, 2, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Kairouz, R.; Daly, R.J. Tyrosine kinase signaling in breast cancer: Modulation of tyrosine kinase signalling in human breast cancer through altered expression of signalling intermediates. Breast Cancer Res. 2000, 2, 197–202. [Google Scholar] [CrossRef]
- Chen, W.S.; Lazar, C.S.; Poenie, M.; Tsien, R.Y.; Gill, G.N.; Rosenfeld, M.G. Requirement for intrinsic protein tyrosine kinase in the immediate and late actions of the EGF receptor. Nature 1987, 328, 820–823. [Google Scholar] [CrossRef]
- Belsches-Jablonski, A.P.; Biscardi, J.S.; Peavy, D.R.; Tice, D.A.; Romney, D.A.; Parsons, S.J. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene 2001, 20, 1465–1475. [Google Scholar] [CrossRef] [Green Version]
- Maa, M.C.; Leu, T.H.; McCarley, D.J.; Schatzman, R.C.; Parsons, S.J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc. Natl. Acad. Sci. USA 1995, 92, 6981–6985. [Google Scholar] [CrossRef]
- Muthuswamy, S.K.; Siegel, P.M.; Dankort, D.L.; Webster, M.A.; Muller, W.J. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol. Cell. Biol. 1994, 14, 735–743. [Google Scholar] [CrossRef]
- Osherov, N.; Levitzki, A. Epidermal-growth-factor-dependent activation of the src-family kinases. Eur. J. Biochem. 1994, 225, 1047–1053. [Google Scholar] [CrossRef]
- Boerner, J.L.; Demory, M.L.; Silva, C.; Parsons, S.J. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol. Cell. Biol. 2004, 24, 7059–7071. [Google Scholar] [CrossRef]
- Irby, R.B.; Mao, W.; Coppola, D.; Kang, J.; Loubeau, J.M.; Trudeau, W.; Karl, R.; Fujita, D.J.; Jove, R.; Yeatman, T.J. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 1999, 21, 187–190. [Google Scholar] [CrossRef]
- Karagianni, P.; Wong, J. HDAC3: Taking the SMRT-N-CoRrect road to repression. Oncogene 2007, 26, 5439–5449. [Google Scholar] [CrossRef]
- Bartling, B.; Hofmann, H.S.; Boettger, T.; Hansen, G.; Burdach, S.; Silber, R.E.; Simm, A. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer 2005, 49, 145–154. [Google Scholar] [CrossRef]
- Cui, Z.; Xie, M.; Wu, Z.; Shi, Y. Relationship between Histone Deacetylase 3 (HDAC3) and Breast Cancer. Med. Sci. Monit. 2018, 24, 2456–2464. [Google Scholar] [CrossRef]
- Tsai, S.C.; Seto, E. Regulation of histone deacetylase 2 by protein kinase CK2. J. Biol. Chem. 2002, 277, 31826–31833. [Google Scholar] [CrossRef]
- Blom, N.; Sicheritz-Ponten, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef]
- Choi, H.K.; Choi, Y.; Kang, H.; Lim, E.J.; Park, S.Y.; Lee, H.S.; Park, J.M.; Moon, J.; Kim, Y.J.; Choi, I.; et al. PINK1 positively regulates HDAC3 to suppress dopaminergic neuronal cell death. Hum. Mol. Genet. 2015, 24, 1127–1141. [Google Scholar] [CrossRef]
- Choi, H.K.; Choi, Y.; Park, E.S.; Park, S.Y.; Lee, S.H.; Seo, J.; Jeong, M.H.; Jeong, J.H.; Lee, P.C.; Choi, K.C.; et al. Programmed cell death 5 mediates HDAC3 decay to promote genotoxic stress response. Nat. Commun. 2015, 6, 7390. [Google Scholar] [CrossRef]
- Zhang, X.; Ozawa, Y.; Lee, H.; Wen, Y.D.; Tan, T.H.; Wadzinski, B.E.; Seto, E. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 2005, 19, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Feng, D.; Fang, B.; Mullican, S.E.; You, S.H.; Lim, H.W.; Everett, L.J.; Nabel, C.S.; Li, Y.; Selvakumaran, V.; et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell 2013, 52, 769–782. [Google Scholar] [CrossRef]
- Lee, H.; Rezai-Zadeh, N.; Seto, E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 2004, 24, 765–773. [Google Scholar] [CrossRef]
- Pflum, M.K.; Tong, J.K.; Lane, W.S.; Schreiber, S.L. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J. Biol. Chem. 2001, 276, 47733–47741. [Google Scholar] [CrossRef]
- Velu, T.J.; Beguinot, L.; Vass, W.C.; Willingham, M.C.; Merlino, G.T.; Pastan, I.; Lowy, D.R. Epidermal-growth-factor-dependent transformation by a human EGF receptor proto-oncogene. Science 1987, 238, 1408–1410. [Google Scholar] [CrossRef]
- Biscardi, J.S.; Maa, M.C.; Tice, D.A.; Cox, M.E.; Leu, T.H.; Parsons, S.J. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 1999, 274, 8335–8343. [Google Scholar] [CrossRef]
- Emiliani, S.; Fischle, W.; Van Lint, C.; Al-Abed, Y.; Verdin, E. Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. USA 1998, 95, 2795–2800. [Google Scholar] [CrossRef]
- Krusche, C.A.; Wulfing, P.; Kersting, C.; Vloet, A.; Bocker, W.; Kiesel, L.; Beier, H.M.; Alfer, J. Histone deacetylase-1 and -3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res. Treat. 2005, 90, 15–23. [Google Scholar] [CrossRef]
- Stojanovic, N.; Hassan, Z.; Wirth, M.; Wensel, P.; Beyer, M.; Schafer, C.; Brand, P.; Kroemer, A.; Staber, R.H.; Arlt, A.; et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 2017, 36, 1804–1815. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef]
- Woo, Y.M. Epigenetic Regulation in Cystogenesis. Adv. Exp. Med. Biol. 2016, 933, 59–68. [Google Scholar]
- Cai, R.; Kwon, P.; Yan-Neale, Y.; Sambuccetti, L.; Fischer, D.; Cohen, D. Mammalian histone deacetylase 1 protein is posttranslationally modified by phosphorylation. Biochem. Biophys. Res. Commun. 2001, 283, 445–453. [Google Scholar] [CrossRef]
- Myoui, A.; Nishimura, R.; Williams, P.J.; Hirage, T.; Tamura, D.; Michigami, T.; Mundy, G.R.; Yoneda, T. C-SRC tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003, 63, 5028–5033. [Google Scholar]
- Hsieh, H.Y.; Chuang, H.C.; Shen, F.H.; Detroja, K.; Hsin, L.W.; Chen, C.S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, X.; Tan, H.; Lu, Y.; Tao, D.; Liu, Y.; Ma, Y. PIWIL2 suppresses Siah2-mediated degradation of HDAC3 and facilitates CK2alpha-mediated HDAC3 phosphorylation. Cell Death Dis. 2018, 9, 423. [Google Scholar] [CrossRef]
- Longworth, M.S.; Laimins, L.A. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene 2006, 25, 4495–4500. [Google Scholar] [CrossRef] [Green Version]
- Nardozzi, J.D.; Lott, K.; Cingolani, G. Phosphorylation meets nuclear import: A review. Cell Commun. Signal. 2010, 8, 32. [Google Scholar] [CrossRef]
- Kitamura, R.; Sekimoto, T.; Ito, S.; Harada, S.; Yamagata, H.; Masai, H.; Yoneda, Y.; Yanagi, K. Nuclear import of Epstein-Barr virus nuclear antigen 1 mediated by NPI-1 (Importin alpha5) is up- and down-regulated by phosphorylation of the nuclear localization signal for which Lys379 and Arg380 are essential. J. Virol. 2006, 80, 1979–1991. [Google Scholar] [CrossRef]
- Kann, M.; Sodeik, B.; Vlachou, A.; Gerlich, W.H.; Helenius, A. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J. Cell Biol. 1999, 145, 45–55. [Google Scholar] [CrossRef]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef] [Green Version]
- Shuai, K.; Stark, G.R.; Kerr, I.M.; Darnell, J.E. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 1993, 261, 1744–1746. [Google Scholar] [CrossRef]
- Wenta, N.; Strauss, H.; Meyer, S.; Vinkemeier, U. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc. Natl. Acad. Sci. USA 2008, 105, 9238–9243. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, K.; Terasawa, K.; Morimoto, H.; Nishida, E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol. Cell. Biol. 2006, 26, 1679–1690. [Google Scholar] [CrossRef]
- Chuderland, D.; Konson, A.; Seger, R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol. Cell 2008, 31, 850–861. [Google Scholar] [CrossRef]
- Hill, C.S. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009, 19, 36–46. [Google Scholar] [CrossRef]
- Smillie, D.A.; Llinas, A.J.; Ryan, J.T.; Kemp, G.D.; Sommerville, J. Nuclear import and activity of histone deacetylase in Xenopus oocytes is regulated by phosphorylation. J. Cell Sci. 2004, 117, 1857–1866. [Google Scholar] [CrossRef] [Green Version]
- Grozinger, C.M.; Schreiber, S.L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 2000, 97, 7835–7840. [Google Scholar] [CrossRef]
- Han, K.A.; Shin, W.H.; Jung, S.; Seol, W.; Seo, H.; Ko, C.; Chung, K.C. Leucine-rich repeat kinase 2 exacerbates neuronal cytotoxicity through phosphorylation of histone deacetylase 3 and histone deacetylation. Hum. Mol. Genet. 2017, 26, 1–18. [Google Scholar] [CrossRef]
- Wee, P.; Shi, H.; Jiang, J.; Wang, Y.; Wang, Z. EGF stimulates the activation of EGF receptors and the selective activation of major signaling pathways during mitosis. Cell Signal. 2015, 27, 638–651. [Google Scholar] [CrossRef]
- Abe, M.; Kuroda, Y.; Hirose, M.; Watanabe, Y.; Nakano, M.; Handa, T. Inhibition of autophosphorylation of epidermal growth factor receptor by small peptides in vitro. Br. J. Pharmacol. 2006, 147, 402–411. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [Green Version]
- Hanigan, T.W.; Aboukhatwa, S.M.; Taha, T.Y.; Frasor, J.; Petukhow, P.A. Divergent JNK Phosphorylation of HDAC3 in Triple-Negative Breast Cancer Cells Determines HDAC Inhibitor Binding and Selectivity. Cell Chem. Biol. 2017, 24, 1356–1367. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwak, S.-M.; Seo, J.; Hwang, J.-T.; Sung, G.-J.; Song, J.-H.; Jeong, J.-H.; Lee, S.-H.; Yoon, H.-G.; Choi, H.-K.; Choi, K.-C. EGFR–c-Src-Mediated HDAC3 Phosphorylation Exacerbates Invasion of Breast Cancer Cells. Cells 2019, 8, 930. https://doi.org/10.3390/cells8080930
Kwak S-M, Seo J, Hwang J-T, Sung G-J, Song J-H, Jeong J-H, Lee S-H, Yoon H-G, Choi H-K, Choi K-C. EGFR–c-Src-Mediated HDAC3 Phosphorylation Exacerbates Invasion of Breast Cancer Cells. Cells. 2019; 8(8):930. https://doi.org/10.3390/cells8080930
Chicago/Turabian StyleKwak, Sung-Min, Jaesung Seo, Jin-Taek Hwang, Gi-Jun Sung, Ji-Hye Song, Ji-Hoon Jeong, Seung-Hyun Lee, Ho-Geun Yoon, Hyo-Kyoung Choi, and Kyung-Chul Choi. 2019. "EGFR–c-Src-Mediated HDAC3 Phosphorylation Exacerbates Invasion of Breast Cancer Cells" Cells 8, no. 8: 930. https://doi.org/10.3390/cells8080930
APA StyleKwak, S.-M., Seo, J., Hwang, J.-T., Sung, G.-J., Song, J.-H., Jeong, J.-H., Lee, S.-H., Yoon, H.-G., Choi, H.-K., & Choi, K.-C. (2019). EGFR–c-Src-Mediated HDAC3 Phosphorylation Exacerbates Invasion of Breast Cancer Cells. Cells, 8(8), 930. https://doi.org/10.3390/cells8080930